
Advanced Electrocatalysts for the Oxygen Evolution Reaction: From Single- to Multielement Materials


 , , and
, , and
Abstract
:
1. Introduction
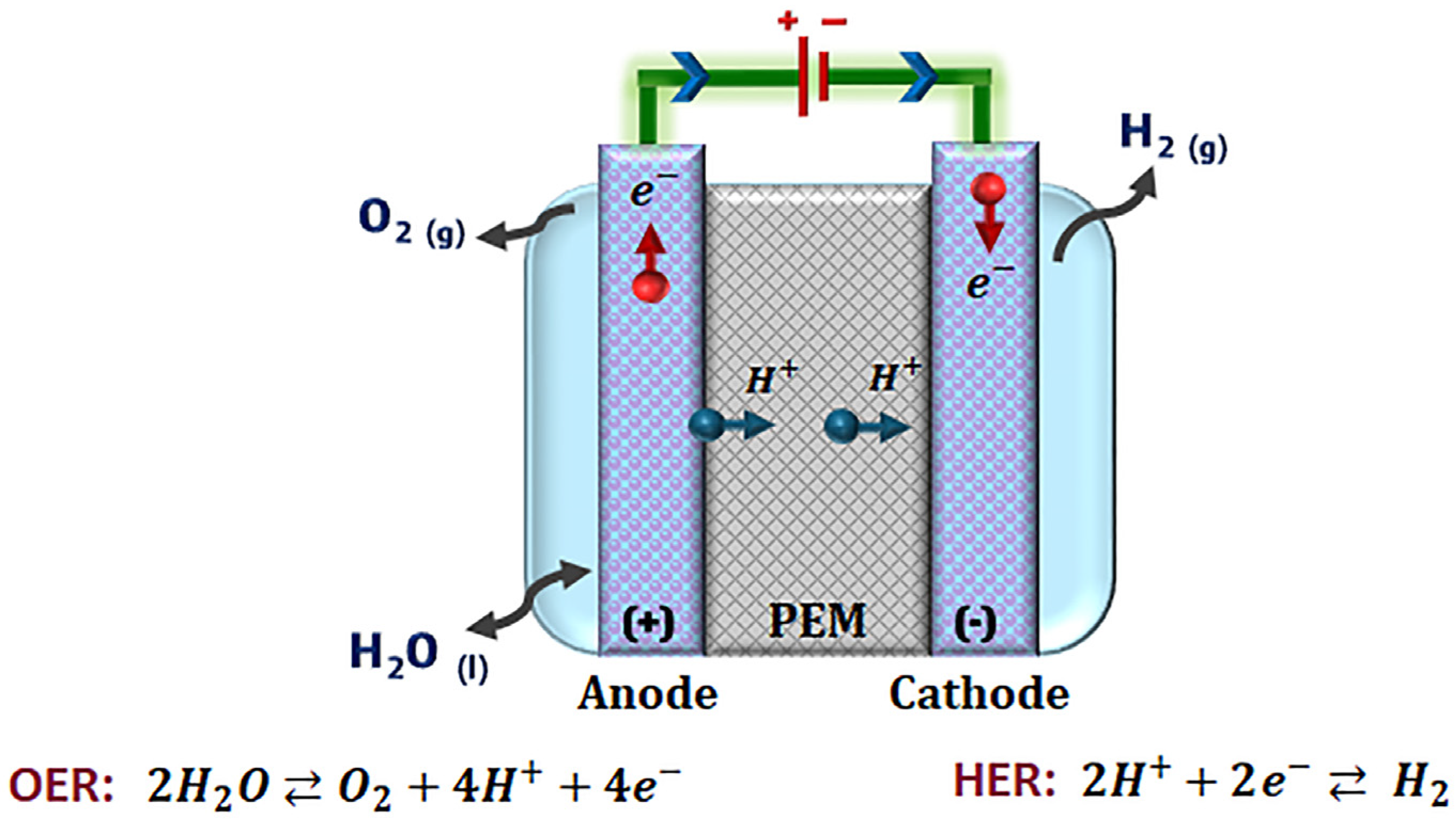
2. Fundamentals of Advanced Electrocatalyst Design for Oxygen Evolution Reaction
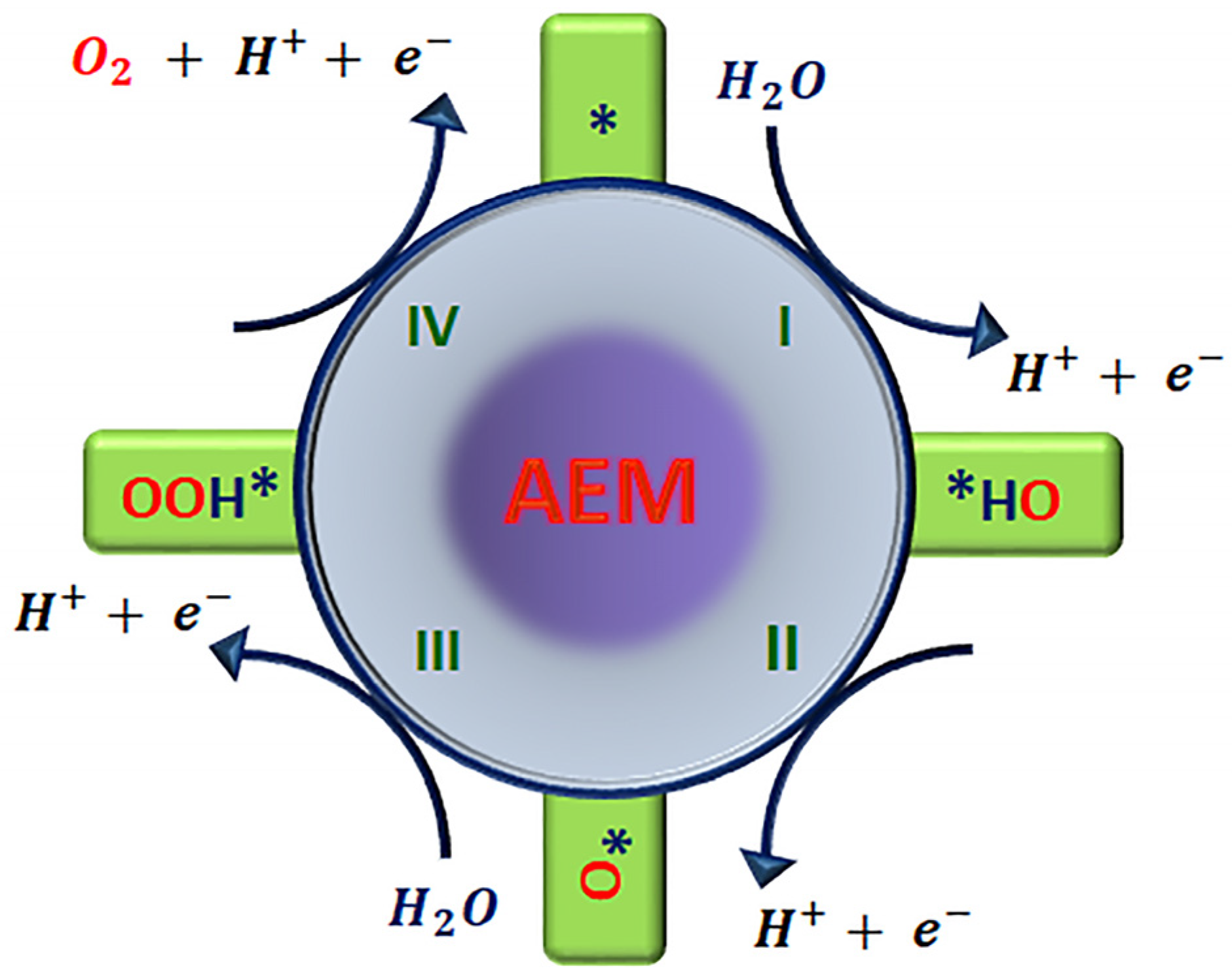
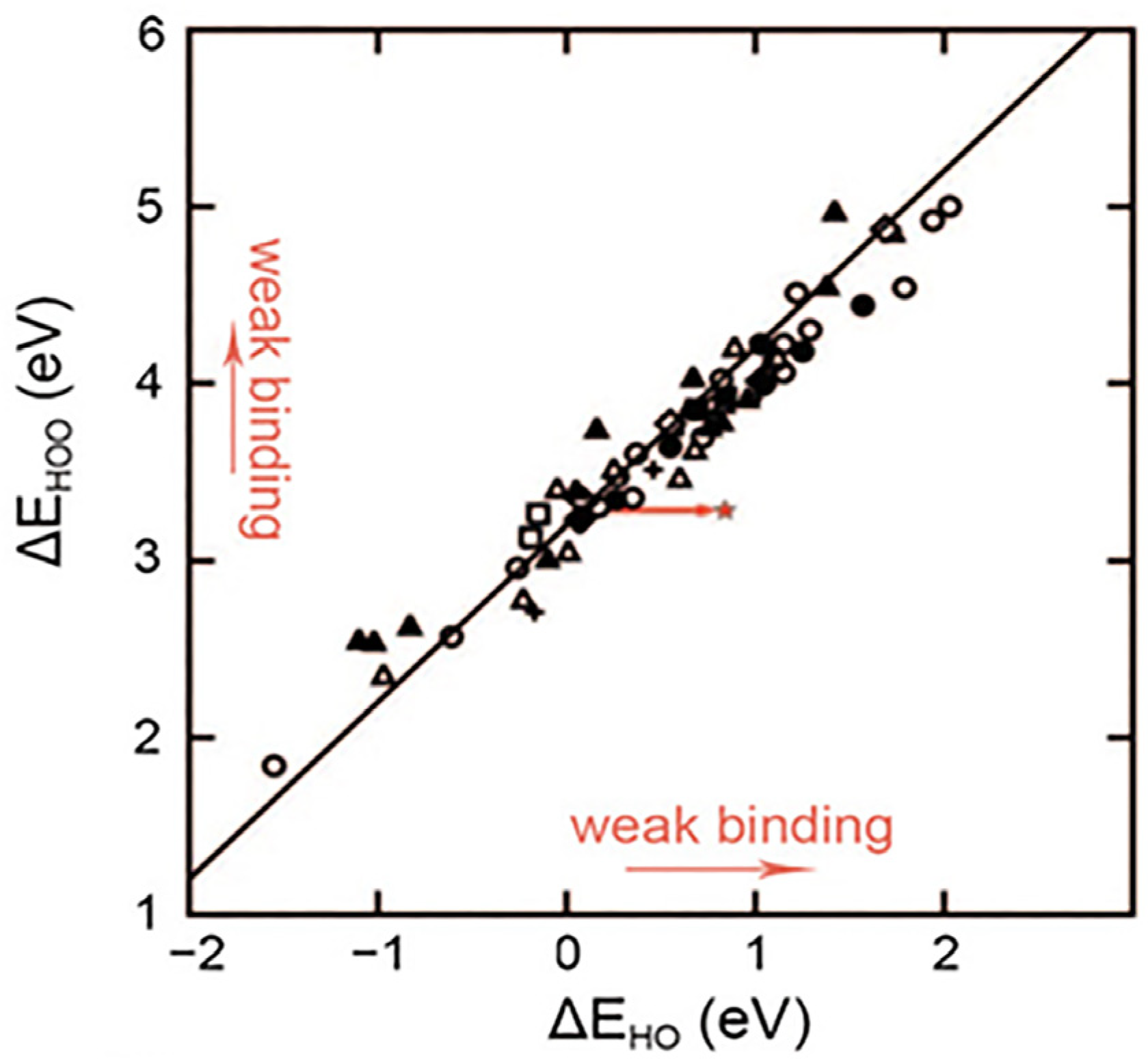
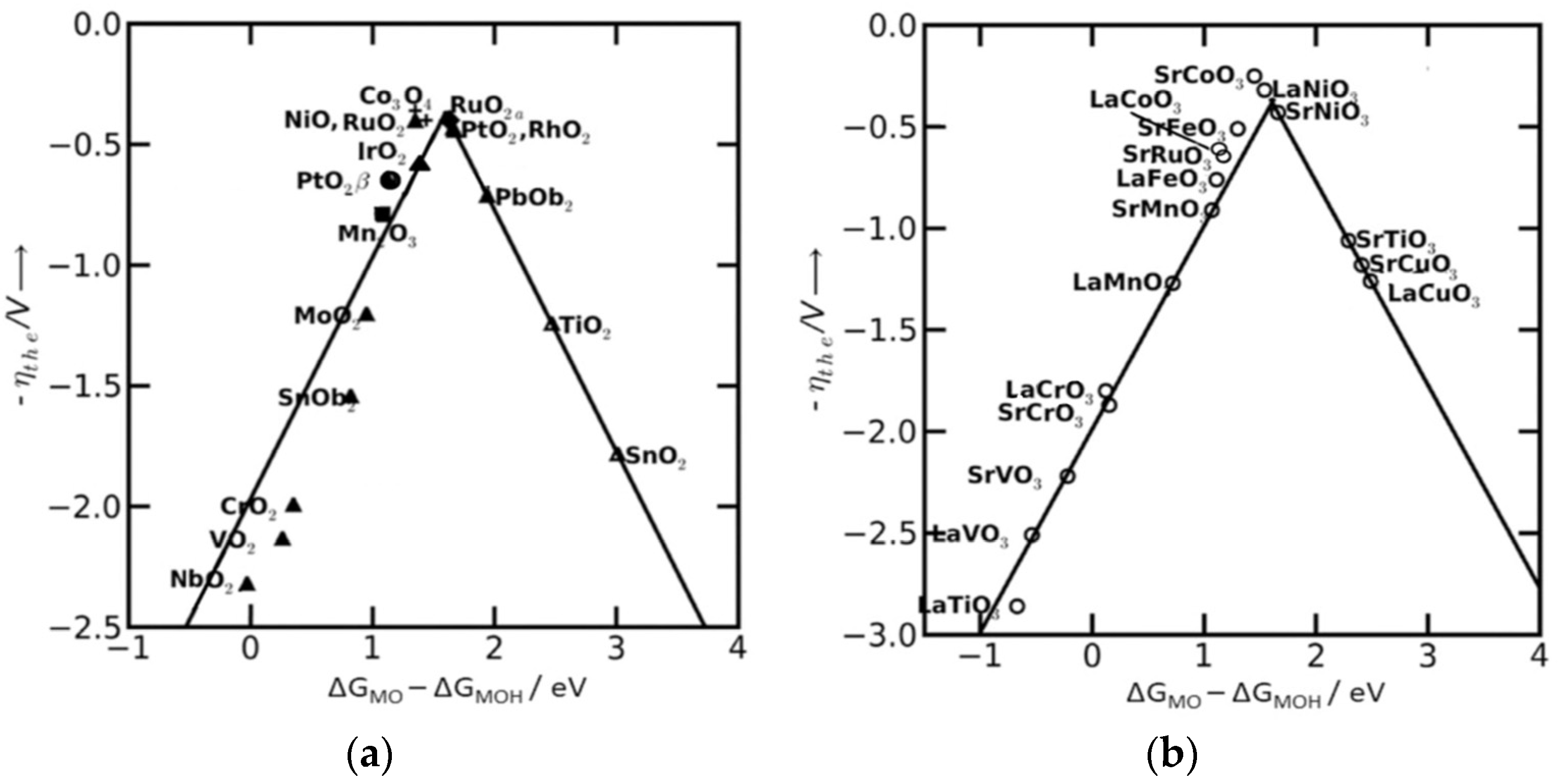
2.1. Mechanism of Adsorbate Evolution and Its Scaling Relationship
2.2. Design of Efficient Electrocatalysts Based on Scaling Relations
2.3. Strategies for Breaking Scaling Relationships
2.3.1. OOH* Stabilization Independent on OH*
Introduction of a Second Adsorption-Relation Site
Formation of Hydrogen Bond (Or Introduction of Nanoscopic Confinement)
Introducing a Proton Acceptor (OO* + H*)
2.3.2. Direct O-O Coupling in the Absence of *OOH
Oxo Radical Coupling
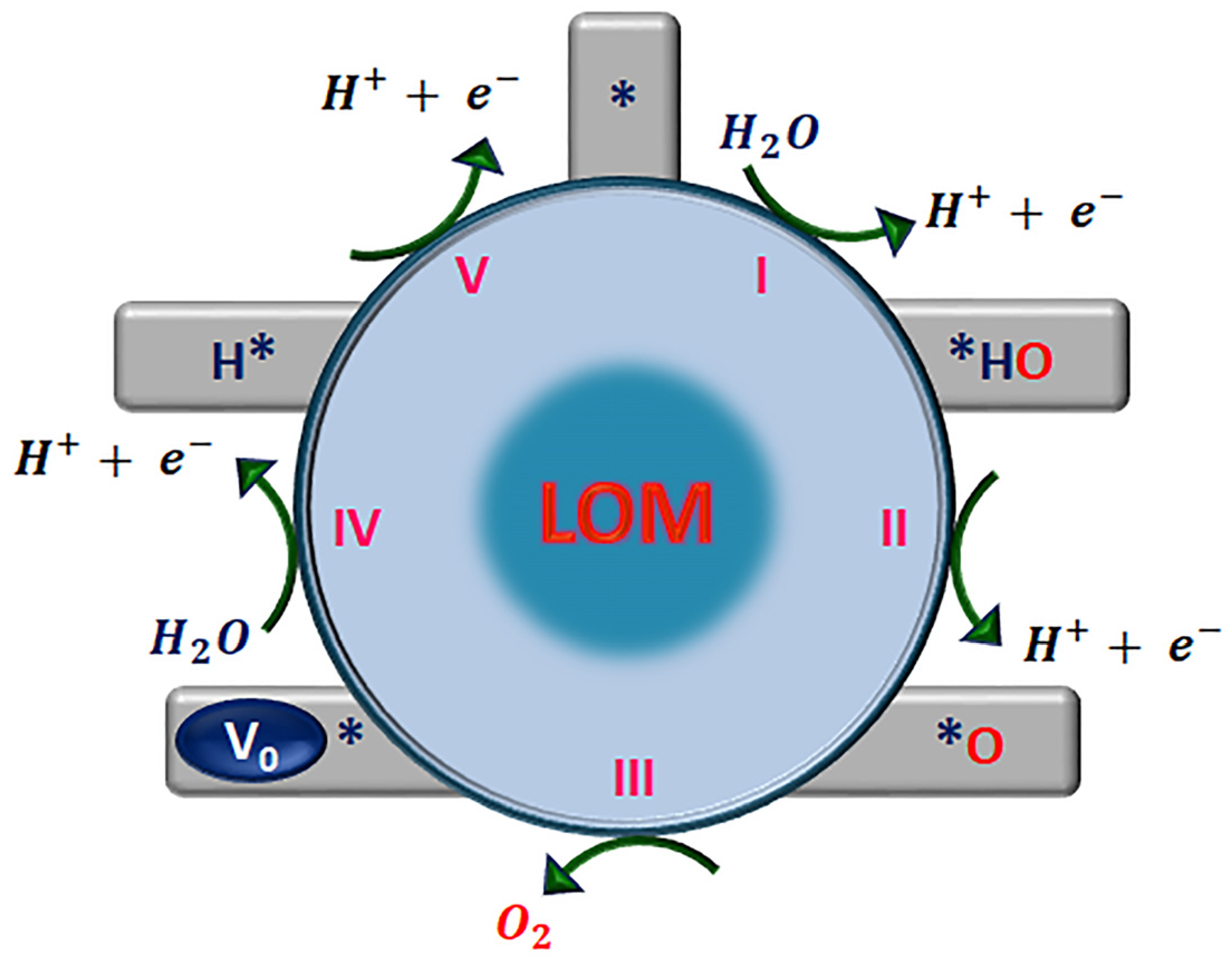
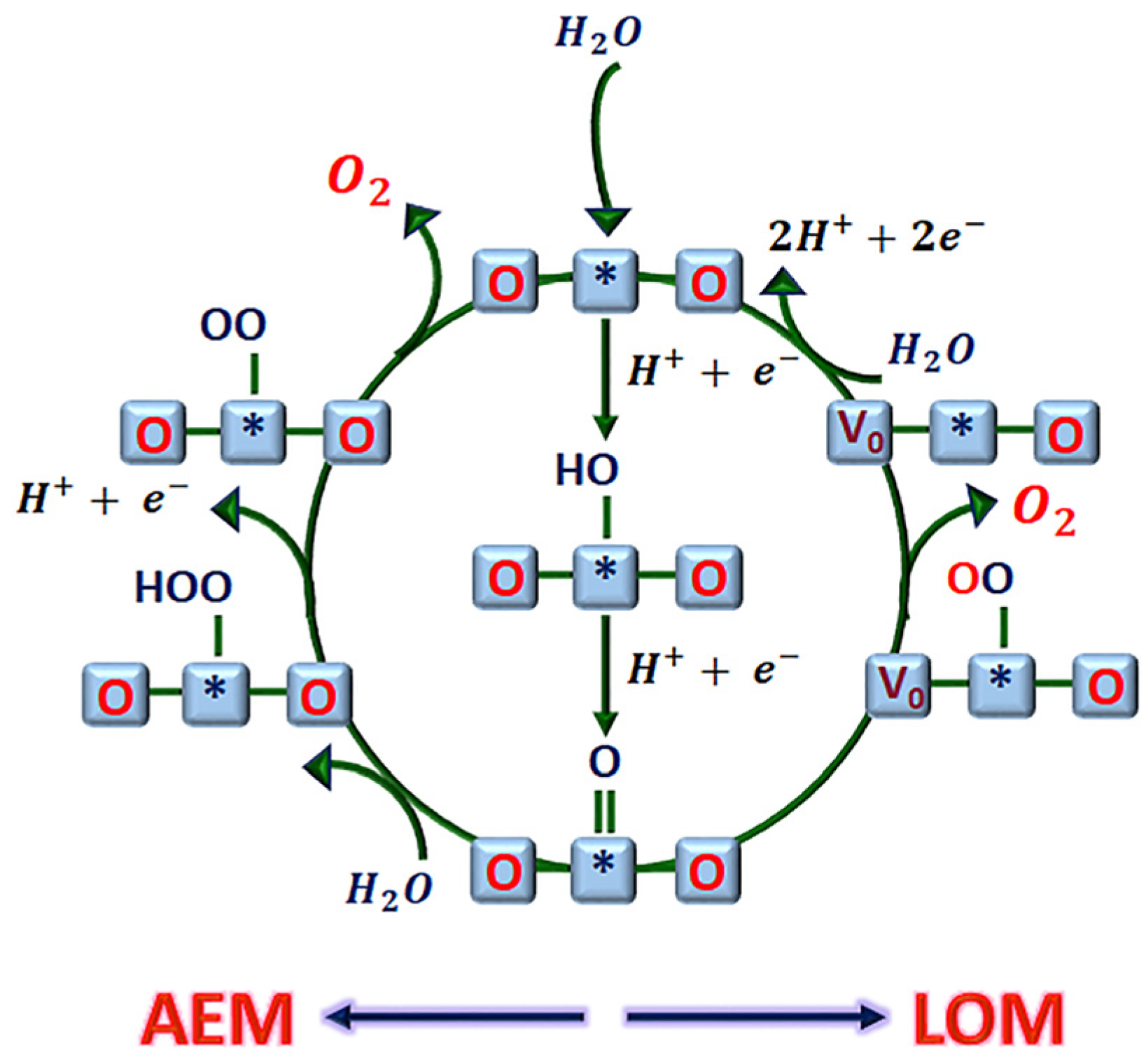
Lattice-Oxygen-Mediated Mechanism
