
A Review of Transition Metal Nitride-Based Catalysts for Electrochemical Nitrogen Reduction to Ammonia

Abstract
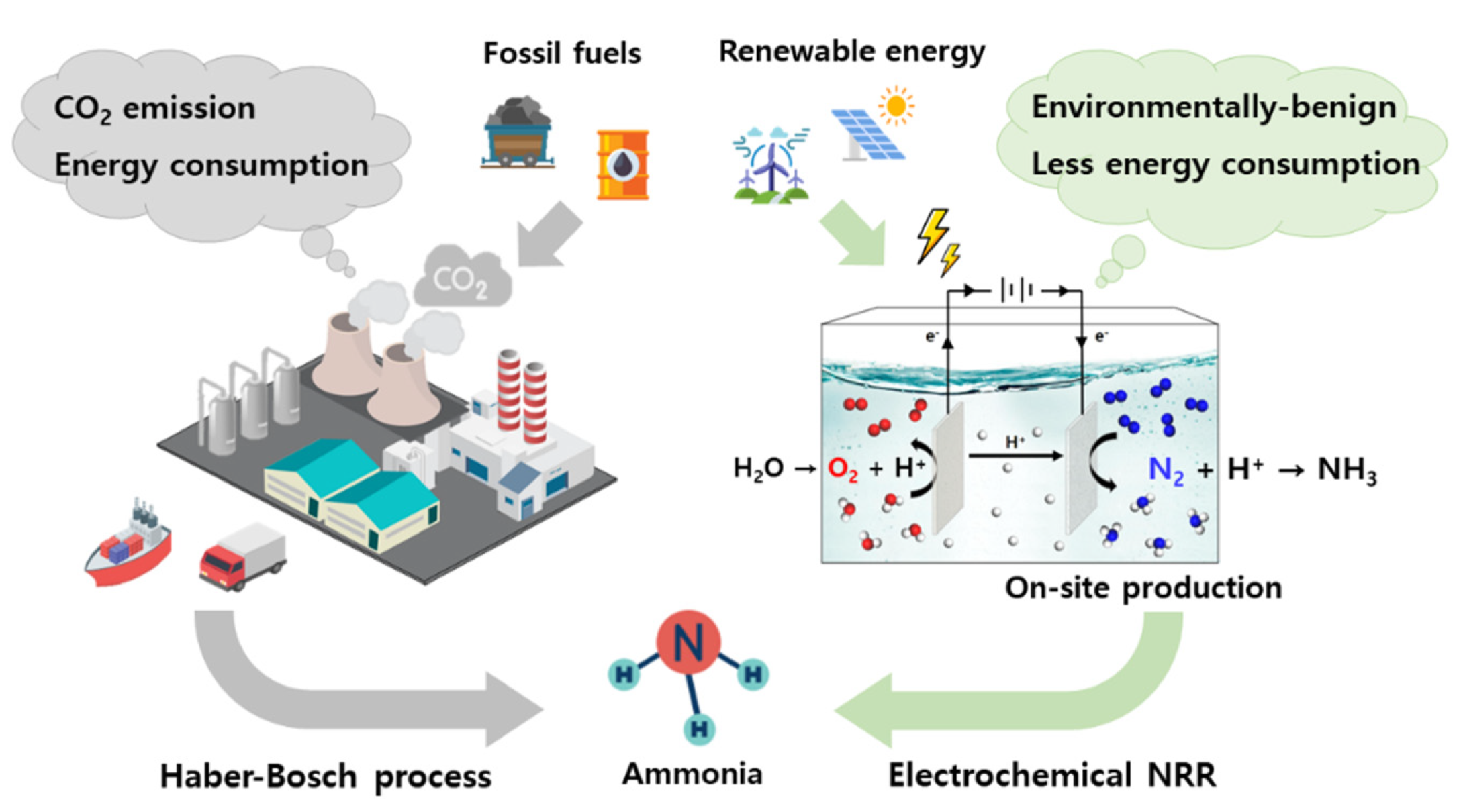
:1. Introduction
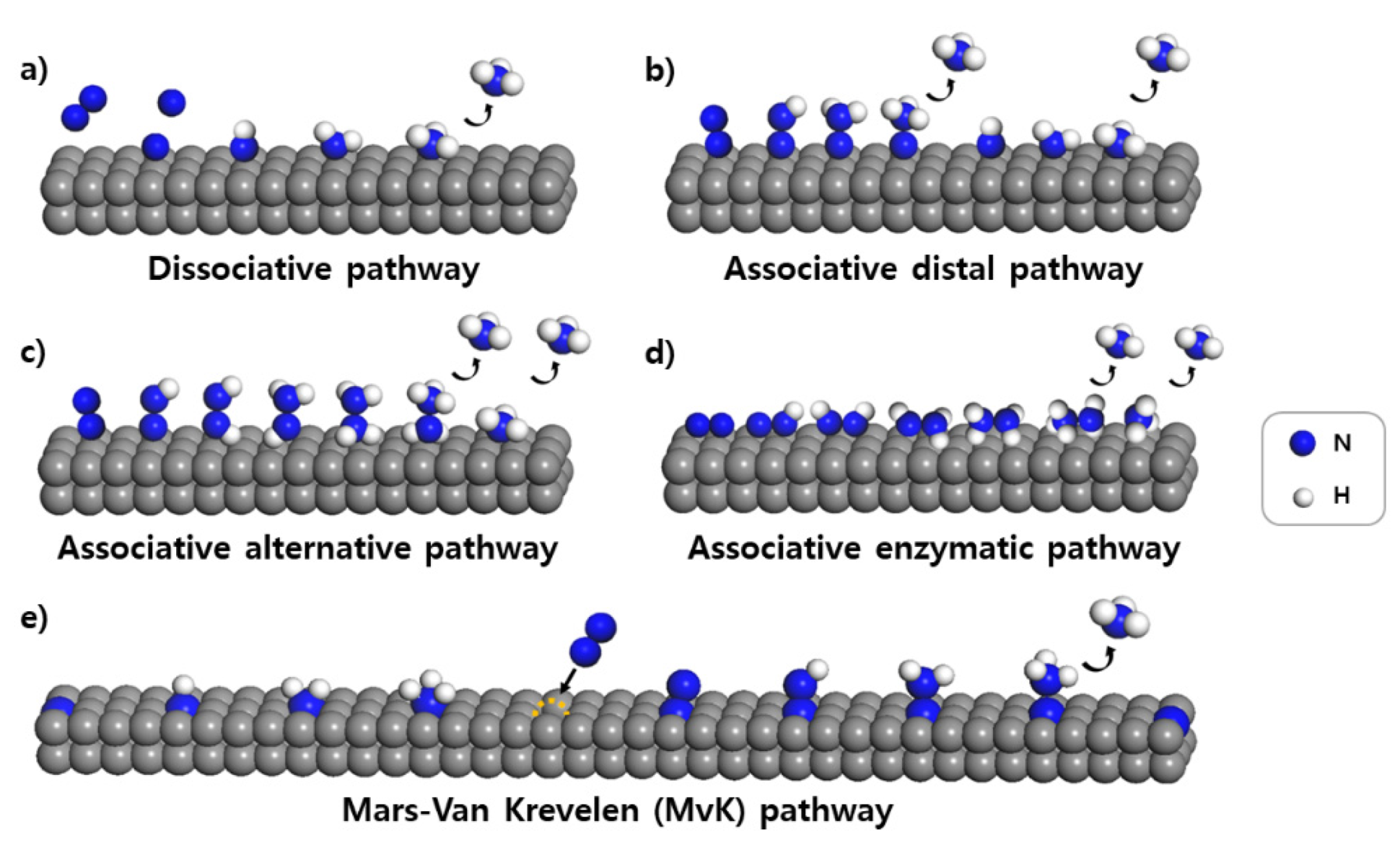
2. Mechanisms of NRR
3. TMN-Based Catalysts
3.1. TMN-Based Catalysts with Catalytic Activity
3.1.1. Vanadium Nitride-Based Catalysts
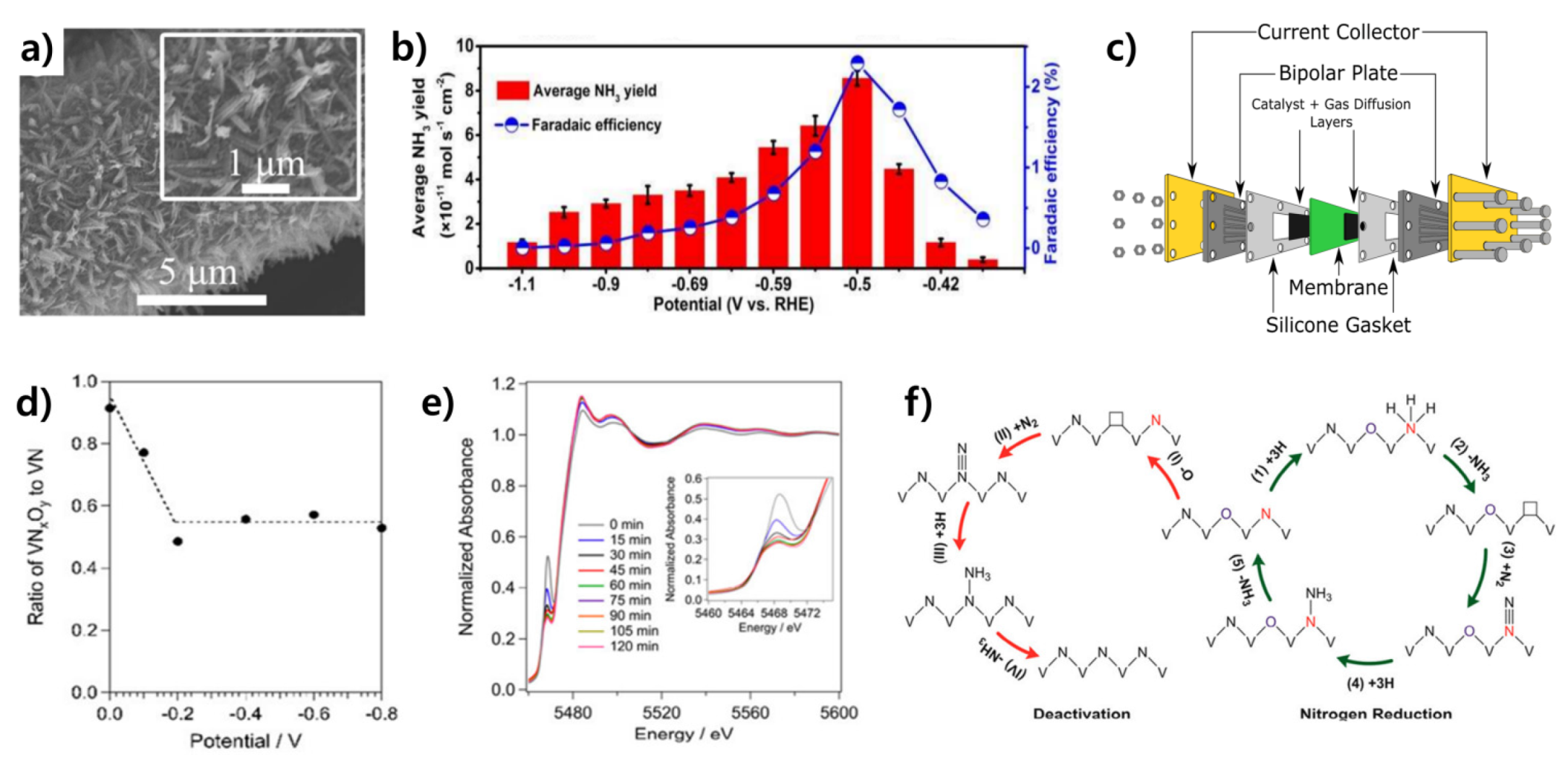
3.1.2. Chromium Nitride-Based Catalysts
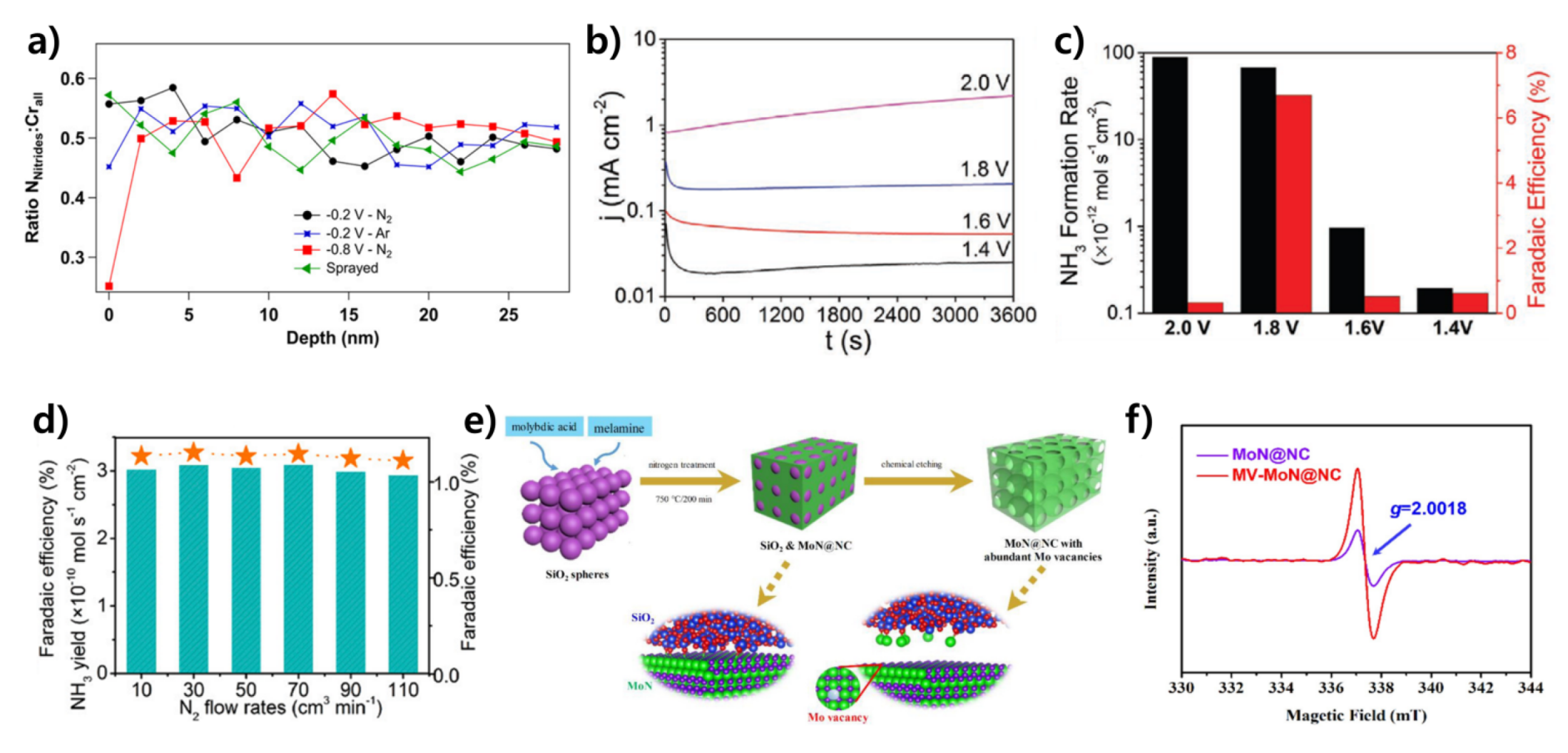
3.1.3. Molybdenum Nitride-Based Catalysts

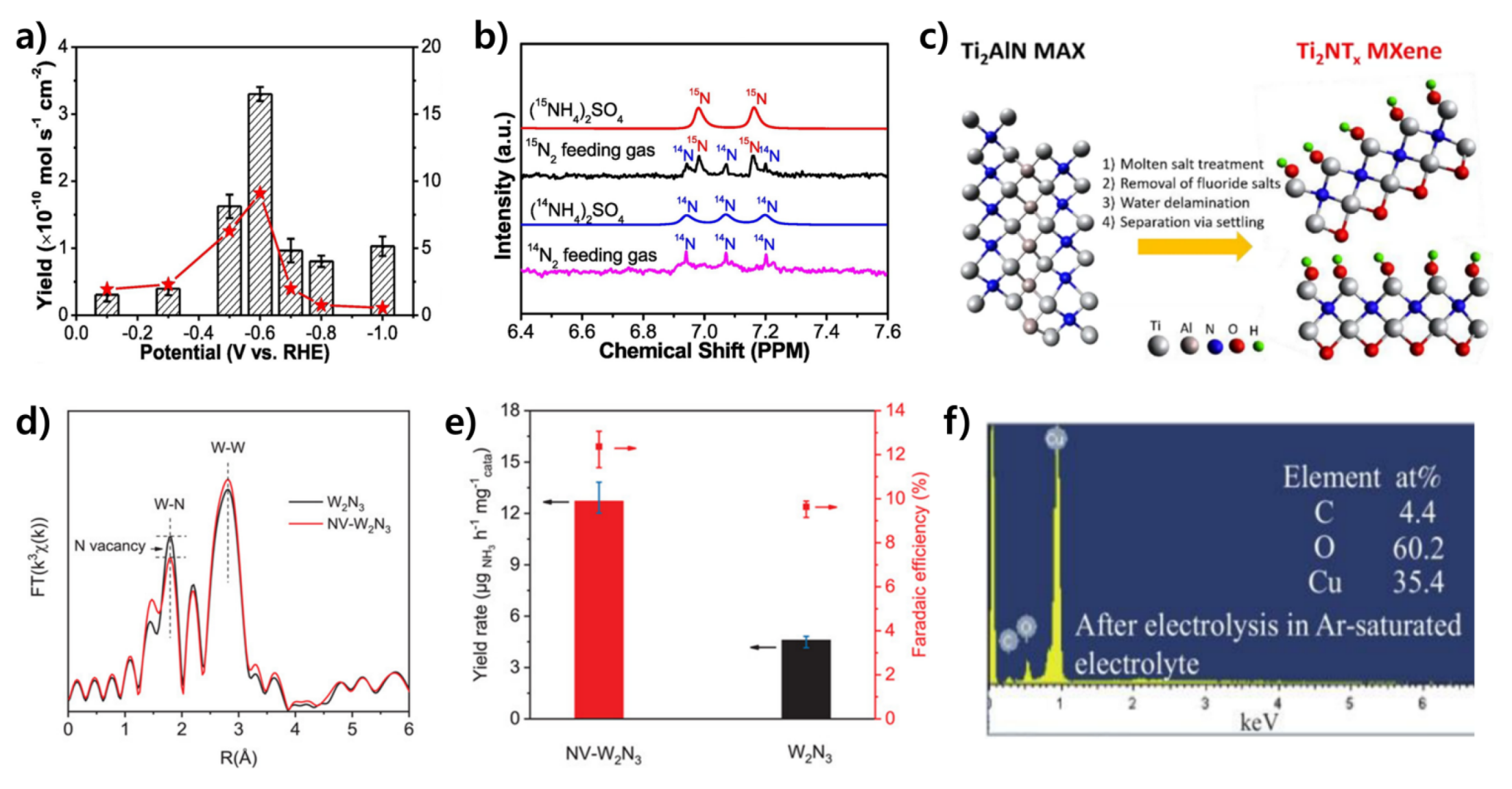
3.1.4. Titanium Nitride-Based Catalysts
3.1.5. Other TMN-Based Catalysts
3.2. TMN-Based Catalysts with Non-Catalytic Activity (Leaching or Decomposition)
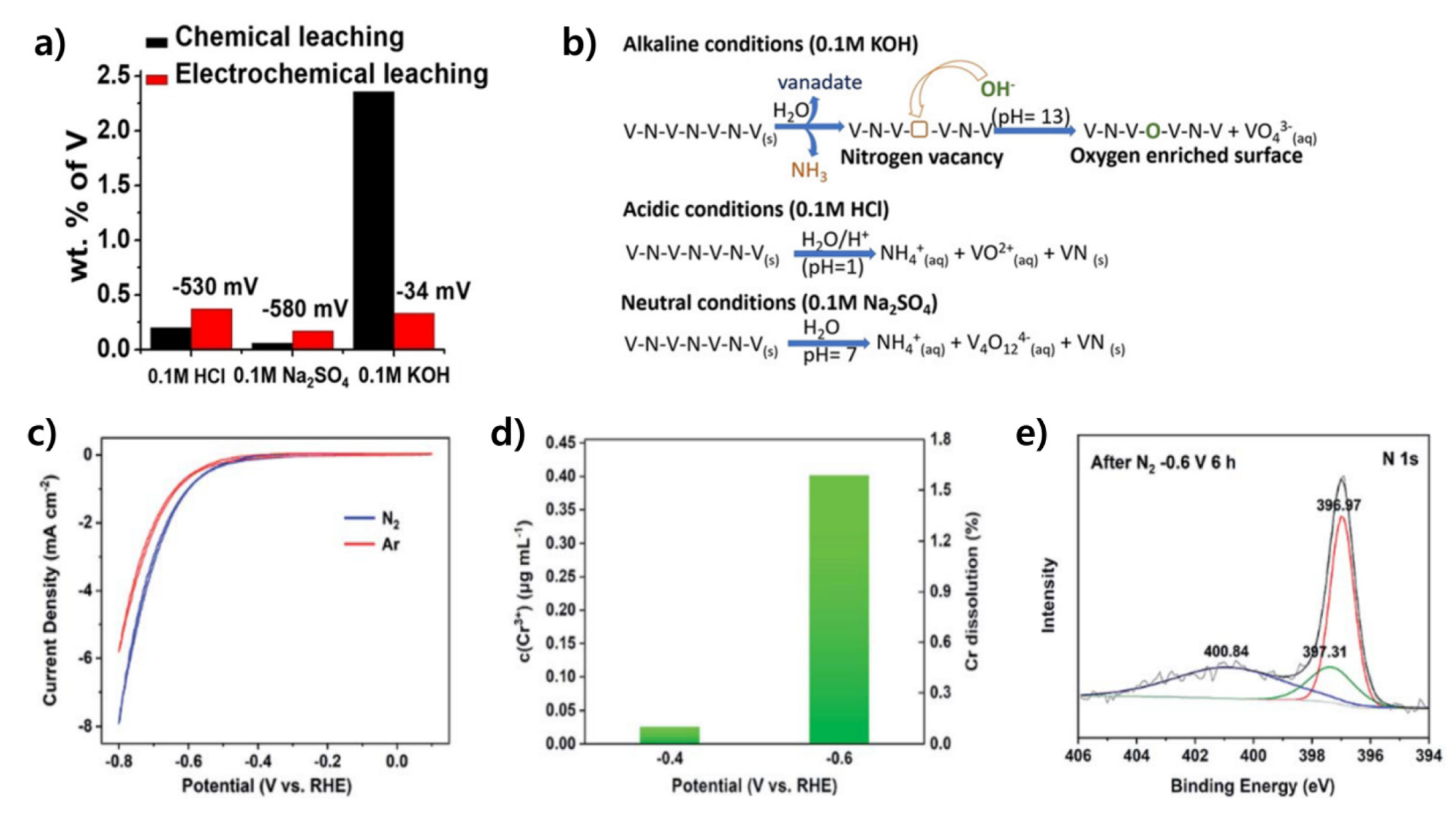
3.2.1. Vanadium Nitride-Based Catalysts

3.2.2. Chromium Nitride-Based Catalysts
3.2.3. Molybdenum Nitride-Based Catalysts
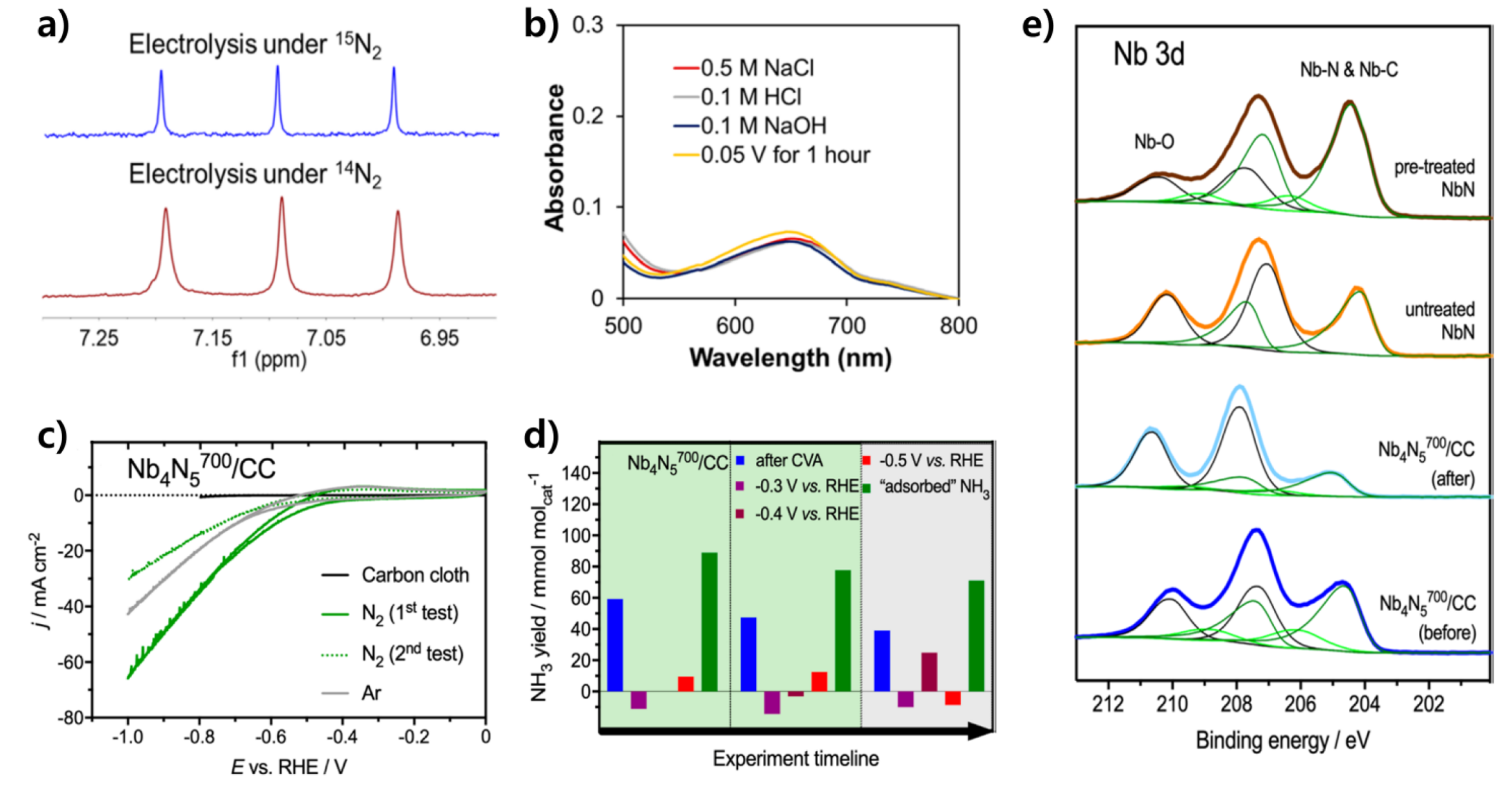
3.2.4. Niobium Nitride-Based Catalysts
4. Summary and Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ghavam, S.; Vahdati, M.; Wilson, I.; Styring, P. Sustainable ammonia production processes. Front. Energy Res. 2021, 9, 34. [Google Scholar] [CrossRef]
- Afif, A.; Radenahmad, N.; Cheok, Q.; Shams, S.; Kim, J.H.; Azad, A.K. Ammonia-fed fuel cells: A comprehensive review. Renew. Sustain. Energy Rev. 2016, 60, 822–835. [Google Scholar] [CrossRef]
- Erisman, J.W.; Sutton, M.A.; Galloway, J.; Klimont, Z.; Winiwarter, W. How a century of ammonia synthesis changed the world. Nat. Geosci. 2008, 1, 636–639. [Google Scholar] [CrossRef]
- Bird, F.; Clarke, A.; Davies, P.; Surkovic, E. Ammonia: Zero-Carbon Fertiliser, Fuel and Energy Store. In The Royal Society; Policy Briefing: London, UK, 2020. [Google Scholar]
- Wang, X.; Yang, J.; Salla, M.; Xi, S.; Yang, Y.; Li, M.; Zhang, F.; Zhu, M.K.; Huang, S.; Huang, S. Redox-Mediated Ambient Electrolytic Nitrogen Reduction for Hydrazine and Ammonia Generation. Angew. Chem. Int. Ed. 2021, 60, 18721–18727. [Google Scholar] [CrossRef]
- Guo, X.; Zhu, Y.; Ma, T. Lowering reaction temperature: Electrochemical ammonia synthesis by coupling various electrolytes and catalysts. J. Energy Chem. 2017, 26, 1107–1116. [Google Scholar] [CrossRef]
- Chatterjee, S.; Parsapur, R.K.; Huang, K.-W. Limitations of Ammonia as a Hydrogen Energy Carrier for the Transportation Sector. ACS Energy Lett. 2021, 6, 4390–4394. [Google Scholar] [CrossRef]
- Yang, B.; Ding, W.; Zhang, H.; Zhang, S. Recent progress in electrochemical synthesis of ammonia from nitrogen: Strategies to improve the catalytic activity and selectivity. Energy Environ. Sci. 2021, 14, 672–687. [Google Scholar] [CrossRef]
- Humphreys, J.; Lan, R.; Tao, S. Development and recent progress on ammonia synthesis catalysts for Haber–Bosch process. Adv. Energy Sustain. Res. 2021, 2, 2000043. [Google Scholar] [CrossRef]
- Qing, G.; Ghazfar, R.; Jackowski, S.T.; Habibzadeh, F.; Ashtiani, M.M.; Chen, C.-P.; Smith III, M.R.; Hamann, T.W. Recent advances and challenges of electrocatalytic N2 reduction to ammonia. Chem. Rev. 2020, 120, 5437–5516. [Google Scholar] [CrossRef]
- Faria, J.A. Renaissance of ammonia synthesis for sustainable production of energy and fertilizers. Curr. Opin. Green Sustain. Chem. 2021, 29, 100466. [Google Scholar] [CrossRef]
