1. Introduction
Water splitting over particulate semiconductor photocatalysts has been studied as a means of generating renewable hydrogen from water using solar energy on a large scale [
1]. However, the practicality of this process requires a solar-to-hydrogen energy conversion efficiency for photocatalytic water splitting of at least 5%. To meet this target, it will be necessary to develop narrow bandgap photocatalysts that can efficiently utilize visible light. Unfortunately, an oxide photocatalyst capable of splitting water into hydrogen and oxygen will typically have a bandgap equal to or greater than 3 eV, and so will not absorb visible light. This insufficient bandgap results from the valence band maximum, which is composed primarily of O 2
p orbitals, being located at an overly positive potential (approximately +3 V vs. a reversible hydrogen electrode, or RHE) with respect to the oxygen evolution potential (+1.23 V vs. RHE) [
2]. In contrast, certain transition metal-based semiconductor oxynitrides have a band gap narrow enough to absorb visible light while being highly chemically stable [
3]. Furthermore, such materials have a bandgap suitable for water splitting into hydrogen and oxygen and have been reported to exhibit activity during photocatalytic water splitting reactions based on one- and two-step excitation schemes [
4,
5]. In these materials, the valence band is mainly composed of hybridized O 2
p and N 2
p orbitals, while the conduction band primarily comprises
d orbitals of the constituent transition metals. Because N 2
p orbitals are located at more negative potentials than O 2
p orbitals, the top of the valence band for an oxynitride will be shifted to a more negative potential compared with that for the corresponding oxide. The potential of the conduction band is not significantly affected, but the narrowed bandgap reduces the energy offset between the band-edge potential and the redox potential for the reactants, such that the driving force for the water splitting reaction is also reduced. It is, therefore, essential to load particulate oxynitride photocatalysts with cocatalysts to enhance their photocatalytic activity by promoting charge separation in the bulk material and to enhance surface redox reactions [
6].
Here, BaTaO
2N is a perovskite-type semiconducting oxynitride with a bandgap energy of 1.9 eV. This bandgap allows for the absorption of visible light at wavelengths up to 650 nm [
7]. This material has been widely studied with regard to applications in photocatalytic water splitting systems and photoelectrochemical water oxidation reactions because it exhibits exceptional stability and has a bandgap that could permit overall water splitting and unassisted photoelectrochemical water oxidation under visible light [
8,
9,
10]. Indeed, it has been experimentally confirmed that the bandgap straddles the redox potentials for hydrogen evolution and oxygen evolution reactions from water [
11]. Recently, single-crystal BaTaO
2N particles with low defect densities have been synthesized by a flux method [
12]. When loaded with highly dispersed Pt nanoparticles serving as a reduction cocatalyst in such a way that there is intimate contact between the Pt nanoparticles and the photocatalyst, single-crystal particulate BaTaO
2N has been shown to evolve hydrogen from an aqueous methanol solution with an apparent quantum yield (AQY) of 6.8% at a wavelength of 420 nm [
13]. Furthermore, BaTaO
2N can also evolve oxygen from an aqueous AgNO
3 solution with an AQY of 0.55% at 420 nm when loaded with cobalt oxide (CoO
x) as an oxygen evolution cocatalyst [
14]. Doping of Mg into Ta sites in this compound has been reported to enhance the oxygen evolution activity of BaTaO
2N by lowering the density of reduced Ta species, positively shifting the valence band edge [
15]. A similar effect was observed by the present authors in a prior study [
16]. Substitution of Ca for Ta was also reported to improve the oxygen evolution activity of the resultant BaTaO
2N (BaCa
x/3Ta
1-x/3O
2+yN
1-y (0 ≤
x,
y ≤ 1)) [
17]. However, the CoO
x was loaded by impregnation followed by heating at a high temperature under either an inert or reducing atmosphere. This process seemingly enhanced the n-type semiconducting properties of the BaTaO
2N and, thus, decreased the hydrogen evolution activity of the material in our preliminary work. Consequently, it would be desirable to develop a means of loading oxygen evolution cocatalysts without the application of high temperatures.