3. Oxygen Evolution Reaction Electrocatalysts
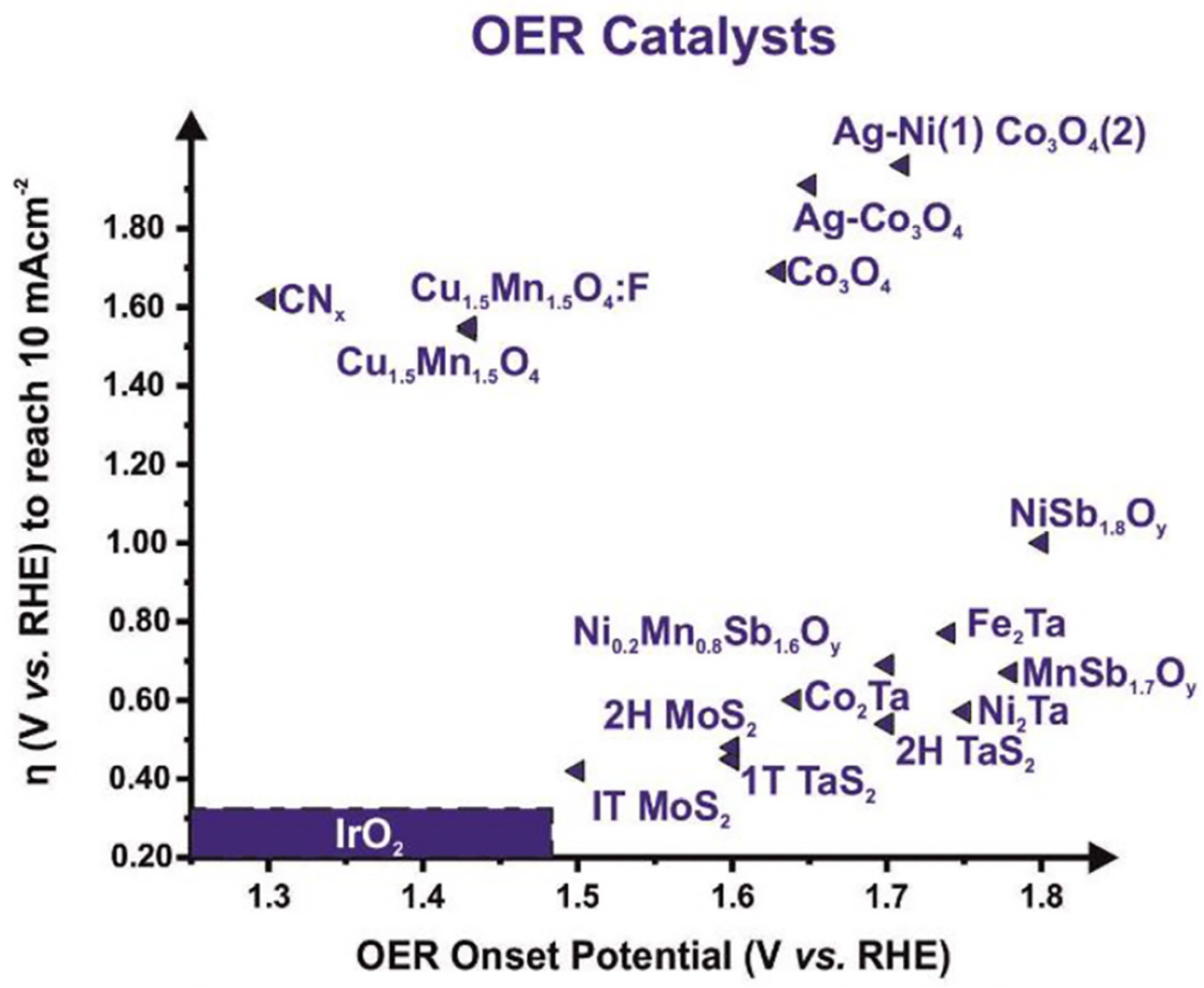
3.1. Single-Component and Binary-Component Electrocatalysts and Effect of Dopants
3.2. Ternary and Quaternary Component Electrocatalysts
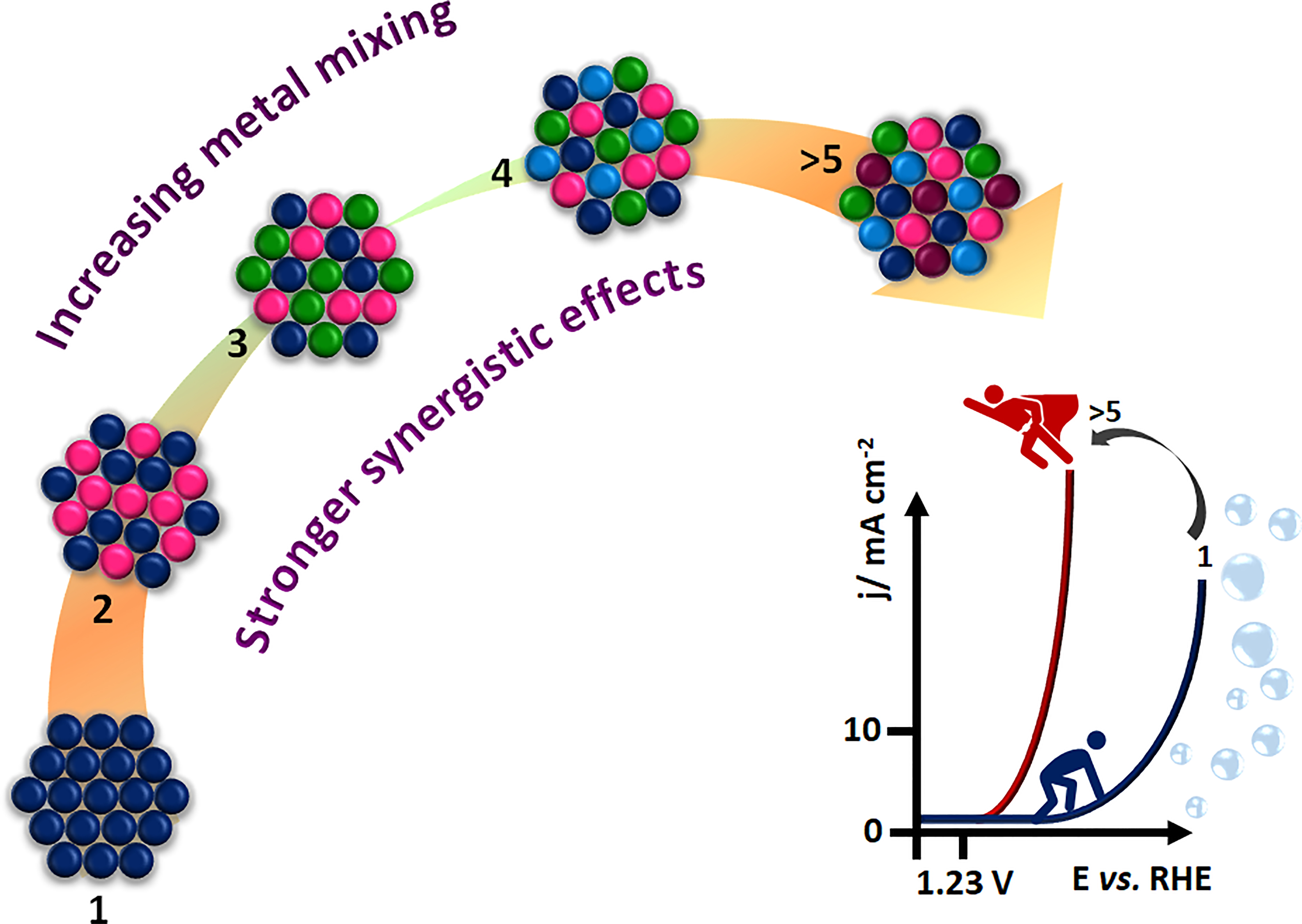
3.3. Multicomponent Electrocatalysts
3.4. Increased Functionality of Single- to Multielement Materials
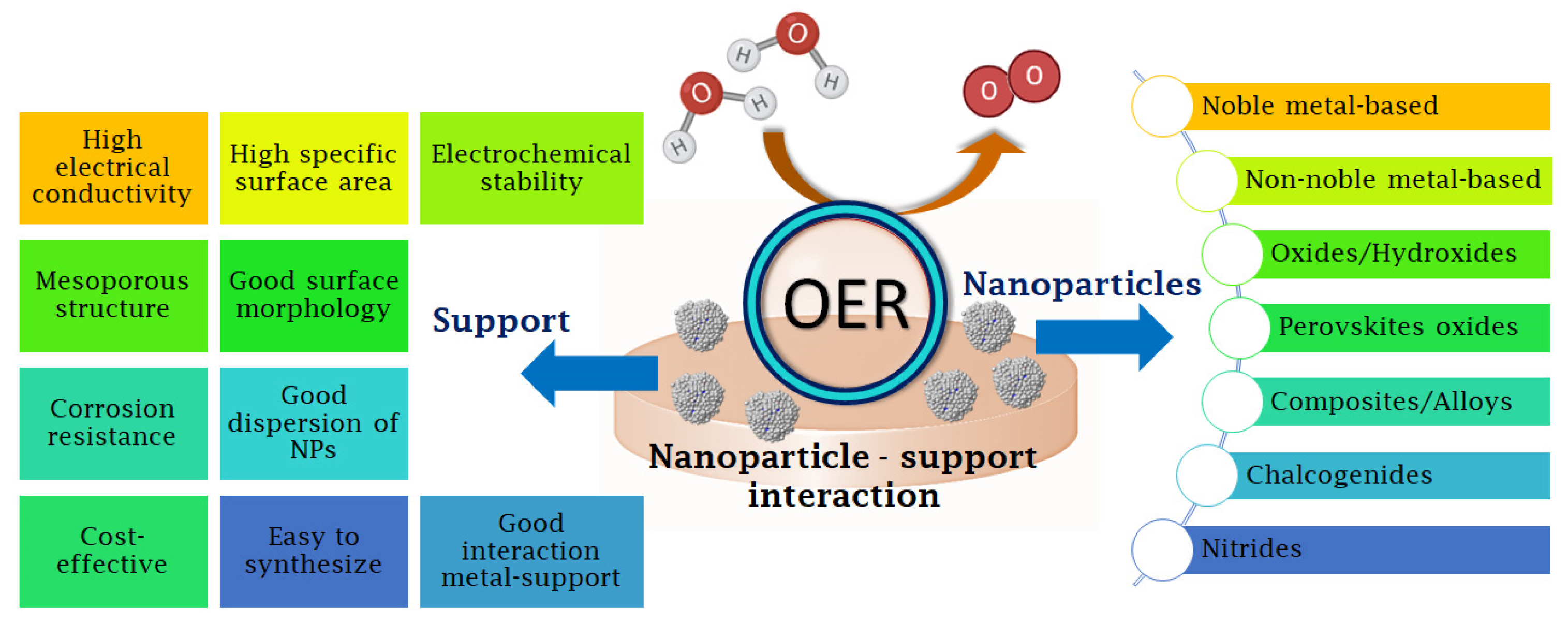
4. Effect of Supports
5. Summary and Outlook
- The need not to lose sight of the objective of having a unifying descriptor of catalytic activity. However, it is equally essential to carry out theoretical and experimental investigations to understand the factors involved in poor stability in strongly acidic and corrosive environments. It is critically necessary to establish structure–stability relationships, so proposing descriptors that relate catalyst structure, activity, and stability is the key to rational design and will represent a valuable advance in the fundamentals of the OER.