- Cao, N.; Zheng, G. Aqueous electrocatalytic N2 reduction under ambient conditions. Nano Res. 2018, 11, 2992–3008. [Google Scholar] [CrossRef]
- Guo, X.; Du, H.; Qu, F.; Li, J. Recent progress in electrocatalytic nitrogen reduction. J. Mater. Chem. A 2019, 7, 3531–3543. [Google Scholar] [CrossRef]
- Chen, J.G.; Crooks, R.M.; Seefeldt, L.C.; Bren, K.L.; Bullock, R.M.; Darensbourg, M.Y.; Holland, P.L.; Hoffman, B.; Janik, M.J.; Jones, A.K. Beyond fossil fuel–driven nitrogen transformations. Science 2018, 360, eaar6611. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.; Hill, A.K.; Torrente-Murciano, L. Current and future role of Haber–Bosch ammonia in a carbon-free energy landscape. Energy Environ. Sci. 2020, 13, 331–344. [Google Scholar] [CrossRef]
- Bicer, Y.; Dincer, I.; Zamfirescu, C.; Vezina, G.; Raso, F. Comparative life cycle assessment of various ammonia production methods. J. Clean. Prod. 2016, 135, 1379–1395. [Google Scholar] [CrossRef]
- Egenhofer, C.; Schrefler, L.; Rizos, V.; Marcu, A.; Genoese, F.; Renda, A.; Wieczorkiewicz, J.; Roth, S.; Infelise, F.; Luchetta, G. The Composition and Drivers of Energy Prices and Costs in Energy-Intensive Industries: The Case of Ceramics, Glass and Chemicals; Centre for European Policy Studies: Brussels, Belgium, 2014. [Google Scholar]
- Service, R.F. Liquid sunshine. Science 2018, 361, 120–123. [Google Scholar] [CrossRef]
- Wang, L.; Xia, M.; Wang, H.; Huang, K.; Qian, C.; Maravelias, C.T.; Ozin, G.A. Greening Ammonia toward the Solar Ammonia Refinery. Joule 2018, 2, 1055–1074. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, S.; Pales, A.F.; McGlade, C.; Remme, U.; Wanner, B.; Varro, L.; D’Ambrosio, D.; Spencer, T. Net Zero by 2050: A Roadmap for the Global Energy Sector; International Energy Agency: Paris, France, 2021. [Google Scholar]
- Tang, C.; Qiao, S.-Z. How to explore ambient electrocatalytic nitrogen reduction reliably and insightfully. Chem. Soc. Rev. 2019, 48, 3166–3180. [Google Scholar] [CrossRef]
- Foster, S.L.; Bakovic, S.I.P.; Duda, R.D.; Maheshwari, S.; Milton, R.D.; Minteer, S.D.; Janik, M.J.; Renner, J.N.; Greenlee, L.F. Catalysts for nitrogen reduction to ammonia. Nat. Catal. 2018, 1, 490–500. [Google Scholar] [CrossRef]
- Hu, L.; Xing, Z.; Feng, X. Understanding the electrocatalytic interface for ambient ammonia synthesis. ACS Energy Lett. 2020, 5, 430–436. [Google Scholar] [CrossRef]
- Li, S.-J.; Bao, D.; Shi, M.-M.; Wulan, B.-R.; Yan, J.-M.; Jiang, Q. Amorphizing of Au Nanoparticles by CeOx–RGO Hybrid Support towards Highly Efficient Electrocatalyst for N2 Reduction under Ambient Conditions. Adv. Mater. 2017, 29, 1700001. [Google Scholar] [CrossRef] [PubMed]
- Bao, D.; Zhang, Q.; Meng, F.-L.; Zhong, H.-X.; Shi, M.-M.; Zhang, Y.; Yan, J.-M.; Jiang, Q.; Zhang, X.-B. Electrochemical Reduction of N2 under Ambient Conditions for Artificial N2 Fixation and Renewable Energy Storage Using N2/NH3 Cycle. Adv. Mater. 2017, 29, 1604799. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Azofra, L.M.; Harb, M.; Cavallo, L.; Zhang, X.; Suryanto, B.H.; MacFarlane, D.R. Energy-efficient nitrogen reduction to ammonia at low overpotential in aqueous electrolyte under ambient conditions. ChemSusChem 2018, 11, 3416–3422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiyi, L.; Keyang, H.; Pengwu, X.; Wendong, W.; Nana, L.; Haiyan, Z.; Zaijun, L.; Xiaohao, L. Synthesis of a ruthenium–graphene quantum dot–graphene hybrid as a promising single-atom catalyst for electrochemical nitrogen reduction with ultrahigh yield rate and selectivity. J. Mater. Chem. A 2021, 9, 24582–24589. [Google Scholar] [CrossRef]