Cobalt oxyhydroxide (CoOOH), intended for use as an oxygen evolution cocatalyst, has been loaded onto oxide photocatalysts, such as Al-doped SrTiO
3 and BiVO
4, by oxidative photodeposition, and has been found to enhance the oxygen evolution activities of these compounds [
18,
19]. This procedure does not involve heat treatment, but the oxidative photodeposition process requires the valence-band edge of the photocatalyst to be more positive than the redox potential of the cocatalyst (CoOOH/Co
2+ in the case of CoOOH). Therefore, it is difficult to effectively photodeposit CoOOH onto an oxynitride with a less positive valence-band edge, especially in the case of those compounds with absorption-edge wavelengths longer than 600 nm.
Among the various other oxygen evolution cocatalysts applicable to photocatalysts and photoanodes, iron oxyhydroxide (FeOOH) can be oxidatively deposited using Fe
2+ ions at a relatively negative potential [
20] and, thus, may be photodeposited onto narrow-bandgap materials, such as BaTaO
2N. In fact, FeOOH was previously photodeposited on an Al-doped SrTiO
3 photocatalyst and was found to promote the oxygen evolution activity of the photocatalyst [
21].
In the present work, methods for the photodeposition of Fe-based cocatalysts (FeOx) on particulate BaTaO2N and Mg-doped BaTaO2N (BaTaO2N:Mg), and the subsequent effects on photocatalytic oxygen evolution activity, were investigated. The prompt removal of photoexcited electrons from these BaTaO2N photocatalysts was determined to be essential for the effective photodeposition of FeOx cocatalysts and was also shown to improve the water oxidation activity.
2. Results and Discussion
The X-ray diffraction (XRD) patterns indicated that the major phase in both the BaTaO
2N and BaTaO
2N:Mg samples was BaTaO
2N, which has a cubic perovskite structure (
Figure S1) [
22]. The BaTaO
2N:Mg was also found to contain small amounts of Ta
3N
5 [
23] as a by-product, because the substitution of Mg for Ta generated an excess of Ta in the material [
16]. The diffuse-reflectance spectroscopy (DRS) data showed that the onset of light absorption by the undoped and Mg-doped BaTaO
2N occurred at approximately 660 and 620 nm, respectively (
Figure S1). The substitution of Mg for Ta in these specimens would be expected to be accompanied by the replacement of N
3− by O
2− to maintain charge neutrality, thus, lowering the nitrogen content in the perovskite-type oxynitride. As a result, the top of the valence band should be shifted positively, and the bandgap should broaden. These effects were previously confirmed to occur in undoped and Mg-doped BaTaO
2N by Zhang et al. using thermogravimetry and impedance spectroscopy [
15]. The undoped BaTaO
2N produced in the present work consisted of faceted particles several hundred nanometers in size with exposed smooth surfaces (
Figure S1). It is also evident that doping with Mg both reduced the particle size and roughened the particle surfaces. Suppression of particle growth and formation of characteristic morphology by doping have often been observed in perovskite-type oxides, such as NaTaO
3 [
24,
25,
26] and SrTiO
3 [
27]. Compared with specimens fabricated in prior studies, the BaTaO
2N:Mg produced in this work exhibited stronger absorption at wavelengths longer than the absorption edge in addition to more distorted morphologies [
16]. This could be ascribed to the deficiency of Ba species and to deviations from the desired temperature during the nitridation, because it was suppressed by adding additional Ba and adjusting the heating conditions. The effect of FeO
x photodeposition (as discussed below) was qualitatively reproducible regardless of variations in the BaTaO
2N:Mg products.