- Future research should try not only to break the scaling relationship but also to optimize the binding energies to achieve a lower overpotential. However, understanding and controlling the competition between different reaction mechanisms remains a major challenge.
- Promote the development of electrocatalysts based on earth-abundant materials, such as low-cost, environmentally friendly transition metals with significant activity, to replace the use of very expensive and scarce iridium in acidic OER electrocatalysts, which is a bottleneck at the high-scale PEM-WE level.
- In the meantime, electrocatalytic processes that are dynamic, surface-active sites, or active species can be generated during the electrochemical process, so it is necessary to pay special attention to the synthesis of the precatalytic material as well as to the operating conditions for its activation. Also, the self-reconstruction of the surface during the OER process can be beneficial or detrimental to the activity and/or stability of the electrocatalyst, usually by agglomeration, dissolution, and detachment of the catalyst. However, the structural reconstruction mechanisms are not entirely clear, so it is necessary to encourage the application of in situ and/or in operando techniques under electrochemical operating conditions, as these are a cornerstone in the development of highly active and stable electrocatalysts that allow a better understanding of the structural evolution of the materials in real time, as well as the nature of the key intermediates and their behavior. Therefore, these techniques allow in-depth understanding of the structure–activity–stability relationship of the OER process.
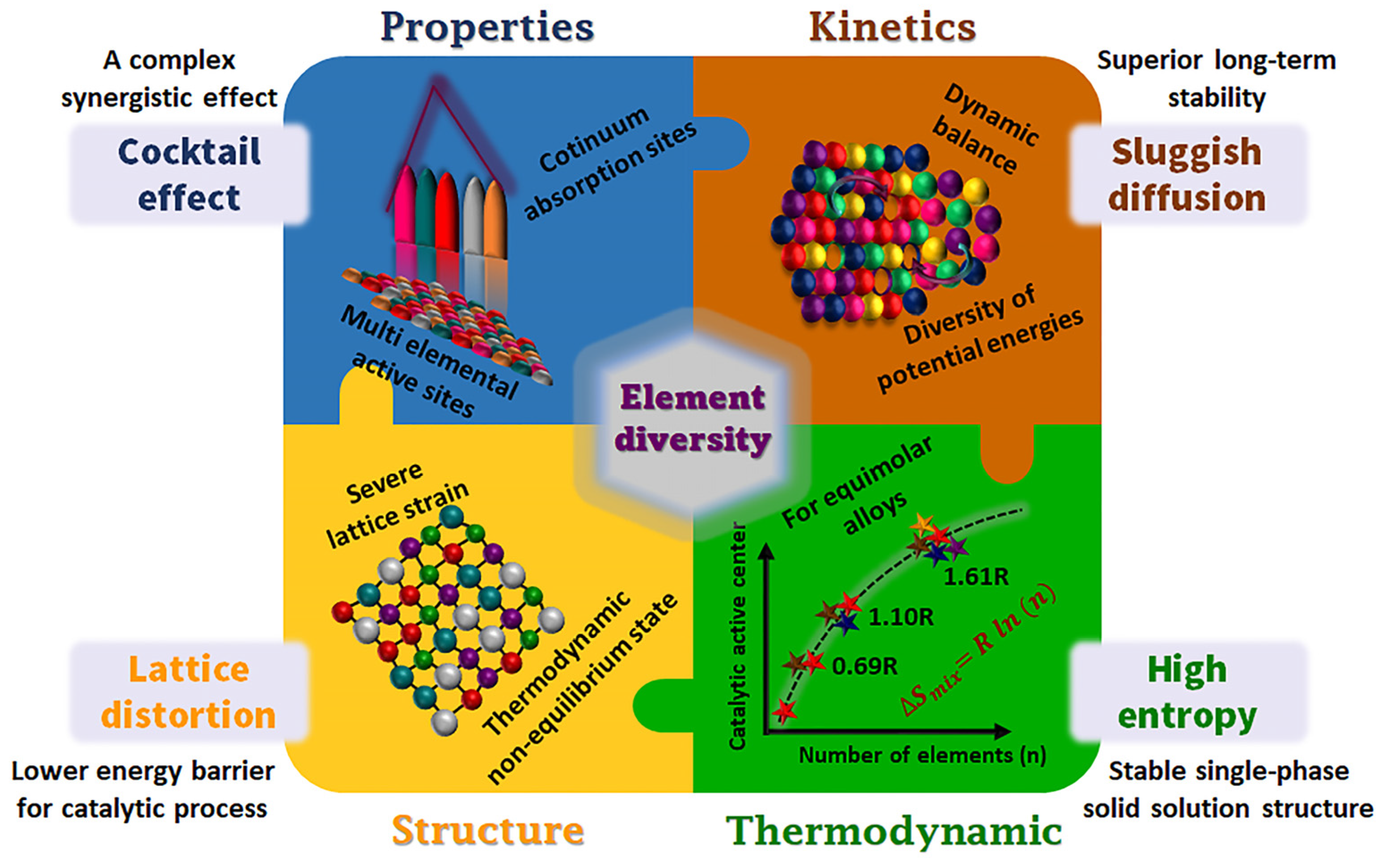
- The recent perspective based on the concept of high entropy to design advanced materials is opening a new space of many possible combinations, leading us to rethink and explore new phenomena, theories, and applications. Certainly, this poses a great challenge, both theoretically and experimentally. Compared to minor component alloys, HEAs are still at a nascent stage. However, HEAs are promising candidates for improving the performance of both AWEs and PEM-WEs. This class of materials is of particular interest for replacing noble metals such as Ir, Ru, and Pt in acidic media. The synergistic effect of HEAs is mainly based on their elemental diversity; this requires new research methods, e.g., statistical methods, machine learning, data-driven combinatorial synthesis, high throughput, and screening techniques, for the efficient exploration of a suitable composition of HEAs as a cost-effective material and, in the not-too-distant future, more attractive properties might be discovered. However, there are still many fundamental concepts to be clarified and many questions and challenges to be addressed in the field of synthesis, characterization, and analysis of electrochemical data, identification of active sites, origin of high activity, etc.
- To improve data quality, standardized measurements of HER and OER properties are necessary as demonstrated by Hung et al. [160]. They recognized that 3D electrodes, such as metal foam materials (Ni, Fe, Co, IrNi-FeNi3 on Ni, and other foams) are an interesting alternative, which exhibit high catalytic activities; however, the impact of parameters such as active area, thickness, porosity, capillarity, and purity need to be better understood. Some of them are not easy to determine experimentally. This contribution presents alternatives for ECSA calculation and highlights that an objective evaluation improves the reliability of the results enriches our fundamental understanding of electrocatalytic activity. Therefore, the implementation of standardized electrochemical parameters to evaluate the performance of electrocatalysts is an area of opportunity to ensure their industrial application.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
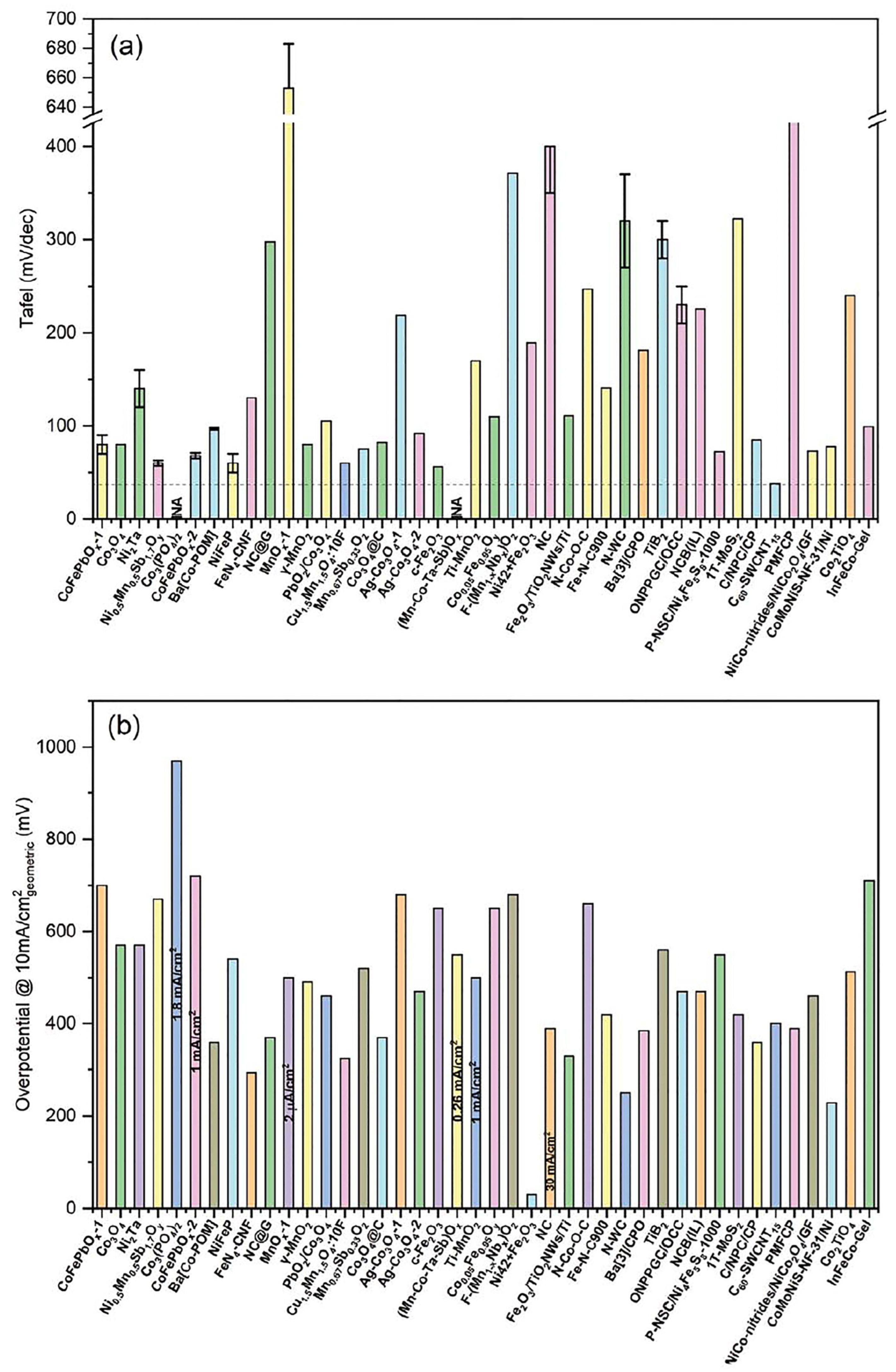
| # | Catalysts | Element(s) | Support Material | Strategy Design | Synthesis Method | Overpotential @ 10 mA cm−2 | Tafel Slope | Stability | Acidic Media | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| mV vs. RHE | mV dec−1 | |||||||||
| Single element | ||||||||||
| 1 | Fe2O3 | Fe | Ti foil | Polymorphism structures | Spray pyrolysis | 650 | 56 | 24 h @ 10 mA cm−2 | 0.5 M H2SO4 | [2] |
| 2 | Co3O4 crystalline | Co | Fluorine-doped tin oxide (FTO) | Engineering interface | Thermal annealing | 570 | 80 | 12 h @ 10 mA cm−2 | 0.5 M H2SO4 | [161] |
| 3 | γ-MnO2 | Mn | FTO | Thermal decomposition | 489 | 79 | 8000 h @ 10 mA cm−2 | 1.0 M H2SO4 | [112] | |
| 4 | Co3O4 nanosheets | Co | Carbon paper | Electroplating and calcination | 370 | 82 | 86.8 h @ 100 mA cm−2 | 0.5 M H2SO4 | [162] | |
| 5 | IrOx@IrO2 | Ir | w/o S | Structural engineering | Adams fusion | 309 | 45 | 6 h @ 10 mA cm−2 | 0.1 M HClO4 | [163] |
| 6 | Mesoporous iridium nanosheets | Ir | Vulcan XC-72 carbon | Mesoporous chemistry | Wet chemical/ micelles | 240 | 49 | 8 h @ 10 mA cm−2 | 0.5 M H2SO4 | [164] |
| 7 | Mn3O4 nanoplates | Mn | --- | Crystal and geometric structure transformation | Rapid thermal annealing | 210 | 54.24 | 20 h @ 10 mA cm−2 | 0.5 M H2SO4 | [113] |
| Binary elements | ||||||||||
| 8 | Fe5Si3 | Fe, Si | w/o S | Multiphase structure | Sintering process | 735 | 381.8 | 1000 cycles | 0.5 M H2SO4 | [165] |
| 9 | Ag-Co3O4 mesoporous | Co, Ag | FTO | Doping with foreign element (Ag) | Electrodeposition, hydrothermal, and calcination | 680 | 219 | 10 h @ 1.6 V Ag/AgCl | 0.5 M H2SO4 | [166] |
| 10 | C3N4-CNT-CF | C, N | Carbon fiber | Metal-free based | Annealing treatment | 570 | 129 | 14 h @ 1.63 V RHE | 0.5 M H2SO4 | [167] |
| 11 | TiO2-modified MnO2 | Mn, Ti | Polycrystalline gold disks | Engineering surface | Sputtering deposition | 510 @ 1 mA cm−2 | 170 | 265 h @ 1.8 V RHE | 0.05 M H2SO4 | [168] |
| 12 | Ag-Co3O4 (400) | Co, Ag | w/o S | Doping with foreign element (Ag) | Hydrothermal and annealed (400 °C) | 470 | 92 | 1000 cycles | 0.5 M H2SO4 | [169] |
| 13 | Co3O4/CeO2 | Co, Ce | FTO | Engineering interface | Electrodeposition | 423 | 88.1 | - | 0.05 M H2SO4 | [170] |
| 14 | 2D MoS2 nanosheets | Mo, S | w/o S | Polymorphism/ heteroatoms | Exfoliation and deposition | 420 | 322 | 2 h @ 10 mA cm−2 | 0.5 M H2SO4 | [171] |
| 15 | W0.57Ir0.43O3-δ | Ir, W | FTO | Structural engineering | Plasma synthesis | 370 | 125 | 0.56 h | 1.0 M H2SO4 | [172] |
| 16 | Cu0.3Ir0.7Oδ | Ir, Cu | Ti plate | Doping with foreign element (Cu) | Hydrothermal and annealing | 351 | 63 | 1.67 h @ 1.68 V RHE | 0.1 M HClO4 | [173] |
| 17 | IrTe NTs | Ir, Te | Vulcan XC-72 carbon | Surface engineering | Galvanic replacement | 290 | 60.3 | 2000 cycles | 0.1 M HClO4 | [174] |
| 18 | IrCu0.77 | Ir, Cu | Vulcan XC-72 carbon | Polyol method | 282 | 78.6 | --- | 0.1 M HClO4 | [175] | |