- Huang, H.; Xia, L.; Shi, X.; Asiri, A.M.; Sun, X. Ag nanosheets for efficient electrocatalytic N2 fixation to NH3 under ambient conditions. ChemComm 2018, 54, 11427–11430. [Google Scholar] [CrossRef]
- Li, W.; Li, K.; Ye, Y.; Zhang, S.; Liu, Y.; Wang, G.; Liang, C.; Zhang, H.; Zhao, H. Efficient electrocatalytic nitrogen reduction to ammonia with aqueous silver nanodots. Commun. Chem. 2021, 4, 10. [Google Scholar] [CrossRef]
- Wang, X.; Luo, M.; Lan, J.; Peng, M.; Tan, Y. Nanoporous Intermetallic Pd3Bi for Efficient Electrochemical Nitrogen Reduction. Adv. Mater. 2021, 33, 2007733. [Google Scholar] [CrossRef]
- Xie, H.; Geng, Q.; Zhu, X.; Luo, Y.; Chang, L.; Niu, X.; Shi, X.; Asiri, A.M.; Gao, S.; Wang, Z. PdP2 nanoparticles–reduced graphene oxide for electrocatalytic N2 conversion to NH3 under ambient conditions. J. Mater. Chem. A 2019, 7, 24760–24764. [Google Scholar] [CrossRef]
- Liu, H.-M.; Han, S.-H.; Zhao, Y.; Zhu, Y.-Y.; Tian, X.-L.; Zeng, J.-H.; Jiang, J.-X.; Xia, B.Y.; Chen, Y. Surfactant-free atomically ultrathin rhodium nanosheet nanoassemblies for efficient nitrogen electroreduction. J. Mater. Chem. A 2018, 6, 3211–3217. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, X.; Zhang, Q.; Tang, T.; Zhang, Y.; Gu, L.; Li, Y.; Bao, J.; Dai, Z.; Hu, J.-S. Engineering Mo/Mo2C/MoC hetero-interfaces for enhanced electrocatalytic nitrogen reduction. J. Mater. Chem. A 2020, 8, 8920–8926. [Google Scholar] [CrossRef]
- Feng, J.; Zhu, X.; Chen, Q.; Xiong, W.; Chen, X.; Luo, Y.; Alshehri, A.A.; Alzahrani, K.A.; Jiang, Z.; Li, W. Ultrasmall V8C7 nanoparticles embedded in conductive carbon for efficient electrocatalytic N2 reduction toward ambient NH3 production. J. Mater. Chem. A 2019, 7, 26227–26230. [Google Scholar] [CrossRef]
- Ren, X.; Zhao, J.; Wei, Q.; Ma, Y.; Guo, H.; Liu, Q.; Wang, Y.; Cui, G.; Asiri, A.M.; Li, B. High-performance N2-to-NH3 conversion electrocatalyzed by Mo2C nanorod. ACS Cent. Sci. 2018, 5, 116–121. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Ji, X.; Ren, X.; Luo, Y.; Shi, X.; Asiri, A.M.; Zheng, B.; Sun, X. Efficient electrochemical N2 reduction to NH3 on MoN nanosheets array under ambient conditions. ACS Sustain. Chem. Eng. 2018, 6, 9550–9554. [Google Scholar] [CrossRef]
- Yang, X.; Nash, J.; Anibal, J.; Dunwell, M.; Kattel, S.; Stavitski, E.; Attenkofer, K.; Chen, J.G.; Yan, Y.; Xu, B. Mechanistic insights into electrochemical nitrogen reduction reaction on vanadium nitride nanoparticles. J. Am. Chem. Soc. 2018, 140, 13387–13391. [Google Scholar] [CrossRef]
- Ma, Z.; Chen, J.; Luo, D.; Thersleff, T.; Dronskowski, R.; Slabon, A. Structural evolution of CrN nanocube electrocatalysts during nitrogen reduction reaction. Nanoscale 2020, 12, 19276–19283. [Google Scholar] [CrossRef]