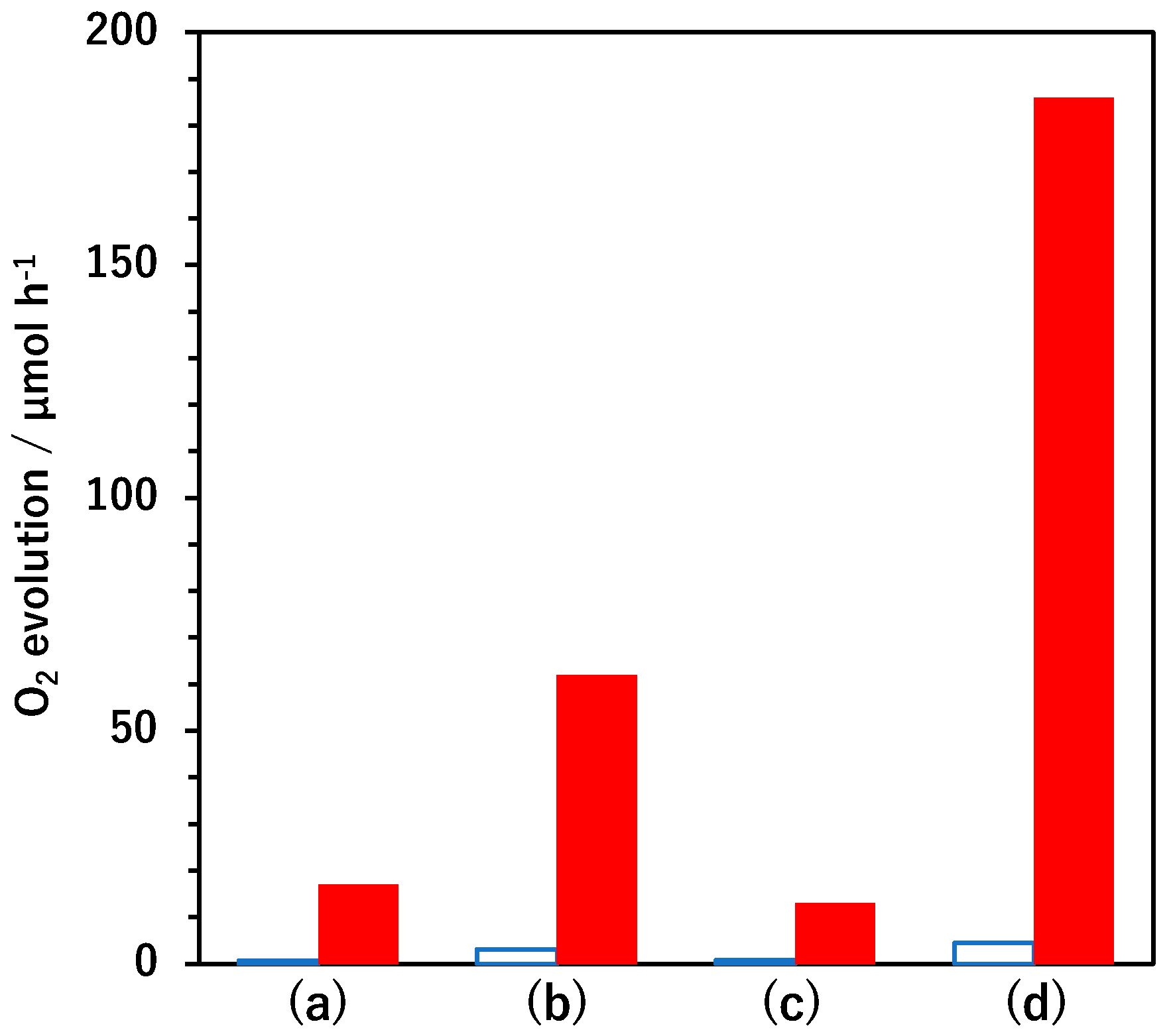
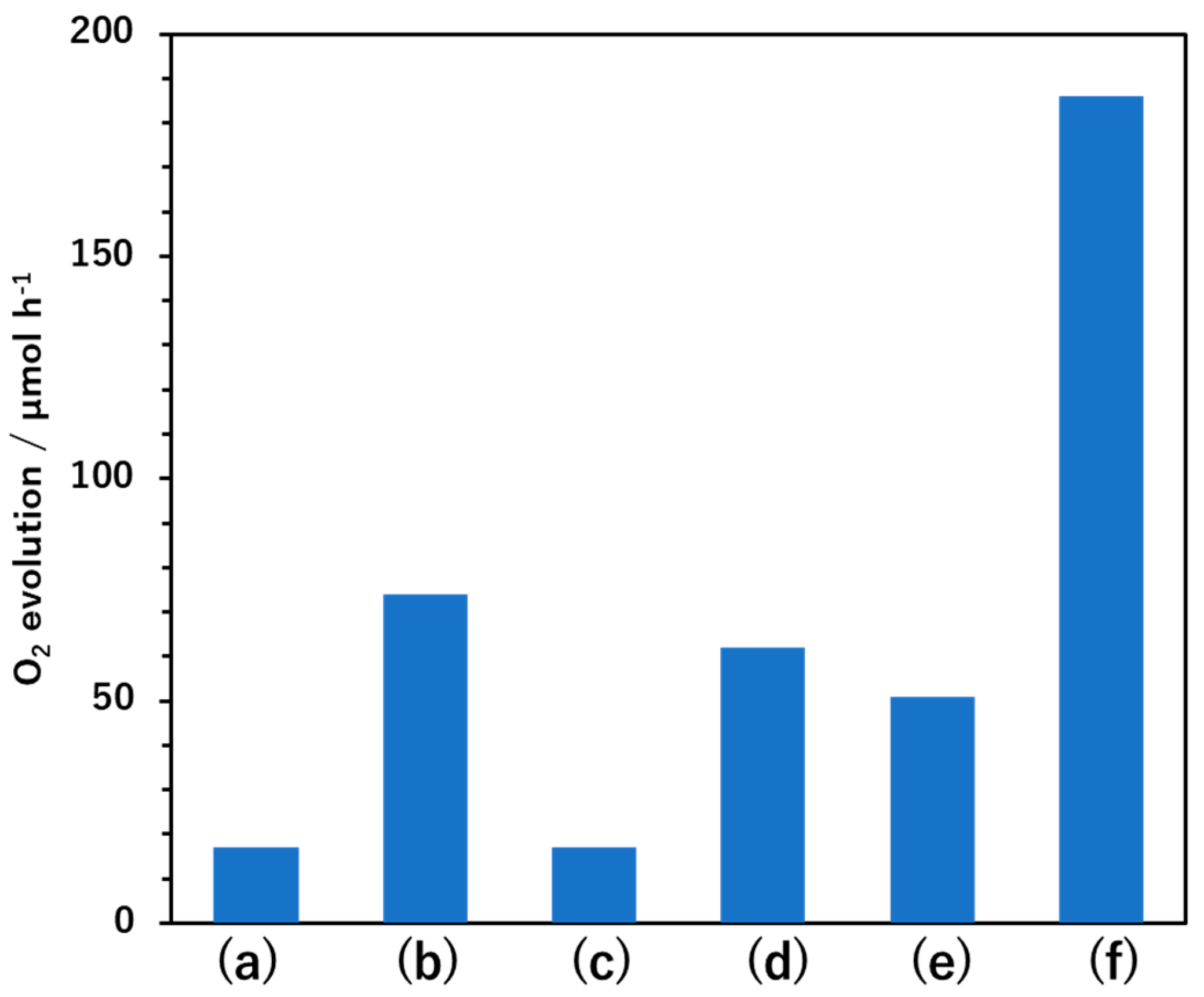
Figure 1 summarizes the initial oxygen evolution rates obtained using BaTaO
2N and BaTaO
2N:Mg loaded with FeO
x and Pt cocatalysts under various conditions in aqueous AgNO
3 solutions. The undoped BaTaO
2N showed a negligible oxygen evolution rate (<1 μmol h
−1), as did the specimen loaded with 0.1 wt% FeO
x by photodeposition. The material loaded with 0.1 wt% Pt by impregnation–reduction and that coloaded sequentially with and 0.1 wt% FeO
x by photodeposition were able to evolve oxygen, although the activity was still low. In contrast, the BaTaO
2N:Mg evolved oxygen at a higher rate even in the absence of the cocatalysts. This performance can possibly be ascribed to a positive shift in the valence band edge that resulted in a stronger driving force for holes involved in the oxygen evolution reaction [
16]. Loading the photocatalyst with Pt by impregnation–reduction increased the oxygen evolution rate by a factor of approximately three, presumably because Pt promoted the extraction of photoexcited electrons from the photocatalyst [
13]. Conversely, photodeposition of FeO
x alone did not appreciably affect the oxygen evolution rate. Notably, in the case that FeO
x was photodeposited after loading Pt by impregnation–reduction, the BaTaO
2N:Mg generated oxygen at a rate that exceeded the sum of the rates obtained for BaTaO
2N:Mg samples loaded with Pt by impregnation–reduction or FeO
x by photodeposition individually. This outcome was indicative of a synergistic effect obtained from coloading the Pt and FeO
x cocatalysts on the water oxidation activity of the BaTaO
2N:Mg.