| 19 | RuNiOx | Ru, Ni | Carbon fiber paper | Dealloying treatment | Dip coating | 280 @ 50 mA cm−2 | 51.91 | 10 h @ 1.5 V RHE | 0.5 M H2SO4 | [176] |
| 20 | IrMoOx nanofibers | Ir, Mo | --- | Electronic modulation | Electrospinning and calcination | 267 | 46.09 | 30 h | 0.5 M H2SO4 | [177] |
| 21 | Fe2O3/TiO2 NWs/Ti | Fe, Ti | Ti foam | Structural engineering | Facile ion exchange process and calcination | 230 @ 1 mA cm−2 | 110.7 | 20 h @ 1.9 V SCE | 0.5 M H2SO4 | [178] |
| 22 | Mn0.73Ru0.27O2-δ | Ru, Mn | w/o S | Oxygen vacancies | Pyrolysis | 208 | 65.3 | 10 h @ 10 mA cm−2 | 0.5 M H2SO4 | [179] |
| 23 | Zn-doped RuO2 hollow nanorod | Ru, Zn | w/o S | Doping with foreign element (Zn) | Annealing process under air | 206 | 45.65 | 30 h @ 10 mA cm−2 | 0.5 M H2SO4 | [180] |
| 24 | Mn-doped RuO2 nanocrystals | Ru, Mn | w/o S | Doping with foreign element (Mn) | Annealing process under air | 158 | 42.94 | 10 h @ 10 mA cm−2 | 0.5 M H2SO4 | [181] |
| 25 | Ru/MnO2 | Ru, Mn | w/o S | Surface engineering/self-reconstruction | One-step cation exchange method | 161 | 29.4 | 10 h @ 10 mA cm−2 | 0.1 M HClO4 | [50] |
| Ternary elements | ||||||||||
| 26 | Ni0.5Mn0.5Sb1.7Oγ | Ni, Mn, Sb | Antimony-doped tin oxide | Phase restructuring | Sputter deposition | 672 | 85 | 168 h @ 10 mA cm−2 | 1.0 M H2SO4 | [182] |
| 27 | Ni40Fe40P20 | Ni, Fe, P | w/o S | Heteroatoms | Melting spinning | 540 | 40 | 30 h @ 10 mA cm−2 | 0.05 M H2SO4 | [183] |
| 28 | Mo-Co9S8 | Mo, Co, S | Carbon cloth | Heteroatoms | solvothermal method | 370 | 90.3 | 24 h @ 1.6 V RHE | 0.5 M H2SO4 | [184] |
| 29 | F-doped Cu1.5Mn1.5O4 nanoparticles | Cu, Mn, F | Porous Ti foil | Doping with foreign element (F) | Chemical synthesis and heat treatment | 320 @ 9.15 mA cm−2 | 60 | 24 h @ 1.55 V RHE | 0.5 M H2SO4 | [185] |
| 30 | CP@NCNT | Co, P, N | N-doped CNT | Heteroatoms | Spray drying | 317 | 75 | 24 h @ 15 mA cm−2 | 0.5 M H2SO4 | [186] |
| 31 | IrNiCu DNF/C | Ir, Ni, Cu | Vulcan XC-72 carbon | Nanoframe structure | Chemical etching | 303 | 48 | 2500 cycles | 0.1 M HClO4 | [187] |
| 32 | FeN4/NF/EG | Fe, N, C | Exfoliated graphene | Dual element-doping (N, C) | Electrodeposition and carbonization | 294 | 129 | 24 h @ 20 mA cm−2 | 0.5 M H2SO4 | [188] |
| 33 | Co-doped IrCu | Ir, Co, Cu | Vulcan XC-72 carbon | Doping with foreign element (Co) | Chemical etching | 293 | 50 | 2000 cycles | 0.1 M HClO4 | [189] |
| 34 | W-Ir-B | Ir, W, B | w/o S | Biphasic structure | Arc melting and drop casting | 291 | 78 | 120 h @ 100 mA cm−2 | 0.5 M H2SO4 | [190] |
| 35 | RuO2/(CoMn)3O4 | Ru, Co, Mn | Carbon cloth | Interface engineering | Hydrothermal process | 270 | 77 | 24 h @ 10 mA cm−2 | 0.5 M H2SO4 | [116] |
| 36 | HNC-Co | Co, N, C | Carbon paper | Dual element-doping (N, C) | Polymerization, reduction, and pyrolysis | 265 | 85 | 100 h | 0.5 M H2SO4 | [191] |
| 37 | SrTi0.67Ir0.33O3 | Ir, Sr, Ti | w/o S | Ir doping | Polymerized complex method | 247 | 43 | 20 h @ 10 mA cm−2 | 0.1 HClO4 | [192] |
| 38 | C-RuO2-RuSe-5 | Ru, Se, C | w/o S | Inducing interstitial atoms | Hydrothermal method | 212 | 49.5 | 30 h @ 10 mA cm−2 | 0.5 M H2SO4 | [193] |
| 39 | Ru NCs/Co2P Hollow microspheres | Ru, Co, P | w/o S | Heteroatoms | Hydrothermal | 197 | 89 | 10 h | 0.5 M H2SO4 | [194] |
| Quaternary elements | ||||||||||
| 40 | InFeCo-CCP | Fe, Co, In, N | w/o S | Organic/ inorganic polymeric | Coordination-substitution polymerization | 710 | 99 | 48 h @ 1.75 V RHE | 0.5 M H2SO4 | [195] |
| 41 | P-NSC/Ni4Fe5S8 | Ni, Fe, N, S | w/o S | Heteroatoms/ porous | Pyrolysis (1000 °C) | 550 | 72.1 | 10,000 cycles | 0.5 M H2SO4 | [196] |
| 42 | Fe35Ni35Co10P20 | Fe, Ni, Co, P | w/o S | Amorphous multialloy | Arc-melting technique | 497 | 79 | 20 h @ 1.73 V RHE | 0.5 M H2SO4 | [197] |
| 43 | Ni42Li2O5 | Steel (Ni, Fe, Mn), Li | AISI Ni42 steel | Surface engineering | Electrooxidation | 445 | 260 | 5.6 h @ 10 mA cm−2 | 0.05 M H2SO4 | [198] |
| 44 | Mn-doped FeP/Co3(PO4)2 | Fe, Co, Mn, P | Carbon cloth | Heteroatoms | Hydrothermal | 390 | 472 | 10,000 cycles | 0.5 M H2SO4 | [199] |
| 45 | Ba[Co-POM]/CP | Co, W, Ba, P | Carbon paste | Polyoxometalate | Metathesis | 361 | 97 | 24 h @ 1.48 V RHE | 1.0 M H2SO4 | [200] |
| 46 | CoMoNiS-NF-31 | Co, Mo, Ni, S | Nickel foam | Heteroatoms/ hierarchical structure | One-pot hydrothermal | 228 | 78 | --- | 0.5 M H2SO4 | [121] |
| Multi-elements | ||||||||||
| 47 | FeCoNiMnW | Fe, Co, Ni, Mn, W | Carbon paper | Multimetal alloy | Electrodeposition | 332 | 145 | 1 h | 0.5 H2SO4 | [201] |
| 48 | Al89Ag1Au1Co1 Cu1Fe1Ir1Ni1Pd1 Pt1Rh1Ru1 | Ir, Ru, Pt, Pd, Au, Rh, Ag, Al, Co, Cu, Fe, Ni, | w/o S | Multimetal alloy | Arc melting and one-step dealloying | 258 | 84.2 | 11.11 h @ 10 mA cm−2 | 0.5 H2SO4 | [202] |
| 49 | FeCoNiIrRu | Ir, Ru, Fe, Co, Ni | Carbon nanofibers | Multimetal alloy | Electrospinning | 241 | 153 | 4 h | 0.5 H2SO4 | [203] |
| 50 | Al96Ni1Co1Ir1Mo1 | Ir, Al, Ni, Co, Mo | Carbon powder | Multimetal alloy | Induction-melting furnace and chemical etching | 233 | 55.2 | 7000 cycles | 0.5 H2SO4 | [126] |
| 51 | IrPdRhMoW | Ir, Pd, Rh, Mo, W | w/o S | Multimetal alloy/structural engineering | Oil-phase method | 188 | --- | 100 h @ 100 mA cm−2 | 0.5 H2SO4 | [127] |
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AEM | Alkaline electrolyte membrane |
| AMO | Amorphous manganese oxides |
| AWE | Alkaline water electrolyzers |
| BEDPs | Binding energy distribution patterns |
| CPET | Concerted proton–electron transfer |
| CUS | Coordinatively unsaturated |
| DEMS | Differential electrochemical mass spectrometry |
| DFT | Density functional theory |
| ESSI | Electrochemical step symmetry index |
| HCP | Hexagonal close-packed structure |
| HEAs | High-entropy alloys |
| HER | Hydrogen evolution reaction |
| IMCs | Intermetallic compounds |
| LDH | Double-layered hydroxides |
| LOM | Lattice oxygen mechanism |
| MOFs | Metalorganic frameworks |
| MD | Molecular dynamics |
| MWCNTs | Multiwalled carbon nanotubes |
| NAP | Near ambient pressure |
| NPs | Nanoparticles |
| OER | Oxygen evolution reaction |
| ONB | Oxygen nonbonding states |
| PEM | Proton exchange membrane |
| PEM-WE | Proton exchange membrane water electrolyzer |
| RDS | Rate-determining step |
| RHE | Reversible hydrogen electrode |
| RPS | Rate-determining potential step |
| SHE | Standard hydrogen electrode |
| SI-SECM | Surface interrogation scanning electrochemical microscopy |
| STEM-EDS | Scanning transmission electron microscopy–energy dispersive X-ray spectroscopy |
| UV-vis | Ultraviolet–visible |
| XAS | X-ray absorption spectroscopy |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray diffraction |
References
- Aizarani, J. Global Primary Energy–Statistics & Facts. Available online: https://www.statista.com/statistics/265598/consumption-of-primary-energy-worldwide/ (accessed on 16 September 2023).