- Zhang, L.; Ji, X.; Ren, X.; Ma, Y.; Shi, X.; Tian, Z.; Asiri, A.M.; Chen, L.; Tang, B.; Sun, X. Electrochemical ammonia synthesis via nitrogen reduction reaction on a MoS2 catalyst: Theoretical and experimental studies. Adv. Mater. 2018, 30, 1800191. [Google Scholar] [CrossRef]
- Kong, J.; Kim, M.-S.; Akbar, R.; Park, H.Y.; Jang, J.H.; Kim, H.; Hur, K.; Park, H.S. Electrochemical nitrogen reduction kinetics on a copper sulfide catalyst for NH3 synthesis at low temperature and atmospheric pressure. ACS Appl. Mater. Interfaces 2021, 13, 24593–24603. [Google Scholar] [CrossRef]
- Zhao, X.; Lan, X.; Yu, D.; Fu, H.; Liu, Z.; Mu, T. Deep eutectic-solvothermal synthesis of nanostructured Fe3S4 for electrochemical N2 fixation under ambient conditions. ChemComm 2018, 54, 13010–13013. [Google Scholar] [CrossRef]
- Han, J.; Ji, X.; Ren, X.; Cui, G.; Li, L.; Xie, F.; Wang, H.; Li, B.; Sun, X. MoO3 nanosheets for efficient electrocatalytic N2 fixation to NH3. J. Mater. Chem. A 2018, 6, 12974–12977. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, X.; Zhang, B.; Luo, Y.; Cui, G.; Xie, F.; Sun, X. Ambient N2 fixation to NH3 electrocatalyzed by a spinel Fe3O4 nanorod. Nanoscale 2018, 10, 14386–14389. [Google Scholar] [CrossRef]
- Kong, J.; Lim, A.; Yoon, C.; Jang, J.H.; Ham, H.C.; Han, J.; Nam, S.; Kim, D.; Sung, Y.-E.; Choi, J.; et al. Electrochemical Synthesis of NH3 at Low Temperature and Atmospheric Pressure Using a γ-Fe2O3 Catalyst. ACS Sustain. Chem. Eng. 2017, 5, 10986–10995. [Google Scholar] [CrossRef]
- Mou, T.; Liang, J.; Ma, Z.; Zhang, L.; Lin, Y.; Li, T.; Liu, Q.; Luo, Y.; Liu, Y.; Gao, S.; et al. High-efficiency electrohydrogenation of nitric oxide to ammonia on a Ni2P nanoarray under ambient conditions. J. Mater. Chem. A 2021, 9, 24268–24275. [Google Scholar] [CrossRef]
- Guo, W.; Liang, Z.; Zhao, J.; Zhu, B.; Cai, K.; Zou, R.; Xu, Q. Hierarchical cobalt phosphide hollow nanocages toward electrocatalytic ammonia synthesis under ambient pressure and room temperature. Small Methods 2018, 2, 1800204. [Google Scholar] [CrossRef]
- Zhu, X.; Wu, T.; Ji, L.; Liu, Q.; Luo, Y.; Cui, G.; Xiang, Y.; Zhang, Y.; Zheng, B.; Sun, X. Unusual electrochemical N2 reduction activity in an earth-abundant iron catalyst via phosphorous modulation. ChemComm 2020, 56, 731–734. [Google Scholar] [CrossRef]
- Lan, J.; Luo, M.; Han, J.; Peng, M.; Duan, H.; Tan, Y. Nanoporous B13C2 towards highly efficient electrochemical nitrogen fixation. Small 2021, 17, 2102814. [Google Scholar] [CrossRef]
- Zhang, M.; Choi, C.; Huo, R.; Gu, G.H.; Hong, S.; Yan, C.; Xu, S.; Robertson, A.W.; Qiu, J.; Jung, Y.; et al. Reduced graphene oxides with engineered defects enable efficient electrochemical reduction of dinitrogen to ammonia in wide pH range. Nano Energy 2020, 68, 104323. [Google Scholar] [CrossRef]
- Song, P.; Wang, H.; Kang, L.; Ran, B.; Song, H.; Wang, R. Electrochemical nitrogen reduction to ammonia at ambient conditions on nitrogen and phosphorus co-doped porous carbon. ChemComm 2019, 55, 687–690. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Cherepanov, P.V.; Choi, J.; Suryanto, B.H.R.; Hodgetts, R.Y.; Bakker, J.M.; Ferrero Vallana, F.M.; Simonov, A.N. A Roadmap to the Ammonia Economy. Joule 2020, 4, 1186–1205. [Google Scholar] [CrossRef]