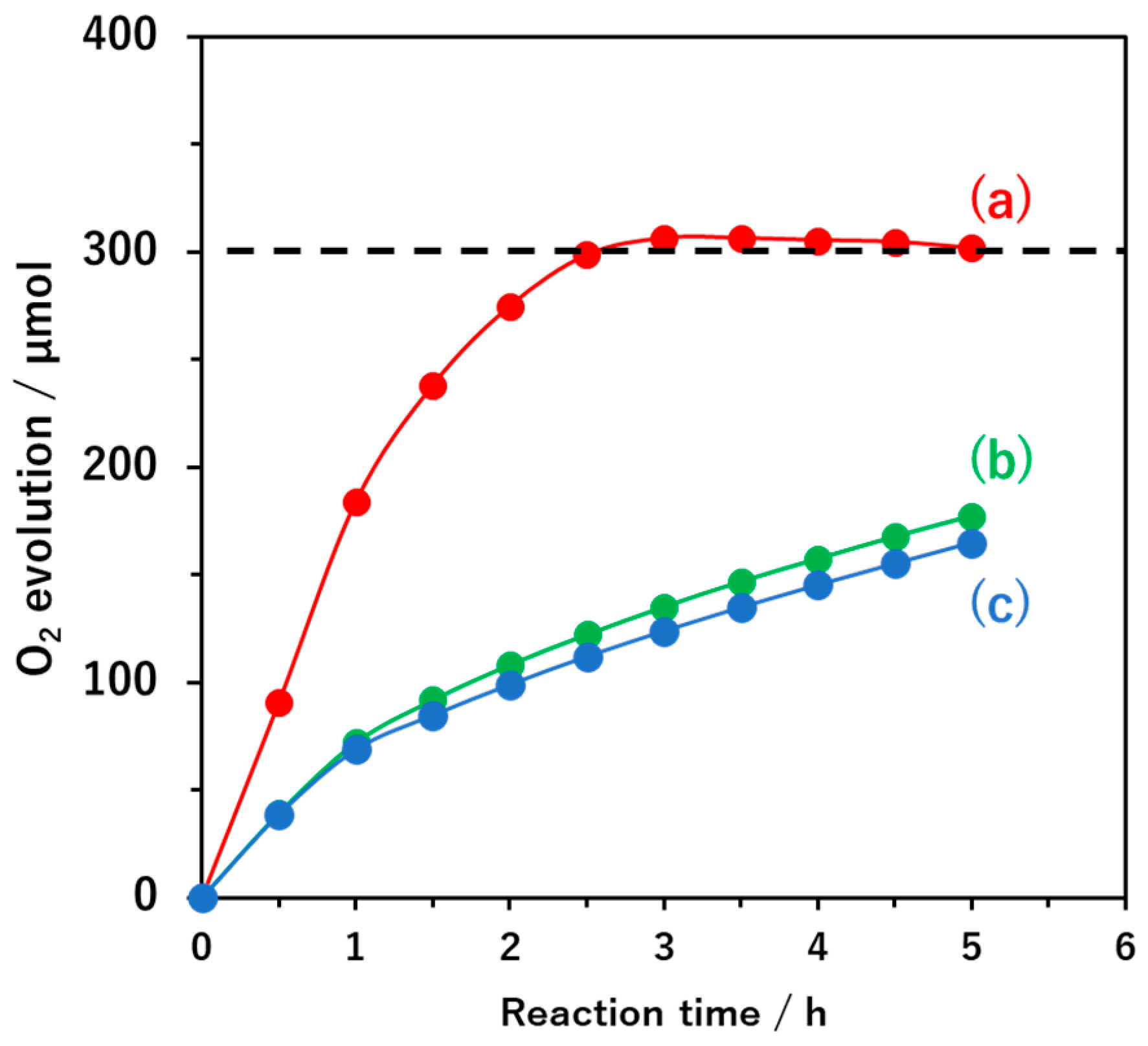
Oxygen evolution was found to cease when the AgNO
3 was depleted (
Figure 2), confirming the stoichiometry of the sacrificial oxygen evolution reaction. Furthermore, there was a negligible production of nitrogen due to photo-oxidation of the oxynitride during these reactions. In the present system, the FeO
x species were evidently oxidatively photodeposited starting from Fe
2+ ions and so the use of FeCl
3 instead of FeCl
2 as the Fe source did not enhance the oxygen evolution activity of the Pt-loaded BaTaO
2N:Mg (
Figure 2). The Fe 2
p X-ray photoelectron spectroscopy (XPS) analysis also suggested that the Fe species on the surface of the FeO
x species oxidatively photodeposited on the BaTaO
2N:Mg was not in the divalent state (
Figure S2), because the binding energy of the Fe 2
p3/2 orbital was evidently greater than 709.5 eV observed for Fe
0.94O [
28]. However, it was not possible to identify the chemical states of the Fe species conclusively using XPS because of the diversity of divalent and trivalent iron oxides and hydroxides. Moreover, it is considered that the FeO
x cocatalysts may have different chemical states during the water oxidation reaction (in water under illumination) and the XPS measurement (in vacuum under darkness). It is desirable to investigate the chemical states of Fe species by operando X-ray absorption spectroscopy [
18] to reveal the working state of the FeO
x cocatalyst. Even so, based on prior research involving Al-doped SrTiO
3, it is assumed that the primary FeO
x species in these materials was FeOOH [
21].