- Kwong, W.L.; Lee, C.C.; Shchukarev, A.; Björn, E.; Messinger, J. High-performance iron (III) oxide electrocatalyst for water oxidation in strongly acidic media. J. Catal. 2018, 365, 29–35. [Google Scholar] [CrossRef]
- Daiyan, R.; MacGill, I.; Amal, R. Opportunities and challenges for renewable power-to-X. ACS Energy Lett. 2020, 5, 3843–3847. [Google Scholar] [CrossRef]
- Ismail, N.; Qin, F.; Fang, C.; Liu, D.; Liu, B.; Liu, X.; Wu, Z.-L.; Chen, Z.; Chen, W. Electrocatalytic acidic oxygen evolution reaction: From nanocrystals to single atoms. Aggregate 2021, 2, e106. [Google Scholar] [CrossRef]
- Shan, J.; Zheng, Y.; Shi, B.; Davey, K.; Qiao, S.-Z. Regulating Electrocatalysts via Surface and Interface Engineering for Acidic Water Electrooxidation. ACS Energy Lett. 2019, 4, 2719–2730. [Google Scholar] [CrossRef]
- Kim, T.; Kim, B.; Kwon, T.; Kim, H.Y.; Kim, J.Y.; Lee, K. Multimetallic nanostructures for electrocatalytic oxygen evolution reaction in acidic media. Mater. Chem. Front. 2021, 5, 4445–4473. [Google Scholar] [CrossRef]
- Reier, T.; Nong, H.N.; Teschner, D.; Schlögl, R.; Strasser, P. Electrocatalytic Oxygen Evolution Reaction in Acidic Environments—Reaction Mechanisms and Catalysts. Adv. Energy Mater. 2017, 7, 1601275. [Google Scholar] [CrossRef]
- An, L.; Wei, C.; Lu, M.; Liu, H.; Chen, Y.; Scherer, G.G.; Fisher, A.C.; Xi, P.; Xu, Z.J.; Yan, C.-H. Recent development of oxygen evolution electrocatalysts in acidic environment. Adv. Mater. 2021, 33, 2006328. [Google Scholar] [CrossRef]
- Buttler, A.; Spliethoff, H. Current status of water electrolysis for energy storage, grid balancing and sector coupling via power-to-gas and power-to-liquids: A review. Renew. Sustain. Energy Rev. 2018, 82, 2440–2454. [Google Scholar] [CrossRef]
- Banham, D.; Ye, S.; Pei, K.; Ozaki, J.-I.; Kishimoto, T.; Imashiro, Y. A review of the stability and durability of non-precious metal catalysts for the oxygen reduction reaction in proton exchange membrane fuel cells. J. Power Sources 2015, 285, 334–348. [Google Scholar] [CrossRef]
- Patra, S.G.; Meyerstein, D. On the mechanism of heterogeneous water oxidation catalysis: A theoretical perspective. Inorganics 2022, 10, 182. [Google Scholar] [CrossRef]
- Exner, K.S. On the lattice oxygen evolution mechanism: Avoiding pitfalls. ChemCatChem 2021, 13, 4066–4074. [Google Scholar] [CrossRef]
- Wei, C.; Rao, R.R.; Peng, J.; Huang, B.; Stephens, I.E.L.; Risch, M.; Xu, Z.J.; Shao-Horn, Y. Recommended practices and benchmark activity for hydrogen and oxygen electrocatalysis in water splitting and fuel cells. Adv. Mater. 2019, 31, 1806296. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Ferrer, I.M.; Chatman, S.M.; Peters, J.C.; Jaramillo, T.F. Benchmarking hydrogen evolving reaction and oxygen evolving reaction electrocatalysts for solar water splitting devices. J. Am. Chem. Soc. 2015, 137, 4347–4357. [Google Scholar] [CrossRef]
- Yan, Z.; Liu, H.; Hao, Z.; Yu, M.; Chen, X.; Chen, J. Electrodeposition of (hydro)oxides for an oxygen evolution electrode. Chem. Sci. 2020, 11, 10614–10625. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Soriano-López, J.; Schmitt, W.; García-Melchor, M. Computational modelling of water oxidation catalysts. Curr. Opin. Electrochem. 2018, 7, 22–30. [Google Scholar] [CrossRef]
- Cherevko, S.; Geiger, S.; Kasian, O.; Kulyk, N.; Grote, J.-P.; Savan, A.; Shrestha, B.R.; Merzlikin, S.; Breitbach, B.; Ludwig, A.; et al. Oxygen and hydrogen evolution reactions on Ru, RuO2, Ir, and IrO2 thin film electrodes in acidic and alkaline electrolytes: A comparative study on activity and stability. Catal. Today 2016, 262, 170–180. [Google Scholar] [CrossRef]
- Yuan, J.; Cheng, X.; Lei, C.; Yang, B.; Li, Z.; Luo, K.; Koko Lam, K.H.; Lei, L.; Hou, Y.; Ostrikov, K.K. Bimetallic oxyhydroxide as a high-performance water oxidation electrocatalyst under industry-relevant conditions. Engineering 2021, 7, 1306–1312. [Google Scholar] [CrossRef]
- Suen, N.-T.; Hung, S.-F.; Quan, Q.; Zhang, N.; Xu, Y.-J.; Chen, H.M. Electrocatalysis for the oxygen evolution reaction: Recent development and future perspectives. Chem. Soc. Rev. 2017, 46, 337–365. [Google Scholar] [CrossRef]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef] [PubMed]
- Lyu, F.; Wang, Q.; Choi, S.M.; Yin, Y. Noble-metal-free electrocatalysts for oxygen evolution. Small 2019, 15, 1804201. [Google Scholar] [CrossRef]
- Song, J.; Wei, C.; Huang, Z.-F.; Liu, C.; Zeng, L.; Wang, X.; Xu, Z.J. A review on fundamentals for designing oxygen evolution electrocatalysts. Chem. Soc. Rev. 2020, 49, 2196–2214. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, Q.; Song, B.; Xu, P. Regulation strategy of transition metal oxide-based electrocatalysts for enhanced oxygen evolution reaction. Acc. Mater. Res. 2022, 3, 1088–1100. [Google Scholar] [CrossRef]
- Xin, Y.; Li, S.; Qian, Y.; Zhu, W.; Yuan, H.; Jiang, P.; Guo, R.; Wang, L. High-entropy alloys as a platform for catalysis: Progress, challenges, and opportunities. ACS Catal. 2020, 10, 11280–11306. [Google Scholar] [CrossRef]
- Bokov, D.; Turki Jalil, A.; Chupradit, S.; Suksatan, W.; Javed Ansari, M.; Shewael, I.H.; Valiev, G.H.; Kianfar, E. Nanomaterial by sol-gel method: Synthesis and application. Adv. Mater. Sci. Eng. 2021, 2021, 5102014. [Google Scholar] [CrossRef]
- Baig, N.; Kammakakam, I.; Falath, W. Nanomaterials: A review of synthesis methods, properties, recent progress, and challenges. Mater. Adv. 2021, 2, 1821–1871. [Google Scholar] [CrossRef]
- Tiwari, J.N.; Tiwari, R.N.; Kim, K.S. Zero-dimensional, one-dimensional, two-dimensional and three-dimensional nanostructured materials for advanced electrochemical energy devices. Prog. Mater. Sci. 2012, 57, 724–803. [Google Scholar] [CrossRef]
- Vazhayil, A.; Vazhayal, L.; Thomas, J.; Ashok, C.S.; Thomas, N. A comprehensive review on the recent developments in transition metal-based electrocatalysts for oxygen evolution reaction. Appl. Surf. Sci. Adv. 2021, 6, 100184. [Google Scholar] [CrossRef]
- Chen, Z.; Guo, L.; Pan, L.; Yan, T.; He, Z.; Li, Y.; Shi, C.; Huang, Z.-F.; Zhang, X.; Zou, J.-J. Advances in oxygen evolution electrocatalysts for proton exchange membrane water electrolyzers. Adv. Energy Mater. 2022, 12, 2103670. [Google Scholar] [CrossRef]
- Li, J. Oxygen evolution reaction in energy conversion and storage: Design strategies under and beyond the energy scaling relationship. Nano-Micro Lett. 2022, 14, 112. [Google Scholar] [CrossRef]
- Sanchez Casalongue, H.G.; Ng, M.L.; Kaya, S.; Friebel, D.; Ogasawara, H.; Nilsson, A. Insitu observation of surface species on iridium oxide nanoparticles during the oxygen evolution reaction. Angew. Chem. 2014, 53, 7169–7172. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Schechter, A.; Feng, L. Iridium-based catalysts for oxygen evolution reaction in acidic media: Mechanism, catalytic promotion effects and recent progress. Nano Res. Energy 2023, 2, e9120056. [Google Scholar] [CrossRef]
- Shi, Z.; Wang, X.; Ge, J.; Liu, C.; Xing, W. Fundamental understanding of the acidic oxygen evolution reaction: Mechanism study and state-of-the-art catalysts. Nanoscale 2020, 12, 13249–13275. [Google Scholar] [CrossRef]
- Huang, Z.-F.; Song, J.; Dou, S.; Li, X.; Wang, J.; Wang, X. Strategies to break the scaling relation toward enhanced oxygen electrocatalysis. Matter 2019, 1, 1494–1518. [Google Scholar] [CrossRef]
- Doyle, A.D.; Montoya, J.H.; Vojvodic, A. Improving oxygen electrochemistry through nanoscopic confinement. ChemCatChem 2015, 7, 738–742. [Google Scholar] [CrossRef]
- Liu, J.; Liu, H.; Chen, H.; Du, X.; Zhang, B.; Hong, Z.; Sun, S.; Wang, W. Progress and challenges toward the rational design of oxygen electrocatalysts based on a descriptor approach. Adv. Sci. 2020, 7, 1901614. [Google Scholar] [CrossRef]
- Wu, Y.-H.; Mehta, H.; Willinger, E.; Yuwono, J.A.; Kumar, P.V.; Abdala, P.M.; Wach, A.; Kierzkowska, A.; Donat, F.; Kuznetsov, D.A.; et al. Altering oxygen binding by redox-inactive metal substitution to control catalytic activity: Oxygen reduction on manganese oxide nanoparticles as a model system. Angew. Chem. 2023, 62, e202217186. [Google Scholar] [CrossRef] [PubMed]
- Koper, M.T.M. Thermodynamic theory of multi-electron transfer reactions: Implications for electrocatalysis. J. Electroanal. Chem. 2011, 660, 254–260. [Google Scholar] [CrossRef]
- Wu, Z.-P.; Lu, X.F.; Zang, S.-Q.; Lou, X.W. Non-noble-metal-based electrocatalysts toward the oxygen evolution reaction. Adv. Funct. Mater. 2020, 30, 1910274. [Google Scholar] [CrossRef]
- Trasatti, S. Electrocatalysis by oxides—Attempt at a unifying approach. J. Electroanal. Chem. Interfacial Electrochem. 1980, 111, 125–131. [Google Scholar] [CrossRef]
- Deeksha; Kour, P.; Ahmed, I.; Sunny; Sharma, S.K.; Yadav, K.; Mishra, Y.K. Transition metal-based perovskite oxides: Emerging electrocatalysts for oxygen evolution reaction. ChemCatChem 2023, 15, e202300040. [Google Scholar] [CrossRef]
- Hong, W.T.; Risch, M.; Stoerzinger, K.A.; Grimaud, A.; Suntivich, J.; Shao-Horn, Y. Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 2015, 8, 1404–1427. [Google Scholar] [CrossRef]
- Craig, M.J.; García-Melchor, M. Reaction descriptors for the oxygen evolution reaction: Recent advances, challenges, and opportunities. Curr. Opin. Electrochem. 2022, 35, 101044. [Google Scholar] [CrossRef]
- Fabbri, E.; Habereder, A.; Waltar, K.; Kötz, R.; Schmidt, T.J. Developments and perspectives of oxide-based catalysts for the oxygen evolution reaction. Catal. Sci. Technol. 2014, 4, 3800–3821. [Google Scholar] [CrossRef]
- Huang, X.; Wang, J.; Tao, H.B.; Tian, H.; Xu, H. An essential descriptor for the oxygen evolution reaction on reducible metal oxide surfaces. Chem. Sci. 2019, 10, 3340–3345. [Google Scholar] [CrossRef]
- Li, Y.; Lu, X.; Li, Y.; Zhang, X. Oxygen evolution reaction in nanoconfined carbon nanotubes. Phys. E 2018, 99, 1–5. [Google Scholar] [CrossRef]
- Shao, Y.; de Groot, H.J.M.; Buda, F. Proton acceptor near the active site lowers dramatically the O-O bond formation energy barrier in photocatalytic water splitting. J. Phys. Chem. Lett. 2019, 10, 7690–7697. [Google Scholar] [CrossRef]
- She, S.; Zhu, Y.; Chen, Y.; Lu, Q.; Zhou, W.; Shao, Z. Realizing ultrafast oxygen evolution by introducing proton acceptor into perovskites. Adv. Energy Mater. 2019, 9, 1900429. [Google Scholar] [CrossRef]
- Lin, C.; Li, J.-L.; Li, X.; Yang, S.; Luo, W.; Zhang, Y.; Kim, S.-H.; Kim, D.-H.; Shinde, S.S.; Li, Y.-F.; et al. In-situ reconstructed Ru atom array on α-MnO2 with enhanced performance for acidic water oxidation. Nat. Catal. 2021, 4, 1012–1023. [Google Scholar] [CrossRef]
- Liang, J.; Gao, X.; Xu, K.; Lu, J.; Liu, D.; Zhao, Z.; Tse, E.C.M.; Peng, Z.; Zhang, W.; Liu, J. Unraveling the Asymmetric O-O Radical Coupling Mechanism on Ru–O–Co for Enhanced Acidic Water Oxidation. Small 2023, 2304889. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Shaffer, D.W.; Concepcion, J.J. O-O radical coupling: From detailed mechanistic understanding to enhanced water oxidation catalysis. Inorg. Chem. 2018, 57, 10533–10542. [Google Scholar] [CrossRef] [PubMed]
- Betley, T.A.; Wu, Q.; Van Voorhis, T.; Nocera, D.G. Electronic design criteria for O-O bond formation via metal−oxo complexes. Inorg. Chem. 2008, 47, 1849–1861. [Google Scholar] [CrossRef]
- Fabbri, E.; Schmidt, T.J. Oxygen evolution reaction—The enigma in water electrolysis. ACS Catal. 2018, 8, 9765–9774. [Google Scholar] [CrossRef]
- Alonso-Vante, N.; Colell, H.; Stimming, U.; Tributsch, H. Anomalous low-temperature kinetic effects for oxygen evolution on RuO2 and Pt electrodes. J. Phys. Chem. 1993, 97, 7381–7384. [Google Scholar] [CrossRef]
- Yoo, J.S.; Liu, Y.; Rong, X.; Kolpak, A.M. Electronic origin and kinetic feasibility of the lattice oxygen participation during the oxygen evolution reaction on perovskites. J. Phys. Chem. Lett. 2018, 9, 1473–1479. [Google Scholar] [CrossRef]