- Chen, J.G. Carbide and Nitride Overlayers on Early Transition Metal Surfaces: Preparation, Characterization, and Reactivities. Chem. Rev. 1996, 96, 1477–1498. [Google Scholar] [CrossRef]
- Yang, M.; Allen, A.J.; Nguyen, M.T.; Ralston, W.T.; MacLeod, M.J.; DiSalvo, F.J. Corrosion behavior of mesoporous transition metal nitrides. J. Solid State Chem. 2013, 205, 49–56. [Google Scholar] [CrossRef]
- Marchand, R.; Tessier, F.; DiSalvo, F.J. New routes to transition metal nitrides: And characterization of new phases. J. Mater. Chem. 1999, 9, 297–304. [Google Scholar] [CrossRef]
- Youn, D.H.; Bae, G.; Han, S.; Kim, J.Y.; Jang, J.-W.; Park, H.; Choi, S.H.; Lee, J.S. A highly efficient transition metal nitride-based electrocatalyst for oxygen reduction reaction: TiN on a CNT–graphene hybrid support. J. Mater. Chem. A 2013, 1, 8007–8015. [Google Scholar] [CrossRef]
- Xie, J.; Xie, Y. Transition Metal Nitrides for Electrocatalytic Energy Conversion: Opportunities and Challenges. Chem. Eur. J. 2016, 22, 3588–3598. [Google Scholar] [CrossRef]
- Dongil, A.B. Recent Progress on Transition Metal Nitrides Nanoparticles as Heterogeneous Catalysts. Nanomaterials 2019, 9, 1111. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Deng, Y.; Zeng, J.; Song, H.; Liao, S. A high-performance composite ORR catalyst based on the synergy between binary transition metal nitride and nitrogen-doped reduced graphene oxide. J. Mater. Chem. A 2017, 5, 5829–5837. [Google Scholar] [CrossRef]
- Luo, J.; Tian, X.; Zeng, J.; Li, Y.; Song, H.; Liao, S. Limitations and Improvement Strategies for Early-Transition-Metal Nitrides as Competitive Catalysts toward the Oxygen Reduction Reaction. ACS Catal. 2016, 6, 6165–6174. [Google Scholar] [CrossRef]
- Go, H.; Akio, I.; Tsuyoshi, T.; Kondo, J.N.; Michikazu, H.; Kazunari, D. Ta3N5 as a Novel Visible Light-Driven Photocatalyst (λ < 600 nm). Chem. Lett. 2002, 31, 736–737. [Google Scholar]
- Kim, J.Y.; Lee, M.H.; Kim, J.-H.; Kim, C.W.; Youn, D.H. Facile nanocrystalline Ta3N5 synthesis for photocatalytic dye degradation under visible light. Chem. Phys. Lett. 2020, 738, 136900. [Google Scholar] [CrossRef]
- Seol, M.; Youn, D.H.; Kim, J.Y.; Jang, J.-W.; Choi, M.; Lee, J.S.; Yong, K. Mo-Compound/CNT-Graphene Composites as Efficient Catalytic Electrodes for Quantum-Dot-Sensitized Solar Cells. Adv. Energy Mater. 2014, 4, 1300775. [Google Scholar] [CrossRef]
- Youn, D.; Seol, M.; Kim, J.; Jang, J.-W.; Choi, Y.; Yong, K.; Lee, J.S. TiN Nanoparticles on CNT-Graphene Hybrid Support as Noble-Metal-Free Counter Electrode for Quantum-Dot-Sensitized Solar Cells. ChemSusChem 2013, 6, 261–267. [Google Scholar] [CrossRef]
- Park, S.H.; Jo, T.H.; Lee, M.H.; Kawashima, K.; Mullins, C.B.; Lim, H.-K.; Youn, D.H. Highly active and stable nickel–molybdenum nitride (Ni2Mo3N) electrocatalyst for hydrogen evolution. J. Mater. Chem. A 2021, 9, 4945–4951. [Google Scholar] [CrossRef]
- Park, S.H.; Kang, S.H.; Youn, D.H. Direct One-Step Growth of Bimetallic Ni2Mo3N on Ni Foam as an Efficient Oxygen Evolution Electrocatalyst. Materials 2021, 14, 4768. [Google Scholar] [CrossRef]
- Abghoui, Y.; Garden, A.L.; Hlynsson, V.F.; Björgvinsdóttir, S.; Ólafsdóttir, H.; Skúlason, E. Enabling electrochemical reduction of nitrogen to ammonia at ambient conditions through rational catalyst design. Phys. Chem. Chem. Phys. 2015, 17, 4909–4918. [Google Scholar] [CrossRef] [PubMed]