Even though the oxygen evolution rates were significantly different between the various specimens, inductively coupled plasma–optical emission spectroscopy (ICP-OES) analyses indicated that the amount of Fe photodeposited on the BaTaO
2N:Mg was essentially similar regardless of whether or not Pt was loaded. Specifically, the Fe concentrations were 0.30 and 0.34 wt% for the pristine material and 0.5 wt% Pt-loaded BaTaO
2N:Mg samples in the case that the nominal Fe loading was 0.5 wt%. This observation suggests that the Pt cocatalyst played a vital role by ensuring that the FeO
x cocatalyst was deposited in an effective state. It is likely that the Pt cocatalyst trapped photoexcited electrons and so promoted charge separation in the specimen that could have favored the photodeposition of FeO
x at oxidation sites on the BaTaO
2N:Mg. The importance of the rapid removal of photoexcited electrons during the photodeposition of FeO
x was also confirmed by studying the effect of molecular oxygen. That is, in the case that the photodeposition of FeO
x was conducted in the absence of molecular oxygen, the oxygen evolution activity of the Pt-loaded BaTaO
2N:Mg was only minimally enhanced (
Figure 2). Likewise, in the absence of Pt, the photodeposition of FeO
x also did not improve the oxygen evolution activity of the BaTaO
2N:Mg (
Figure 1). These data suggest that molecular oxygen acted as an electron acceptor to promote the oxidative photodeposition of the FeO
x cocatalyst species. However, it should be noted that the photodeposition site of FeO
x was not controlled in the present samples unlike in the case of SrTiO
3:Al [
21].
Figure 3 shows scanning electron microscope (SEM) images of the BaTaO
2N:Mg samples before and after loading 0.5 wt% Pt by impregnation–reduction and an additional 0.4 wt% FeO
x by photodeposition, where the cocatalyst loading was increased for the ease of observation. The Pt cocatalyst was mostly observed as nanoparticles smaller than 10 nm in size, although somewhat larger nanoparticles and aggregates also formed. After the subsequent photodeposition of FeO
x, additional nanoparticles and aggregates appeared to be deposited, often locally on some specific photocatalyst particles. However, it was difficult to identify the FeO
x cocatalyst or to distinguish the cocatalyst species. At least, the FeO
x cocatalyst did not appear to be photodeposited preferentially on oxidation sites of the BaTaO
2N:Mg particles, partly due to the non-uniform and distorted particle morphology. It is also probable that the flow of photoexcited charge carriers in BaTaO
2N:Mg was not well rectified due to defect levels characteristic of (oxy)nitride materials. Further refinement in the photocatalyst preparation will be required to define the loading sites and structures of FeO
x photodeposited on BaTaO
2N: Mg.