- Qin, P.; Zhang, S.-Q.; Yung, K.-K.-L.; Huang, Z.-F.; Gao, B. Disclosure of charge storage mechanisms in molybdenum oxide nanobelts with enhanced supercapacitive performance induced by oxygen deficiency. Rare Met. 2021, 40, 2447–2454. [Google Scholar] [CrossRef]
- Huang, Z.; Liu, Z.; Liao, M.; Wang, L.; Luo, Z.; Isimjan, T.T.; Yang, X. Synergistically improved hydrogen evolution by interface engineering of monodispersed Co5.47N/CoMoOx hybrid particles on carbon cloth with rich oxygen vacancies. Chem. Eng. J. 2023, 462, 142281. [Google Scholar] [CrossRef]
- Qu, H.-Y.; He, X.; Wang, Y.; Hou, S. Electrocatalysis for the oxygen evolution reaction in acidic media: Progress and challenges. Appl. Sci. 2021, 11, 4320. [Google Scholar] [CrossRef]
- Rong, X.; Parolin, J.; Kolpak, A.M. A fundamental relationship between reaction mechanism and stability in metal oxide catalysts for oxygen evolution. ACS Catal. 2016, 6, 1153–1158. [Google Scholar] [CrossRef]
- Kasian, O.; Grote, J.-P.; Geiger, S.; Cherevko, S.; Mayrhofer, K.J.J. The common intermediates of oxygen evolution and dissolution reactions during water electrolysis on iridium. Angew. Chem. 2018, 57, 2488–2491. [Google Scholar] [CrossRef]
- Macounova, K.; Makarova, M.; Krtil, P. Oxygen evolution on nanocrystalline RuO2 and Ru0.9Ni0.1O2−δ electrodes—DEMS approach to reaction mechanism determination. Electrochem. Commun. 2009, 11, 1865–1868. [Google Scholar] [CrossRef]
- Govindarajan, N.; García-Lastra, J.M.; Meijer, E.J.; Calle-Vallejo, F. Does the breaking of adsorption-energy scaling relations guarantee enhanced electrocatalysis? Curr. Opin. Electrochem. 2018, 8, 110–117. [Google Scholar] [CrossRef]
- Feng, C.; Faheem, M.B.; Fu, J.; Xiao, Y.; Li, C.; Li, Y. Fe-based electrocatalysts for oxygen evolution reaction: Progress and perspectives. ACS Catal. 2020, 10, 4019–4047. [Google Scholar] [CrossRef]
- Lee, Y.; Suntivich, J.; May, K.J.; Perry, E.E.; Shao-Horn, Y. Synthesis and activities of rutile IrO2 and RuO2 nanoparticles for oxygen evolution in acid and alkaline solutions. J. Phys. Chem. Lett. 2012, 3, 399–404. [Google Scholar] [CrossRef]
- Hughes, J.P.; Clipsham, J.; Chavushoglu, H.; Rowley-Neale, S.J.; Banks, C.E. Polymer electrolyte electrolysis: A review of the activity and stability of non-precious metal hydrogen evolution reaction and oxygen evolution reaction catalysts. Renew. Sustain. Energy Rev. 2021, 139, 110709. [Google Scholar] [CrossRef]
- Siracusano, S.; Hodnik, N.; Jovanovic, P.; Ruiz-Zepeda, F.; Šala, M.; Baglio, V.; Aricò, A.S. New insights into the stability of a high performance nanostructured catalyst for sustainable water electrolysis. Nano Energy 2017, 40, 618–632. [Google Scholar] [CrossRef]
- Osgood, H.; Devaguptapu, S.V.; Xu, H.; Cho, J.; Wu, G. Transition metal (Fe, Co, Ni, and Mn) oxides for oxygen reduction and evolution bifunctional catalysts in alkaline media. Nano Today 2016, 11, 601–625. [Google Scholar] [CrossRef]
- Kibsgaard, J.; Chorkendorff, I. Considerations for the scaling-up of water splitting catalysts. Nat. Energy 2019, 4, 430–433. [Google Scholar] [CrossRef]
- Zeradjanin, A.R.; Masa, J.; Spanos, I.; Schlögl, R. Activity and stability of oxides during oxygen evolution reaction—From mechanistic controversies toward relevant electrocatalytic descriptors. Front. Energy Res. 2021, 8, 613092. [Google Scholar] [CrossRef]
- Chen, Z.; Duan, X.; Wei, W.; Wang, S.; Ni, B.-J. Electrocatalysts for acidic oxygen evolution reaction: Achievements and perspectives. Nano Energy 2020, 78, 105392. [Google Scholar] [CrossRef]
- Poienar, M.; Svera, P.; Taranu, B.-O.; Ianasi, C.; Sfirloaga, P.; Buse, G.; Veber, P.; Vlazan, P. Electrochemical investigation of the OER activity for nickel phosphite-based compositions and its morphology-dependent fluorescence properties. Crystals 2022, 12, 1803. [Google Scholar] [CrossRef]
- Plevová, M.; Hnát, J.; Bouzek, K. Electrocatalysts for the oxygen evolution reaction in alkaline and neutral media. A comparative review. J. Power Sources 2021, 507, 230072. [Google Scholar] [CrossRef]
- Zhu, K.; Liu, H.; Li, M.; Li, X.; Wang, J.; Zhu, X.; Yang, W. Atomic-scale topochemical preparation of crystalline Fe3+-doped β-Ni(OH)2 for an ultrahigh-rate oxygen evolution reaction. J. Mater. Chem. A 2017, 5, 7753–7758. [Google Scholar] [CrossRef]
- Zhu, K.; Zhu, X.; Yang, W. Application of In Situ Techniques for the Characterization of NiFe-Based Oxygen Evolution Reaction (OER) Electrocatalysts. Angew. Chem. Int. Ed. 2019, 58, 1252–1265. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.S.; Enman, L.J.; Batchellor, A.S.; Zou, S.; Boettcher, S.W. Oxygen Evolution Reaction Electrocatalysis on Transition Metal Oxides and (Oxy)hydroxides: Activity Trends and Design Principles. Chem. Mater. 2015, 27, 7549–7558. [Google Scholar] [CrossRef]
- Hunter, B.M.; Winkler, J.R.; Gray, H.B. Iron Is the Active Site in Nickel/Iron Water Oxidation Electrocatalysts. Molecules 2018, 23, 903. [Google Scholar] [CrossRef]
- Zhang, J.; Winkler, J.R.; Gray, H.B.; Hunter, B.M. Mechanism of Nickel–Iron Water Oxidation Electrocatalysts. Energy Fuels 2021, 35, 19164–19169. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, Y.; Li, L.; Wei, L.; Su, J.; Guo, L. Enhanced oxygen reduction activity and stability of double-layer nitrogen-doped carbon catalyst with abundant Fe-Co dual-atom sites. Nano Energy 2023, 117, 108854. [Google Scholar] [CrossRef]
- Li, X.; Wang, Z.; Su, Z.; Zhao, Z.; Cai, Q.; Zhao, J. Phthalocyanine-supported single-atom catalysts as a promising bifunctional electrocatalyst for ORR/OER: A computational study. ChemPhysMater. 2022, 1, 237–245. [Google Scholar] [CrossRef]
- Guo, D.; Chi, J.; Yu, H.; Jiang, G.; Shao, Z. Self-supporting NiFe layered double hydroxide “Nanoflower” cluster anode electrode for an efficient alkaline anion exchange membrane water electrolyzer. Energies 2022, 15, 4645. [Google Scholar] [CrossRef]
- Praveen Kumar, M.; Sasikumar, M.; Arulraj, A.; Rajasudha, V.; Murugadoss, G.; Kumar, M.R.; Gouse Peera, S.; Mangalaraja, R.V. NiFe layered double hydroxide electrocatalyst prepared via an electrochemical deposition method for the oxygen evolution reaction. Catalysts 2022, 12, 1470. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Huang, Y. Electron-rich NiFe layered double hydroxides via interface engineering for boosting electrocatalytic oxygen evolution. Appl. Catal. B-Environ. 2021, 297, 120453. [Google Scholar] [CrossRef]
- Hunter, B.M.; Blakemore, J.D.; Deimund, M.; Gray, H.B.; Winkler, J.R.; Müller, A.M. Highly Active Mixed-Metal Nanosheet Water Oxidation Catalysts Made by Pulsed-Laser Ablation in Liquids. J. Am. Chem. Soc. 2014, 136, 13118–13121. [Google Scholar] [CrossRef]
- Wilhelm, M.; Bastos, A.; Neves, C.; Martins, R.; Tedim, J. Ni-Fe layered double hydroxides for oxygen evolution Reaction: Impact of Ni/Fe ratio and crystallinity. Mater. Des. 2021, 212, 110188. [Google Scholar] [CrossRef]
- Shi, G.; Arata, C.; Tryk, D.A.; Tano, T.; Yamaguchi, M.; Iiyama, A.; Uchida, M.; Iida, K.; Watanabe, S.; Kakinuma, K. NiFe Alloy Integrated with Amorphous/Crystalline NiFe Oxide as an Electrocatalyst for Alkaline Hydrogen and Oxygen Evolution Reactions. ACS Omega 2023, 8, 13068–13077. [Google Scholar] [CrossRef] [PubMed]
- Axmann, P.; Glemser, O. Nickel hydroxide as a matrix for unusual valencies: The electrochemical behaviour of metal(III)-ion-substituted nickel hydroxides of the pyroaurite type. J. Alloys Compd. 1997, 246, 232–241. [Google Scholar] [CrossRef]
- Yu, N.; Cao, W.; Huttula, M.; Kayser, Y.; Hoenicke, P.; Beckhoff, B.; Lai, F.; Dong, R.; Sun, H.; Geng, B. Fabrication of FeNi hydroxides double-shell nanotube arrays with enhanced performance for oxygen evolution reaction. Appl. Catal. B-Environ. 2020, 261, 118193. [Google Scholar] [CrossRef]
- Qiao, X.; Kang, H.; Li, Y.; Cui, K.; Jia, X.; Liu, H.; Qin, W.; Pupucevski, M.; Wu, G. Porous Fe-doped β-Ni(OH)2 nanopyramid array electrodes for water splitting. ACS Appl. Mater. Interfaces 2020, 12, 36208–36219. [Google Scholar] [CrossRef] [PubMed]
- Görlin, M.; Ferreira de Araújo, J.; Schmies, H.; Bernsmeier, D.; Dresp, S.; Gliech, M.; Jusys, Z.; Chernev, P.; Kraehnert, R.; Dau, H.; et al. Tracking catalyst redox states and reaction dynamics in Ni–Fe oxyhydroxide oxygen evolution reaction electrocatalysts: The role of catalyst support and electrolyte pH. J. Am. Chem. Soc. 2017, 139, 2070–2082. [Google Scholar] [CrossRef]
- Kou, T.; Wang, S.; Hauser, J.L.; Chen, M.; Oliver, S.R.J.; Ye, Y.; Guo, J.; Li, Y. Ni Foam-supported Fe-doped β-Ni(OH)2 nanosheets show ultralow overpotential for oxygen evolution reaction. ACS Energy Lett. 2019, 4, 622–628. [Google Scholar] [CrossRef]
- Gong, M.; Li, Y.; Wang, H.; Liang, Y.; Wu, J.Z.; Zhou, J.; Wang, J.; Regier, T.; Wei, F.; Dai, H. An Advanced Ni–Fe Layered Double Hydroxide Electrocatalyst for Water Oxidation. J. Am. Chem. Soc. 2013, 135, 8452–8455. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, C.; Shi, Y.; Liang, Y.; Yu, Y.; Zhang, B. Temperature-regulated reversible transformation of spinel-to-oxyhydroxide active species for electrocatalytic water oxidation. J. Mater. Chem. A 2020, 8, 1631–1635. [Google Scholar] [CrossRef]
- Ehelebe, K.; Escalera-López, D.; Cherevko, S. Limitations of aqueous model systems in the stability assessment of electrocatalysts for oxygen reactions in fuel cell and electrolyzers. Curr. Opin. Electrochem. 2021, 29, 100832. [Google Scholar] [CrossRef]
- Liu, X.; Meng, J.; Ni, K.; Guo, R.; Xia, F.; Xie, J.; Li, X.; Wen, B.; Wu, P.; Li, M.; et al. ; et al. Complete reconstruction of hydrate pre-catalysts for ultrastable water electrolysis in industrial-concentration alkali media. Cell Rep. Phys. Sci. 2020, 1, 100241. [Google Scholar] [CrossRef]
- Wang, J.; Hu, J.; Niu, S.; Li, S.; Du, Y.; Xu, P. Crystalline-amorphous Ni2P4O12/NiMoOx nanoarrays for alkaline water electrolysis: Enhanced catalytic activity via in situ surface reconstruction. Small 2022, 18, 2105972. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, A.; Martinez-Moreno, E.; Teschner, D.; Chernev, P.; Gliech, M.; de Araújo, J.F.; Reier, T.; Dau, H.; Strasser, P. Reversible amorphization and the catalytically active state of crystalline Co3O4 during oxygen evolution. Nat. Commun. 2015, 6, 8625. [Google Scholar] [CrossRef]
- Hu, C.; Wang, X.; Yao, T.; Gao, T.; Han, J.; Zhang, X.; Zhang, Y.; Xu, P.; Song, B. Enhanced electrocatalytic oxygen evolution activity by tuning both the oxygen vacancy and orbital occupancy of B-site metal cation in NdNiO3. Adv. Funct. Mater. 2019, 29, 1902449. [Google Scholar] [CrossRef]
- Kwong, W.L.; Lee, C.C.; Shchukarev, A.; Messinger, J. Cobalt-doped hematite thin films for electrocatalytic water oxidation in highly acidic media. Chem. Commun. 2019, 55, 5017–5020. [Google Scholar] [CrossRef]
- Gao, J.; Tao, H.; Liu, B. Progress of nonprecious-metal-based electrocatalysts for oxygen evolution in acidic media. Adv. Mater. 2021, 33, 2003786. [Google Scholar] [CrossRef]
- Jiao, Y.; Yang, C.; Wang, H.; Zhong, Y.; Hu, Y. Optimization strategies on the advanced engineering of Co-based nanomaterials for electrochemical oxygen evolution. J. Alloys Compd. 2022, 890, 161929. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Fan, H.; Ignaszak, A.; Zhang, L.; Shao, S.; Wilkinson, D.P.; Zhang, J. Compositing doped-carbon with metals, non-metals, metal oxides, metal nitrides and other materials to form bifunctional electrocatalysts to enhance metal-air battery oxygen reduction and evolution reactions. Chem. Eng. J. 2018, 348, 416–437. [Google Scholar] [CrossRef]
- Hou, Y.; Wen, Z.; Cui, S.; Ci, S.; Mao, S.; Chen, J. An advanced nitrogen-doped graphene/cobalt-embedded porous carbon polyhedron hybrid for efficient catalysis of oxygen reduction and water splitting. Adv. Funct. Mater. 2015, 25, 872–882. [Google Scholar] [CrossRef]