- Abghoui, Y.; Skúlason, E. Onset potentials for different reaction mechanisms of nitrogen activation to ammonia on transition metal nitride electro-catalysts. Catal. Today 2017, 286, 69–77. [Google Scholar] [CrossRef]
- Abghoui, Y.; Skúlasson, E. Transition Metal Nitride Catalysts for Electrochemical Reduction of Nitrogen to Ammonia at Ambient Conditions. Procedia Comput. Sci. 2015, 51, 1897–1906. [Google Scholar] [CrossRef] [Green Version]
- Abghoui, Y.; Skúlason, E. Electrochemical synthesis of ammonia via Mars-van Krevelen mechanism on the (111) facets of group III–VII transition metal mononitrides. Catal. Today 2017, 286, 78–84. [Google Scholar] [CrossRef]
- Abghoui, Y.; Garden, A.L.; Howalt, J.G.; Vegge, T.; Skúlason, E. Electroreduction of N2 to Ammonia at Ambient Conditions on Mononitrides of Zr, Nb, Cr, and V: A DFT Guide for Experiments. ACS Catal. 2016, 6, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Gambarotta, S.; Scott, J. Multimetallic Cooperative Activation of N2. Angew. Chem. Int. Ed. 2004, 43, 5298–5308. [Google Scholar] [CrossRef]
- Kitano, M.; Inoue, Y.; Yamazaki, Y.; Hayashi, F.; Kanbara, S.; Matsuishi, S.; Yokoyama, T.; Kim, S.-W.; Hara, M.; Hosono, H. Ammonia synthesis using a stable electride as an electron donor and reversible hydrogen store. Nat. Chem. 2012, 4, 934–940. [Google Scholar] [CrossRef]
- Zhang, X.; Kong, R.-M.; Du, H.; Xia, L.; Qu, F. Highly efficient electrochemical ammonia synthesis via nitrogen reduction reactions on a VN nanowire array under ambient conditions. ChemComm 2018, 54, 5323–5325. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, Y.; Ren, X.; Cui, G.; Asiri, A.M.; Zheng, B.; Sun, X. High-Efficiency Electrosynthesis of Ammonia with High Selectivity under Ambient Conditions Enabled by VN Nanosheet Array. ACS Sustain. Chem. Eng. 2018, 6, 9545–9549. [Google Scholar] [CrossRef]
- Nash, J.; Yang, X.; Anibal, J.; Dunwell, M.; Yao, S.; Attenkofer, K.; Chen, J.G.; Yan, Y.; Xu, B. Elucidation of the Active Phase and Deactivation Mechanisms of Chromium Nitride in the Electrochemical Nitrogen Reduction Reaction. J. Phys. Chem. C 2019, 123, 23967–23975. [Google Scholar] [CrossRef]
- Yao, Y.; Feng, Q.; Zhu, S.; Li, J.; Yao, Y.; Wang, Y.; Wang, Q.; Gu, M.; Wang, H.; Li, H.; et al. Chromium Oxynitride Electrocatalysts for Electrochemical Synthesis of Ammonia Under Ambient Conditions. Small Methods 2019, 3, 1800324. [Google Scholar] [CrossRef]
- Ren, X.; Cui, G.; Chen, L.; Xie, F.; Wei, Q.; Tian, Z.; Sun, X. Electrochemical N2 fixation to NH3 under ambient conditions: Mo2N nanorod as a highly efficient and selective catalyst. ChemComm 2018, 54, 8474–8477. [Google Scholar]
- Yang, X.; Ling, F.; Su, J.; Zi, X.; Zhang, H.; Zhang, H.; Li, J.; Zhou, M.; Wang, Y. Insights into the role of cation vacancy for significantly enhanced electrochemical nitrogen reduction. Appl. Catal. B 2020, 264, 118477. [Google Scholar] [CrossRef]
- Kang, S.; Wang, J.; Zhang, S.; Zhao, C.; Wang, G.; Cai, W.; Zhang, H. Plasma-etching enhanced titanium oxynitride active phase with high oxygen content for ambient electrosynthesis of ammonia. Electrochem. Commun. 2019, 100, 90–95. [Google Scholar] [CrossRef]