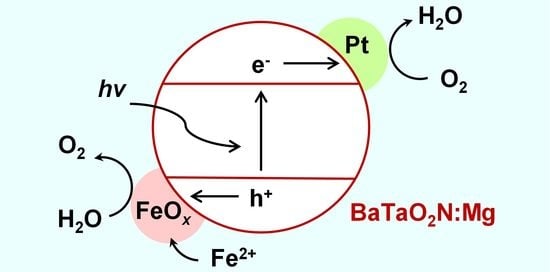
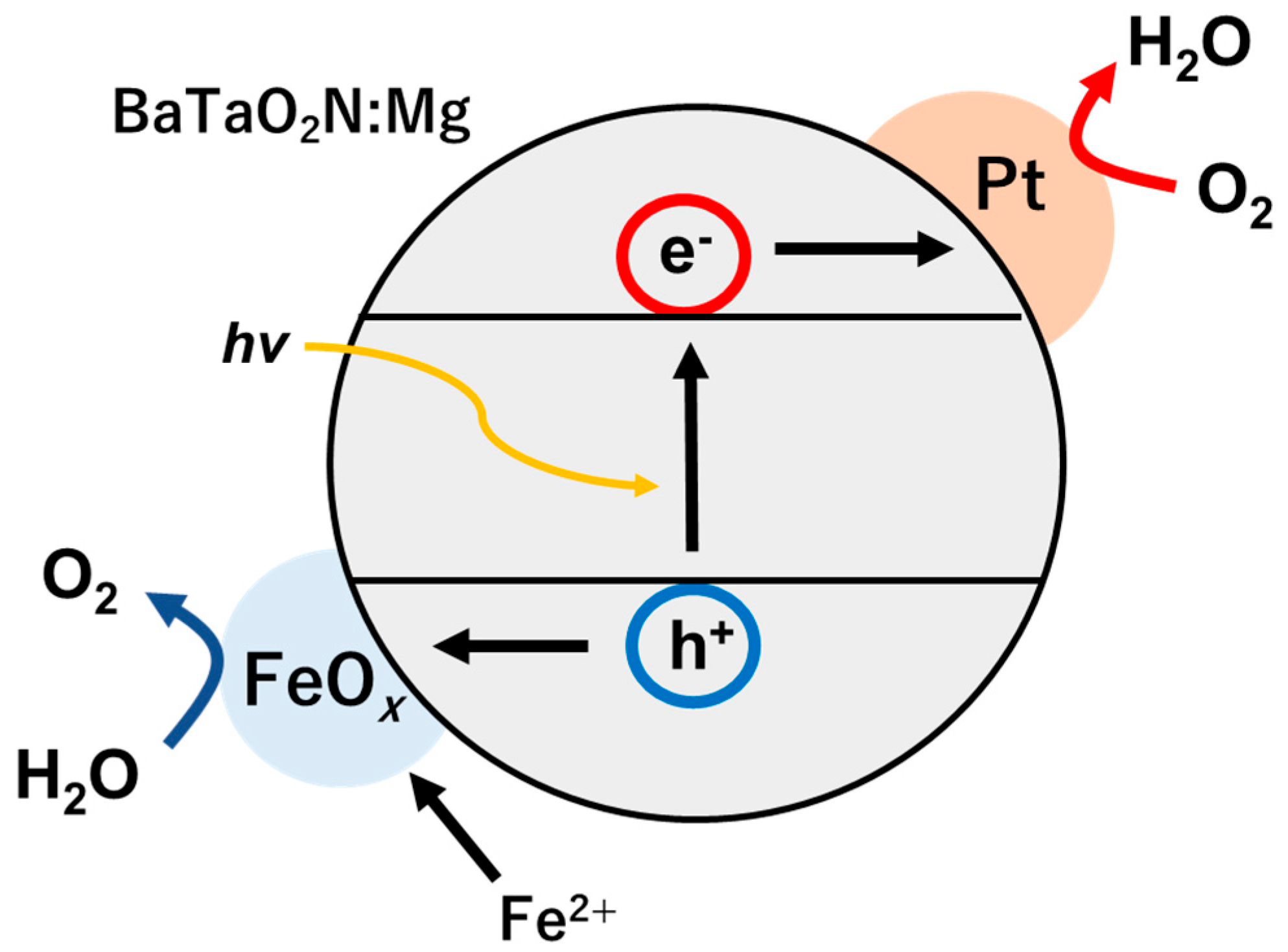
Figure 4 presents a diagram showing a proposed mechanism by which the photodeposition of FeO
x promotes the oxygen evolution activity of BaTaO
2N:Mg. In this mechanism, in response to irradiation, electrons and holes are excited in the photocatalyst. The electrons are subsequently captured by the Pt cocatalyst and consumed in the oxygen reduction reaction in the presence of dissolved oxygen. Simultaneously, the holes oxidize Fe
2+ ions in the solution to allow the oxidative deposition of FeO
x on the BaTaO
2N:Mg photocatalyst. In the absence of the Pt cocatalyst or molecular oxygen, the efficiency of charge separation will be lowered, which presumably prevents the photodeposition of FeO
x in active states on the BaTaO
2N:Mg. Further investigations will be required to establish the chemical states and structure of the FeO
x cocatalyst. Nevertheless, this is a rare example of an oxygen evolution cocatalyst photodeposited onto a narrow bandgap non-oxide photocatalyst that effectively promotes oxygen evolution activity. This process was enabled by loading Pt in advance and providing molecular oxygen during the photodeposition process to ensure the rapid removal of photoexcited electrons. From
Figure 1, it could be construed that doping with Mg induced a positive shift of the valence band edge that was a necessary perquisite for photodeposition of the FeO
x cocatalyst. However, the observed potential shift was as small as 0.1 eV, and so the impact of this shift on the photodeposition process is uncertain. Moreover, the oxygen evolution activity of undoped BaTaO
2N coloaded with the Pt and FeO
x cocatalysts was enhanced to a greater extent when the oxynitride was re-nitrided for 1 h prior to cocatalyst loading. This observation suggests that Mg doping was not essential for effective FeO
x photodeposition but instead enhanced the intrinsic oxygen evolution activity of BaTaO
2N.