- Ling, T.; Yan, D.-Y.; Jiao, Y.; Wang, H.; Zheng, Y.; Zheng, X.; Mao, J.; Du, X.-W.; Hu, Z.; Jaroniec, M.; et al. Engineering surface atomic structure of single-crystal cobalt (II) oxide nanorods for superior electrocatalysis. Nat. Commun. 2016, 7, 12876. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, G.; Qu, J.; Liu, H. Disordering the atomic structure of Co(II) oxide via B-doping: An efficient oxygen vacancy introduction approach for high oxygen evolution reaction electrocatalysts. Small 2018, 14, 1802760. [Google Scholar] [CrossRef]
- Hunter, B.M.; Gray, H.B.; Müller, A.M. Earth-Abundant Heterogeneous Water Oxidation Catalysts. Chem. Rev. 2016, 116, 14120–14136. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Min, Y.; Li, L.; Lian, Y.; Sun, H.; Wang, D.; Rummeli, M.H.; Guo, J.; Zhong, J.; Xu, L.; et al. Crystal splintering of β-MnO2 induced by interstitial Ru doping toward reversible oxygen conversion. Chem. Mater. 2021, 33, 4135–4145. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, S.; Wang, Z.; Mo, Y.; Luo, X.; Yang, F.; Lv, M.; Li, Z.; Liu, X. Manganese-based oxide electrocatalysts for the oxygen evolution reaction: A review. J. Mater. Chem. A. 2023, 11, 5476–5494. [Google Scholar] [CrossRef]
- Meng, Y.; Song, W.; Huang, H.; Ren, Z.; Chen, S.-Y.; Suib, S.L. Structure–property relationship of bifunctional MnO2 nanostructures: Highly efficient, ultra-stable electrochemical water oxidation and oxygen reduction reaction catalysts identified in alkaline media. J. Am. Chem. Soc. 2014, 136, 11452–11464. [Google Scholar] [CrossRef]
- Kamenskii, M.A.; Volkov, F.S.; Eliseeva, S.N.; Tolstopyatova, E.G.; Kondratiev, V.V. Enhancement of Electrochemical Performance of Aqueous Zinc Ion Batteries by Structural and Interfacial Design of MnO2 Cathodes: The Metal Ion Doping and Introduction of Conducting Polymers. Energies 2023, 16, 3221. [Google Scholar] [CrossRef]
- Takashima, T.; Hashimoto, K.; Nakamura, R. Mechanisms of pH-dependent activity for water oxidation to molecular oxygen by MnO2 electrocatalysts. J. Am. Chem. Soc. 2012, 134, 1519–1527. [Google Scholar] [CrossRef]
- Li, A.; Ooka, H.; Bonnet, N.; Hayashi, T.; Sun, Y.; Jiang, Q.; Li, C.; Han, H.; Nakamura, R. Stable potential windows for long-term electrocatalysis by manganese oxides under acidic conditions. Angew. Chem. 2019, 58, 5054–5058. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, B.; Fan, D.; Wang, Y.; Yang, H.; Huang, K.; Pan, X.; Zhang, R.; Tang, H.; Lei, M. Tailoring manganese oxide nanoplates enhances oxygen evolution catalysis in acid. J. Catal. 2022, 405, 265–272. [Google Scholar] [CrossRef]
- Thenuwara, A.C.; Shumlas, S.L.; Attanayake, N.H.; Aulin, Y.V.; McKendry, I.G.; Qiao, Q.; Zhu, Y.; Borguet, E.; Zdilla, M.J.; Strongin, D.R. Intercalation of cobalt into the interlayer of birnessite improves oxygen evolution catalysis. ACS Catal. 2016, 6, 7739–7743. [Google Scholar] [CrossRef]
- Wu, T.; Sun, S.; Song, J.; Xi, S.; Du, Y.; Chen, B.; Sasangka, W.A.; Liao, H.; Gan, C.L.; Scherer, G.G.; et al. Iron-facilitated dynamic active-site generation on spinel CoAl2O4 with self-termination of surface reconstruction for water oxidation. Nat. Catal. 2019, 2, 763–772. [Google Scholar] [CrossRef]
- Niu, S.; Kong, X.-P.; Li, S.; Zhang, Y.; Wu, J.; Zhao, W.; Xu, P. Low Ru loading RuO2/(Co,Mn)3O4 nanocomposite with modulated electronic structure for efficient oxygen evolution reaction in acid. Appl. Catal. B-Environ. 2021, 297, 120442. [Google Scholar] [CrossRef]
- Huynh, M.; Ozel, T.; Liu, C.; Lau, E.C.; Nocera, D.G. Design of template-stabilized active and earth-abundant oxygen evolution catalysts in acid. Chem. Sci. 2017, 8, 4779–4794. [Google Scholar] [CrossRef]
- Mondschein, J.S.; Kumar, K.; Holder, C.F.; Seth, K.; Kim, H.; Schaak, R.E. Intermetallic Ni2Ta electrocatalyst for the oxygen evolution reaction in highly acidic electrolytes. Inorg. Chem. 2018, 57, 6010–6015. [Google Scholar] [CrossRef]
- Antonyshyn, I.; Barrios Jiménez, A.M.; Sichevych, O.; Burkhardt, U.; Veremchuk, I.; Schmidt, M.; Ormeci, A.; Spanos, I.; Tarasov, A.; Teschner, D.; et al. Al2Pt for oxygen evolution in water splitting: A strategy for creating multifunctionality in electrocatalysis. Angew. Chem. 2020, 59, 16770–16776. [Google Scholar] [CrossRef]
- Barrios Jiménez, A.M.; Burkhardt, U.; Cardoso-Gil, R.; Höfer, K.; Altendorf, S.G.; Schlögl, R.; Grin, Y.; Antonyshyn, I. Hf2B2Ir5: A self-optimizing catalyst for the oxygen evolution reaction. ACS Appl. Energy Mater. 2020, 3, 11042–11052. [Google Scholar] [CrossRef]
- Yang, Y.; Yao, H.; Yu, Z.; Islam, S.M.; He, H.; Yuan, M.; Yue, Y.; Xu, K.; Hao, W.; Sun, G.; et al. Hierarchical nanoassembly of MoS2/Co9S8/Ni3S2/Ni as a highly efficient electrocatalyst for overall water splitting in a wide pH range. J. Am. Chem. Soc. 2019, 141, 10417–10430. [Google Scholar] [CrossRef]
- Yeh, J.-W.; Chen, S.-K.; Lin, S.-J.; Gan, J.-Y.; Chin, T.-S.; Shun, T.-T.; Tsau, C.-H.; Chang, S.-Y. Nanostructured high-entropy alloys with multiple principal elements: Novel alloy design concepts and outcomes. Adv. Eng. Mater. 2004, 6, 299–303. [Google Scholar] [CrossRef]
- Yeh, J.-W. Physical metallurgy of high-entropy alloys. JOM 2015, 67, 2254–2261. [Google Scholar] [CrossRef]
- Zhang, G.; Ming, K.; Kang, J.; Huang, Q.; Zhang, Z.; Zheng, X.; Bi, X. High entropy alloy as a highly active and stable electrocatalyst for hydrogen evolution reaction. Electrochim. Acta 2018, 279, 19–23. [Google Scholar] [CrossRef]
- Qiao, H.; Wang, X.; Dong, Q.; Zheng, H.; Chen, G.; Hong, M.; Yang, C.-P.; Wu, M.; He, K.; Hu, L. A high-entropy phosphate catalyst for oxygen evolution reaction. Nano Energy 2021, 86, 106029. [Google Scholar] [CrossRef]
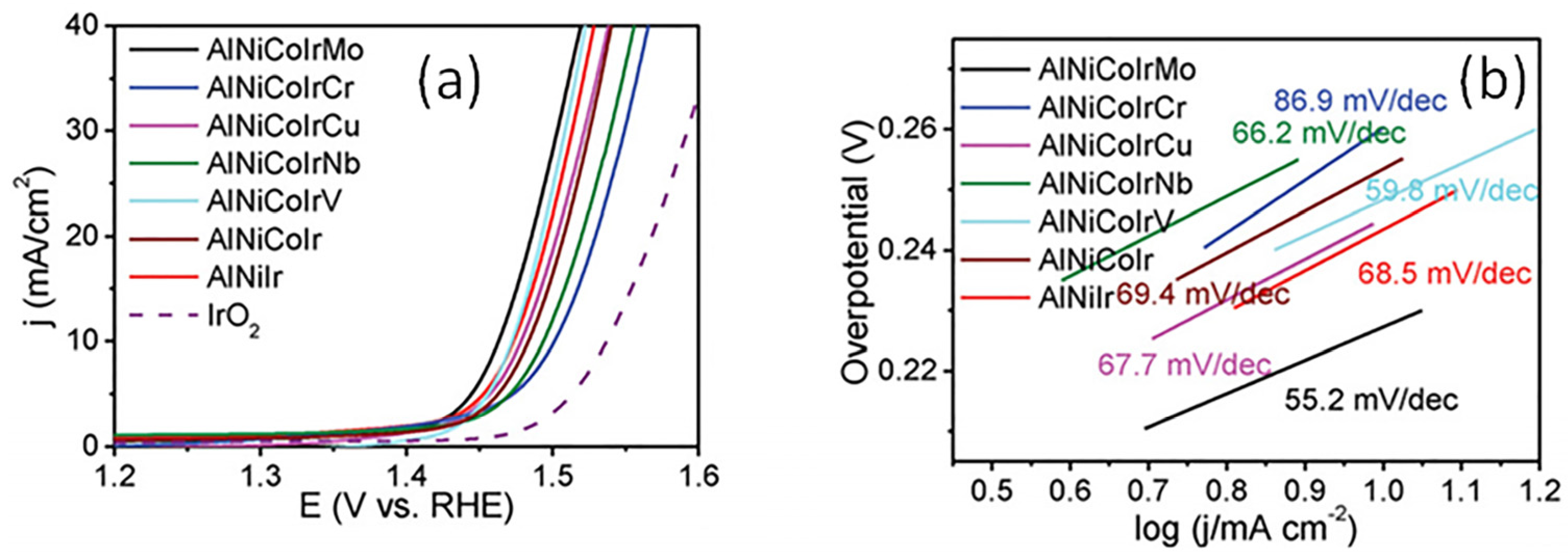
- Jin, Z.; Lv, J.; Jia, H.; Liu, W.; Li, H.; Chen, Z.; Lin, X.; Xie, G.; Liu, X.; Sun, S.; et al. Nanoporous Al-Ni-Co-Ir-Mo high-entropy alloy for record-high water splitting activity in acidic environments. Small 2019, 15, 1904180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Shi, Y.; Chen, X.; Lai, J.; Huang, B.; Wang, L. High-entropy alloy metallene for highly efficient overall water splitting in acidic media. Chin. J. Catal. 2023, 45, 174–183. [Google Scholar] [CrossRef]
- Löffler, T.; Savan, A.; Garzón-Manjón, A.; Meischein, M.; Scheu, C.; Ludwig, A.; Schuhmann, W. Toward a Paradigm Shift in Electrocatalysis Using Complex Solid Solution Nanoparticles. ACS Energy Lett. 2019, 4, 1206–1214. [Google Scholar] [CrossRef]
- Löffler, T.; Savan, A.; Meyer, H.; Meischein, M.; Strotkötter, V.; Ludwig, A.; Schuhmann, W. Design of Complex Solid-Solution Electrocatalysts by Correlating Configuration, Adsorption Energy Distribution Patterns, and Activity Curves. Angew. Chem. Int. Ed. 2020, 59, 5844–5850. [Google Scholar] [CrossRef]
- Yu, L.; Zeng, K.; Li, C.; Lin, X.; Liu, H.; Shi, W.; Qiu, H.-J.; Yuan, Y.; Yao, Y. High-entropy alloy catalysts: From bulk to nano toward highly efficient carbon and nitrogen catalysis. Carbon Energy 2022, 4, 731–761. [Google Scholar] [CrossRef]
- Löffler, T.; Ludwig, A.; Rossmeisl, J.; Schuhmann, W. What Makes High-Entropy Alloys Exceptional Electrocatalysts? Angew. Chem. Int. Ed. 2021, 60, 26894–26903. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, T.A.A.; Löffler, T.; Xiao, B.; Krysiak, O.A.; Strotkötter, V.; Pedersen, J.K.; Clausen, C.M.; Savan, A.; Li, Y.; Schuhmann, W.; et al. Complex-Solid-Solution Electrocatalyst Discovery by Computational Prediction and High-Throughput Experimentation. Angew. Chem. Int. Ed. 2021, 60, 6932–6937. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhu, D.; Yan, J.; Wang, C.-A. Strong metal-support interactions induced by an ultrafast laser. Nat. Commun. 2021, 12, 6665. [Google Scholar] [CrossRef] [PubMed]
- Campos-Roldán, C.A.; González-Huerta, R.G.; Alonso-Vante, N. The oxophilic and electronic effects on anchored platinum nanoparticles on sp2 carbon sites: The hydrogen evolution and oxidation reactions in alkaline medium. Electrochim. Acta 2018, 283, 1829–1834. [Google Scholar] [CrossRef]
- Roldan Cuenya, B.; Behafarid, F. Nanocatalysis: Size- and shape-dependent chemisorption and catalytic reactivity. Surf. Sci. Rep. 2015, 70, 135–187. [Google Scholar] [CrossRef]
- Sharma, S.; Pollet, B.G. Support materials for PEMFC and DMFC electrocatalysts—A review. J. Power Sources 2012, 208, 96–119. [Google Scholar] [CrossRef]
- Bhuvanendran, N.; Ravichandran, S.; Jayaseelan, S.S.; Xu, Q.; Khotseng, L.; Su, H. Improved bi-functional oxygen electrocatalytic performance of Pt–Ir alloy nanoparticles embedded on mwcnt with Pt-enriched surfaces. Energy 2020, 211, 118695. [Google Scholar] [CrossRef]
- Luo, Y.; Alonso-Vante, N. The effect of support on advanced Pt-based cathodes towards the oxygen reduction reaction. State of the art. Electrochim. Acta 2015, 179, 108–118. [Google Scholar] [CrossRef]
- Ma, J.; Habrioux, A.; Morais, C.; Lewera, A.; Vogel, W.; Verde-Gómez, Y.; Ramos-Sanchez, G.; Balbuena, P.B.; Alonso-Vante, N. Spectroelectrochemical probing of the strong interaction between platinum nanoparticles and graphitic domains of carbon. ACS Catal. 2013, 3, 1940–1950. [Google Scholar] [CrossRef]
- Olowoyo, J.O.; Kriek, R.J. Mono- and bimetallic oxides as photo-electrocatalysts for the oxygen evolution reaction—A review. J. Phys. Chem. Solids 2022, 169, 110868. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, Y.; Zhang, R.; Ma, J.; Alonso-Vante, N. Highly active oxygen evolution reaction electrocatalyst based on defective-CeO2-x decorated MOF(Ni/Fe). Electrochim. Acta 2022, 403, 139630. [Google Scholar] [CrossRef]
- Estudillo-Wong, L.A.; Ramos-Sanchez, G.; Calvillo, L.; Granozzi, G.; Alonso-Vante, N. Support interaction effect of platinum nanoparticles on Non-, Y-, Ce-doped anatase and its implication on the ORR in acid and alkaline media. ChemElectroChem 2017, 4, 3264–3275. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, J.; Wang, S.; Jiang, Q.; Feng, R.; Ju, S.; Zhang, W.; Song, F. Silver decorated hydroxides electrocatalysts for efficient oxygen evolution reaction. Chem. Eng. J. 2022, 442, 136168. [Google Scholar] [CrossRef]
- Kumar, P.; Murthy, A.P.; Bezerra, L.S.; Martini, B.K.; Maia, G.; Madhavan, J. Carbon supported nickel phosphide as efficient electrocatalyst for hydrogen and oxygen evolution reactions. Int. J. Hydrog. Energy 2021, 46, 622–632. [Google Scholar] [CrossRef]
- Batool, M.; Waseem, A.; Nadeem, M.A.; Nadeem, M.A. Phase pure synthesis of iron-nickel nitride nanoparticles: A low cost electrocatalyst for oxygen evolution reaction. Int. J. Hydrog. Energy 2023, 48, 18280–18290. [Google Scholar] [CrossRef]
- Bi, J.; Ying, H.; Hao, J.; Li, Z. Application of metal chalcogenide-based anodic electrocatalyst toward substituting oxygen evolution reaction in water splitting. Curr. Opin. Electrochem. 2022, 33, 100963. [Google Scholar] [CrossRef]
- Campos-Roldán, C.A.; Alonso-Vante, N. The hydrogen evolution reaction on nanostructured molybdenum disulfide. J. Mex. Chem. Soc. 2019, 63, 28–38. [Google Scholar]
- Yang, P.; Jiang, Z.; Shi, Y.; Ren, X.; Liang, L.; Shao, Q.; Zhu, K. Enhancement of oxygen evolution reaction performance of FeCoNiCrMn high entropy alloy thin film electrodes through in-situ reconstruction. J. Alloys Compd. 2023, 947, 169699. [Google Scholar] [CrossRef]
- Dakave, S.S.; Bhinge, G.A.; Kengar, N.N.; Teli, A.D.; Kanamadi, C.M. Electrocatalytic performance of cobalt oxide and hybrid composite thin films of cobalt Oxide@Polyaniline for oxygen evolution reaction. Mater. Today-Proc. 2023; in press. [Google Scholar] [CrossRef]
- Luo, Y.; Estudillo-Wong, L.A.; Cavillo, L.; Granozzi, G.; Alonso-Vante, N. An easy and cheap chemical route using a MOF precursor to prepare Pd–Cu electrocatalyst for efficient energy conversion cathodes. J. Catal. 2016, 338, 135–142. [Google Scholar] [CrossRef]
- Liu, C.; Qian, J.; Ye, Y.; Zhou, H.; Sun, C.-J.; Sheehan, C.; Zhang, Z.; Wan, G.; Liu, Y.-S.; Guo, J.; et al. Oxygen evolution reaction over catalytic single-site Co in a well-defined brookite TiO2 nanorod surface. Nat. Catal. 2021, 4, 36–45. [Google Scholar] [CrossRef]
- İlayda Nur, U.R. Development of IrO₂–WO₃ Composite Catalysts from Waste WC–Co Wire Drawing Die for PEM Water Electrolyzers’ Oxygen Evolution Reactions. ACS Sustain. Chem. Eng. 2022, 10, 13100–13111. [Google Scholar]