- Johnson, D.; Hunter, B.; Christie, J.; King, C.; Kelley, E.; Djire, A. Ti2N nitride MXene evokes the Mars-van Krevelen mechanism to achieve high selectivity for nitrogen reduction reaction. Sci. Rep. 2022, 12, 657. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Li, L.; Liu, X.; Tang, C.; Xu, W.; Chen, S.; Song, L.; Zheng, Y.; Qiao, S.-Z. Nitrogen Vacancies on 2D Layered W2N3: A Stable and Efficient Active Site for Nitrogen Reduction Reaction. Adv. Mater. 2019, 31, 1902709. [Google Scholar] [CrossRef]
- He, H.-y.; Wang, S.; Ji, L.-L. Fabrication of self-supported Cu3N electrode for electrocatalytic nitrogen reduction reaction. J. Fuel Chem. Technol. 2022, 50, 484–493. [Google Scholar] [CrossRef]
- Yesudoss, D.K.; Lee, G.; Shanmugam, S. Strong catalyst support interactions in defect-rich γ-Mo2N nanoparticles loaded 2D-h-BN hybrid for highly selective nitrogen reduction reaction. Appl. Catal. B Environ. 2021, 287, 119952. [Google Scholar] [CrossRef]
- Xi, Z.; Shi, K.; Xu, X.; Jing, P.; Liu, B.; Gao, R.; Zhang, J. Boosting Nitrogen Reduction Reaction via Electronic Coupling of Atomically Dispersed Bismuth with Titanium Nitride Nanorods. Adv. Sci. 2022, 9, 2104245. [Google Scholar] [CrossRef]
- Manjunatha, R.; Karajić, A.; Teller, H.; Nicoara, K.; Schechter, A. Electrochemical and Chemical Instability of Vanadium Nitride in the Synthesis of Ammonia Directly from Nitrogen. ChemCatChem 2020, 12, 438–443. [Google Scholar] [CrossRef]
- Du, H.-L.; Gengenbach, T.R.; Hodgetts, R.; MacFarlane, D.R.; Simonov, A.N. Critical Assessment of the Electrocatalytic Activity of Vanadium and Niobium Nitrides toward Dinitrogen Reduction to Ammonia. ACS Sustain. Chem. Eng. 2019, 7, 6839–6850. [Google Scholar] [CrossRef]
- Guo, W.; Liang, Z.; Tang, Y.; Cai, K.; Qiu, T.; Wu, Y.; Zhang, K.; Gao, S.; Zou, R. Understanding the lattice nitrogen stability and deactivation pathways of cubic CrN nanoparticles in the electrochemical nitrogen reduction reaction. J. Mater. Chem. A 2021, 9, 8568–8575. [Google Scholar] [CrossRef]
- Hu, B.; Hu, M.; Seefeldt, L.; Liu, T.L. Electrochemical Dinitrogen Reduction to Ammonia by Mo2N: Catalysis or Decomposition? ACS Energy Lett. 2019, 4, 1053–1054. [Google Scholar] [CrossRef]
- Andersen, S.Z.; Čolić, V.; Yang, S.; Schwalbe, J.A.; Nielander, A.C.; McEnaney, J.M.; Enemark-Rasmussen, K.; Baker, J.G.; Singh, A.R.; Rohr, B.A.; et al. A rigorous electrochemical ammonia synthesis protocol with quantitative isotope measurements. Nature 2019, 570, 504–508. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Suryanto, B.H.R.; Wang, D.; Du, H.-L.; Hodgetts, R.Y.; Ferrero Vallana, F.M.; MacFarlane, D.R.; Simonov, A.N. Identification and elimination of false positives in electrochemical nitrogen reduction studies. Nat. Commun. 2020, 11, 5546. [Google Scholar] [CrossRef]
- Wei, J.; Jing, Y.; Zhao, Z.; Fan, Z.; Liang, Z.; Huang, J.; Wu, H.; Xie, Z.; Liu, D.; Qu, D.; et al. Catalyst-Support interactions enhanced electrochemical nitrogen reduction on Au/ZrO2. Electrochim. Acta 2021, 381, 138222. [Google Scholar] [CrossRef]