To date, oxygen evolution cocatalysts have typically been loaded on oxynitride photocatalysts using impregnation methods and have been found to significantly improve oxygen evolution activity [
14,
15,
16,
17]. Therefore, in the present work, FeO
x cocatalysts were loaded on BaTaO
2N:Mg by impregnation followed by calcination under various conditions.
Figure 5 compares the initial oxygen evolution rates over BaTaO
2N:Mg loaded with FeO
x cocatalysts using various processes from aqueous AgNO
3 solutions. The loading of FeO
x by impregnation followed by nitrogen annealing or hydrogen reduction and coloading of Pt and Fe by co-impregnation–hydrogen reduction enhanced the oxygen evolution activity of BaTaO
2N:Mg. However, photodeposition of FeO
x following Pt loading was more effective. It is also important to stress that this photodeposition procedure did not require any heat treatment, unlike the impregnation methods. This new process is, therefore, expected to provide a practical approach to the coloading of hydrogen and oxygen evolution cocatalysts on thermally unstable non-oxide photocatalysts. In our previous work, one-step excitation overall water splitting was achieved by BaTaO
2N:Mg coloaded with Cr
2O
3-coated Rh as a hydrogen evolution cocatalyst and IrO
2 as a cocatalyst, where the former was loaded by impregnation–reduction and reductive photodeposition and the latter by adsorption [
16]. It is expected that the FeO
x cocatalyst would be applicable as an oxygen evolution cocatalyst alternative to IrO
2. However, our preliminary trials were unsuccessful. The FeO
x cocatalyst appeared to degrade during the photodeposition of Cr
2O
3 in aqueous methanol solution. On the other hand, it seemed difficult to photodeposit FeO
x after Cr
2O
3 coating because the oxygen reduction reaction would be suppressed. To utilize the FeO
x cocatalyst effectively in the overall water splitting reaction, it is necessary to investigate the cocatalyst loading procedure carefully.
Considering that the sequential coloading of Pt by impregnation–reduction and FeO
x by oxidative photodeposition promoted the oxygen evolution activity of BaTaO
2N:Mg, the FeO
x loading and the reaction conditions were optimized (
Table 1). The oxygen evolution rate obtained from specimens in 10 mM AgNO
3 aqueous solutions was found to increase with an increasing FeO
x loading amount up to 0.2 wt%. However, the oxygen evolution was observed to decrease after the Ag
+ cations in the solution were rapidly consumed, and so the concentration of AgNO
3 was increased from 10 to 30 mM. At this higher concentration, the oxygen production rate plateaued at approximately 230 µmol h
−1 for an FeO
x loading of 0.4 wt%. This FeO
x/Pt/BaTaO
2N:Mg sample exhibited an AQY of 1.2% at 420 nm during the oxygen evolution reaction. This value is lower than the AQY of 2.59% at 420 nm obtained for BaTaO
2N:Mg produced by nitridation of an amorphous oxide and loaded with CoO
x by impregnation–nitridation [
15], but greater than that for BaTaO
2N prepared by flux-assisted nitridation and loaded with CoO
x by impregnation–annealing (0.55% at 420 nm) [
14]. Notably, nitridation of BaTaO
2N:Mg as a pretreatment prior to loading of the Pt and FeO
x cocatalysts was found to improve the oxygen evolution activity further. This suggests that modifying the BaTaO
2N:Mg itself, such as by varying the degree of nitridation, crystallinity, or surface states, could also change the efficacy of the photodeposited FeO
x cocatalyst. Photodeposition of oxygen evolution cocatalysts is, therefore, a valid means of improving the BaTaO
2N photocatalysts, and there is evidently much room for further improvement and for new applications.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}