- Wang, J.; Wei, J.; An, C.; Tang, H.; Deng, Q.; Li, J. Electrocatalyst design for the conversion of energy molecules: Electronic state modulation and mass transport regulation. Chem. Commun. 2022, 58, 10907–10924. [Google Scholar] [CrossRef]
- Ologunagba, D.; Kattel, S. A Density Functional Theory Study of Electrochemical Nitrogen Reduction to Ammonia on the (100) Surface of Transition-Metal Oxynitrides. J. Phys. Chem. C 2022, 126, 17045–17055. [Google Scholar] [CrossRef]
- Wei, X.; Vogel, D.; Keller, L.; Kriescher, S.; Wessling, M. Microtubular Gas Diffusion Electrode Based on Ruthenium-Carbon Nanotubes for Ambient Electrochemical Nitrogen Reduction to Ammonia. ChemElectroChem 2020, 7, 4679–4684. [Google Scholar] [CrossRef]
- Kolen, M.; Antoniadis, G.; Schreuders, H.; Boshuizen, B.; van Noordenne, D.D.; Ripepi, D.; Smith, W.A.; Mulder, F.M. Combinatorial Screening of Bimetallic Electrocatalysts for Nitrogen Reduction to Ammonia Using a High-Throughput Gas Diffusion Electrode Cell Design. J. Electrochem. Soc. 2022, 169, 124506. [Google Scholar] [CrossRef]
- Kolen, M.; Ripepi, D.; Smith, W.A.; Burdyny, T.; Mulder, F.M. Overcoming Nitrogen Reduction to Ammonia Detection Challenges: The Case for Leapfrogging to Gas Diffusion Electrode Platforms. ACS Catal. 2022, 12, 5726–5735. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TMNs Catalyst | Electrolyte | Potential /V vs. RHE | Production Rate /mol s−1 cm−2 | FE /% | Isotope | Test Cell Condition | Ref |
|---|---|---|---|---|---|---|---|
| VN/CC | 0.1 M HCl | −0.3 | 2.48 × 10−10 | 3.58 | X | H-cell | [73] |
| VN/TM | 0.1 M HCl | −0.5 | 8.40 × 10−11 | 2.25 | O | H-cell | [74] |
| VN nanoparticles | - | −0.1 | 3.3 × 10−10 | 6.0 | O | MEA | [37] |
| Cr2N | - | −0.2 | 1.4 × 10−11 | 0.58 | O | MEA | [75] |
| CrN | 0.1 M HCl | −0.5 | 6.1 × 10−11 | 16.6 | X | H-cell | [38] |
| CrO0.66N0.56 | - | 2.0 | 8.94 × 10−11 | 6.7 (at 1.8 V) | X | PEMEL | [76] |
| MoN NA/CC | 0.1 M HCl | −0.3 | 3.01 × 10−10 | 1.15 | O | H-cell | [36] |
| Mo2N/GCE | 0.1 M HCl | −0.3 | 78.4 μg mgcat−1 h−1 | 4.5 | X | H-cell | [77] |
| MV-MoN@NC | 0.1 M HCl | −0.2 | 5.02 × 10−10 | 6.9 | O | H-cell | [78] |
| TiN-PE | 0.1 M Na2SO4 | −0.6 | 3.32 × 10−10 | 9.1 | O | Two-compartment | [79] |
| Ti2N MXene | 0.1 M HCl | −0.25 | 1.85 × 10−10 | 19.85 | X | H-cell | [80] |
| 2D layered W2N3 | 0.1 M KOH | −0.2 | 3.8 ± 0.32 × 10−11 | 11.67 ± 0.93 | O | H-cell | [81] |
| Cu3N/CF | 0.1 M Na2SO4 | −0.2 | 1.12 × 10−10 | 1.5 | X | H-cell | [82] |
| γ-Mo2N on 2D-h-BN | 0.1 M Na2SO4 | −0.3 | 35.9 μg mg−1 h−1 | 61.5 | X | H-cell | [83] |
| NC/Bi SAs/TiN/CC | 0.1 M Na2SO4 | −0.8 | 75.15 μg mgcat−1 h−1 | 24.6 | O | H-cell | [84] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.Y.; Jang, Y.J.; Youn, D.H. A Review of Transition Metal Nitride-Based Catalysts for Electrochemical Nitrogen Reduction to Ammonia. Catalysts 2023, 13, 639. https://doi.org/10.3390/catal13030639
Park SY, Jang YJ, Youn DH. A Review of Transition Metal Nitride-Based Catalysts for Electrochemical Nitrogen Reduction to Ammonia. Catalysts. 2023; 13(3):639. https://doi.org/10.3390/catal13030639
Chicago/Turabian StylePark, So Young, Youn Jeong Jang, and Duck Hyun Youn. 2023. "A Review of Transition Metal Nitride-Based Catalysts for Electrochemical Nitrogen Reduction to Ammonia" Catalysts 13, no. 3: 639. https://doi.org/10.3390/catal13030639