- Liang, Y.; Li, Y.; Wang, H.; Zhou, J.; Wang, J.; Regier, T.; Dai, H. Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction. Nat. Mater. 2011, 10, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Kundu, M.K.; Mishra, R.; Bhowmik, T.; Kanrar, S.; Barman, S. Three-dimensional hierarchically porous iridium oxide-nitrogen doped carbon hybrid: An efficient bifunctional catalyst for oxygen evolution and hydrogen evolution reaction in acid. Int. J. Hydrog. Energy 2020, 45, 6036–6046. [Google Scholar] [CrossRef]
- Estudillo-Wong, L.A.; Luo, Y.; Díaz-Real, J.A.; Alonso-Vante, N. Enhanced oxygen reduction reaction stability on platinum nanoparticles photo-deposited onto oxide-carbon composites. Appl. Catal. B-Environ. 2016, 187, 291–300. [Google Scholar] [CrossRef]
- Hazarika, K.K.; Goswami, C.; Saikia, H.; Borah, B.J.; Bharali, P. Cubic Mn2O3 nanoparticles on carbon as bifunctional electrocatalyst for oxygen reduction and oxygen evolution reactions. Mol. Catal. 2017, 451, 153–160. [Google Scholar] [CrossRef]
- Wen, X.; Bai, L.; Li, M.; Guan, J. Ultrafine iridium oxide supported on carbon nanotubes for efficient catalysis of oxygen evolution and oxygen reduction reactions. Mater. Today Energy 2018, 10, 153–160. [Google Scholar] [CrossRef]
- Huang, C.; Wu, D.; Qin, P.; Ding, K.; Pi, C.; Ruan, Q.; Song, H.; Gao, B.; Chen, H.; Chu, P.K. Ultrafine Co nanodots embedded in N-doped carbon nanotubes grafted on hexagonal VN for highly efficient overall water splitting. Nano Energy 2020, 73, 104788. [Google Scholar] [CrossRef]
- Joshi, P.; Yadav, R.; Hara, M.; Inoue, T.; Motoyama, Y.; Yoshimura, M. Contribution of B,N-co-doped reduced graphene oxide as a catalyst support to the activity of iridium oxide for oxygen evolution reaction. J. Mater. Chem. A 2021, 9, 9066–9080. [Google Scholar] [CrossRef]
- Huang, C.; Chu, P.K. Recommended practices and benchmarking of foam electrodes in water splitting. Trends Chem. 2022, 4, 1065–1077. [Google Scholar] [CrossRef]
- Mondschein, J.S.; Callejas, J.F.; Read, C.G.; Chen, J.Y.C.; Holder, C.F.; Badding, C.K.; Schaak, R.E. Crystalline cobalt oxide films for sustained electrocatalytic oxygen evolution under strongly acidic conditions. Chem. Mater. 2017, 29, 950–957. [Google Scholar] [CrossRef]
- Yang, X.; Li, H.; Lu, A.-Y.; Min, S.; Idriss, Z.; Hedhili, M.N.; Huang, K.-W.; Idriss, H.; Li, L.-J. Highly acid-durable carbon coated Co3O4 nanoarrays as efficient oxygen evolution electrocatalysts. Nano Energy 2016, 25, 42–50. [Google Scholar] [CrossRef]
- Baik, C.; Lee, S.W.; Pak, C. Glycine-induced ultrahigh-surface-area IrO2@IrOx catalyst with balanced activity and stability for efficient water splitting. Electrochim. Acta 2021, 390, 138885. [Google Scholar] [CrossRef]
- Jiang, B.; Guo, Y.; Kim, J.; Whitten, A.E.; Wood, K.; Kani, K.; Rowan, A.E.; Henzie, J.; Yamauchi, Y. Mesoporous metallic iridium nanosheets. J. Am. Chem. Soc. 2018, 140, 12434–12441. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; He, Y.; He, Z.; Wang, Z.; Jiang, Y.; Gao, H. Porous Fe5Si3 intermetallic anode for the oxygen evolution reaction in acidic electrolytes. J. Colloid Interface Sci. 2022, 605, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.-L.; Chi, J.-Q.; Xie, J.-Y.; Dong, B.; Liu, Z.-Z.; Gao, W.-K.; Lin, J.-H.; Chai, Y.-M.; Liu, C.-G. Mesoporous Ag-doped Co3O4 nanowire arrays supported on FTO as efficient electrocatalysts for oxygen evolution reaction in acidic media. Renew. Energy 2018, 119, 54–61. [Google Scholar] [CrossRef]
- Peng, Z.; Yang, S.; Jia, D.; Da, P.; He, P.; Al-Enizi, A.M.; Ding, G.; Xie, X.; Zheng, G. Homologous metal-free electrocatalysts grown on three-dimensional carbon networks for overall water splitting in acidic and alkaline media. J. Mater. Chem. A 2016, 4, 12878–12883. [Google Scholar] [CrossRef]
- Frydendal, R.; Paoli, E.A.; Chorkendorff, I.; Rossmeisl, J.; Stephens, I.E.L. Toward an active and stable catalyst for oxygen evolution in acidic media: Ti-stabilized MnO2. Adv. Energy Mater 2015, 5, 1500991. [Google Scholar] [CrossRef]
- Yan, K.-L.; Qin, J.-F.; Lin, J.-H.; Dong, B.; Chi, J.-Q.; Liu, Z.-Z.; Dai, F.-N.; Chai, Y.-M.; Liu, C.-G. Probing the active sites of Co3O4 for the acidic oxygen evolution reaction by modulating the Co2+/Co3+ ratio. J. Mater. Chem. A 2018, 6, 5678–5686. [Google Scholar] [CrossRef]
- Huang, J.; Sheng, H.; Ross, R.D.; Han, J.; Wang, X.; Song, B.; Jin, S. Modifying redox properties and local bonding of Co3O4 by CeO2 enhances oxygen evolution catalysis in acid. Nat. Commun. 2021, 12, 3036. [Google Scholar] [CrossRef]
- Wu, J.; Liu, M.; Chatterjee, K.; Hackenberg, K.P.; Shen, J.; Zou, X.; Yan, Y.; Gu, J.; Yang, Y.; Lou, J.; et al. Exfoliated 2D transition metal disulfides for enhanced electrocatalysis of oxygen evolution reaction in acidic medium. Adv. Mater. Interfaces 2016, 3, 1500669. [Google Scholar] [CrossRef]
- Kumari, S.; Ajayi, B.P.; Kumar, B.; Jasinski, J.B.; Sunkara, M.K.; Spurgeon, J.M. A low-noble-metal W1−xIrxO3−δ water oxidation electrocatalyst for acidic media via rapid plasma synthesis. Energy Environ. Sci. 2017, 10, 2432–2440. [Google Scholar] [CrossRef]
- Sun, W.; Song, Y.; Gong, X.-Q.; Cao, L.-m.; Yang, J. An efficiently tuned d-orbital occupation of IrO2 by doping with Cu for enhancing the oxygen evolution reaction activity. Chem. Sci. 2015, 6, 4993–4999. [Google Scholar] [CrossRef]
- Shi, Q.; Zhu, C.; Du, D.; Wang, J.; Xia, H.; Engelhard, M.H.; Feng, S.; Lin, Y. Ultrathin dendritic IrTe nanotubes for an efficient oxygen evolution reaction in a wide pH range. J. Mater. Chem. A 2018, 6, 8855–8859. [Google Scholar] [CrossRef]
- Liu, X.; Li, Z.; Zhou, L.; Wang, K.; Zhao, X.; Li, Q.; Deng, Y. Facile synthesis of IrCu microspheres based on polyol method and study on their electro-catalytic performances to oxygen evolution reaction. Nanomaterials 2019, 9, 1145. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Cai, X.; Shen, S.; Yin, J.; Jiang, K.; Zhang, J. Dealloyed RuNiOx as a robust electrocatalyst for the oxygen evolution reaction in acidic media. Dalton Trans. 2021, 50, 5124–5127. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, W.; Song, N.; Zhong, M.; Yan, S.; Xu, J.; Zhu, W.; Wang, C.; Lu, X. Electronic modulation of iridium-molybdenum oxides with a low crystallinity for high-efficiency acidic oxygen evolution reaction. Chem. Eng. J. 2022, 440, 135851. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, Q.; Wang, A.; Duan, J.; Zhou, W.; Sang, Y.; Liu, H. Iron oxide embedded titania nanowires—An active and stable electrocatalyst for oxygen evolution in acidic media. Nano Energy 2018, 45, 118–126. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Y.; Yang, B.; Li, Z.; Qin, X.; Zhang, Q.; Lei, L.; Qiu, M.; Wu, G.; Hou, Y. Highly active ruthenium sites stabilized by modulating electron-feeding for sustainable acidic oxygen-evolution electrocatalysis. Energy Environ. Sci. 2022, 15, 2356–2365. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, B.; Su, J.; Zhao, K.; Chen, L. MOF-Derived zinc-doped ruthenium oxide hollow nanorods as highly active and stable electrocatalysts for oxygen evolution in acidic media. ChemNanoMat 2021, 7, 117–121. [Google Scholar] [CrossRef]
- Chen, S.; Huang, H.; Jiang, P.; Yang, K.; Diao, J.; Gong, S.; Liu, S.; Huang, M.; Wang, H.; Chen, Q. Mn-doped RuO2 nanocrystals as highly active electrocatalysts for enhanced oxygen evolution in acidic media. ACS Catal. 2020, 10, 1152–1160. [Google Scholar] [CrossRef]
- Moreno-Hernandez, I.A.; MacFarland, C.A.; Read, C.G.; Papadantonakis, K.M.; Brunschwig, B.S.; Lewis, N.S. Crystalline nickel manganese antimonate as a stable water-oxidation catalyst in aqueous 1.0 m H2SO4. Energy Environ. Sci. 2017, 10, 2103–2108. [Google Scholar] [CrossRef]
- Hu, F.; Zhu, S.; Chen, S.; Li, Y.; Ma, L.; Wu, T.; Zhang, Y.; Wang, C.; Liu, C.; Yang, X.; et al. Amorphous metallic NiFeP: A conductive bulk material achieving high activity for oxygen evolution reaction in both alkaline and acidic media. Adv. Mater. 2017, 29, 1606570. [Google Scholar] [CrossRef]
- Wang, L.; Duan, X.; Liu, X.; Gu, J.; Si, R.; Qiu, Y.; Qiu, Y.; Shi, D.; Chen, F.; Sun, X.; et al. Atomically dispersed Mo supported on metallic Co9S8 nanoflakes as an advanced noble-metal-free bifunctional water splitting catalyst working in universal pH conditions. Adv. Energy Mater. 2020, 10, 1903137. [Google Scholar] [CrossRef]
- Patel, P.P.; Datta, M.K.; Velikokhatnyi, O.I.; Kuruba, R.; Damodaran, K.; Jampani, P.; Gattu, B.; Shanthi, P.M.; Damle, S.S.; Kumta, P.N. Noble metal-free bifunctional oxygen evolution and oxygen reduction acidic media electro-catalysts. Sci. Rep. 2016, 6, 28367. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Hou, W.; Lu, Y.; Zhang, W.; Chen, Y. Cobalt phosphide nanoparticles supported within network of N-doped carbon nanotubes as a multifunctional and scalable electrocatalyst for water splitting. J. Energy Chem. 2021, 52, 130–138. [Google Scholar] [CrossRef]
- Park, J.; Sa, Y.J.; Baik, H.; Kwon, T.; Joo, S.H.; Lee, K. Iridium-based multimetallic nanoframe@nanoframe structure: An efficient and robust electrocatalyst toward oxygen evolution reaction. ACS Nano 2017, 11, 5500–5509. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Chen, H.; Cao, J.; Yang, J.; Qiu, M.; Xia, Y.; Yuan, C.; Yang, B.; Li, Z.; Zhang, X.; et al. Fe-N4 sites embedded into carbon nanofiber integrated with electrochemically exfoliated graphene for oxygen evolution in acidic medium. Adv. Energy Mater. 2018, 8, 1801912. [Google Scholar] [CrossRef]
- Kwon, T.; Hwang, H.; Sa, Y.J.; Park, J.; Baik, H.; Joo, S.H.; Lee, K. Cobalt assisted synthesis of IrCu hollow octahedral nanocages as highly active electrocatalysts toward oxygen evolution reaction. Adv. Funct. Mater. 2017, 27, 1604688. [Google Scholar] [CrossRef]
- Li, R.; Wang, H.; Hu, F.; Chan, K.C.; Liu, X.; Lu, Z.; Wang, J.; Li, Z.; Zeng, L.; Li, Y.; et al. IrW nanochannel support enabling ultrastable electrocatalytic oxygen evolution at 2 a cm−2 in acidic media. Nat. Commun. 2021, 12, 3540. [Google Scholar] [CrossRef]
- Su, H.; Zhao, X.; Cheng, W.; Zhang, H.; Li, Y.; Zhou, W.; Liu, M.; Liu, Q. Hetero-N-coordinated Co single sites with high turnover frequency for efficient electrocatalytic oxygen evolution in an acidic medium. ACS Energy Lett. 2019, 4, 1816–1822. [Google Scholar] [CrossRef]
- Liang, X.; Shi, L.; Liu, Y.; Chen, H.; Si, R.; Yan, W.; Zhang, Q.; Li, G.-D.; Yang, L.; Zou, X. Activating inert, nonprecious perovskites with iridium dopants for efficient oxygen evolution reaction under acidic conditions. Angew. Chem. 2019, 58, 7631–7635. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cheng, C.; Yuan, Q.; Yang, H.; Meng, F.; Zhang, Q.; Gu, L.; Cao, J.; Li, L.; Haw, S.-C.; et al. Exceptionally active and stable RuO2 with interstitial carbon for water oxidation in acid. Chem 2022, 8, 1673–1687. [Google Scholar] [CrossRef]
- Deng, Y.; Yang, L.; Wang, Y.; Zeng, L.; Yu, J.; Chen, B.; Zhang, X.; Zhou, W. Ruthenium nanoclusters anchored on cobalt phosphide hollow microspheres by green phosphating process for full water splitting in acidic electrolyte. Chin. Chem. Lett. 2021, 32, 511–515. [Google Scholar] [CrossRef]
- Fang, Z.; Wu, P.; Yu, K.; Li, Y.; Zhu, Y.; Ferreira, P.J.; Liu, Y.; Yu, G. Hybrid organic–inorganic gel electrocatalyst for stable acidic water oxidation. ACS Nano 2019, 13, 14368–14376. [Google Scholar] [CrossRef]
- Hu, Q.; Li, G.; Liu, X.; Zhu, B.; Li, G.; Fan, L.; Chai, X.; Zhang, Q.; Liu, J.; He, C. Coupling pentlandite nanoparticles and dual-doped carbon networks to yield efficient and stable electrocatalysts for acid water oxidation. J. Mater. Chem. A 2019, 7, 461–468. [Google Scholar] [CrossRef]
- Jiang, S.; Zhu, L.; Yang, Z.; Wang, Y. Enhanced electrocatalytic performance of FeNiCoP amorphous alloys as oxygen-evolving catalysts for electrolytic water splitting application. Electrochim. Acta 2021, 368, 137618. [Google Scholar] [CrossRef]
- Schäfer, H.; Küpper, K.; Schmidt, M.; Müller-Buschbaum, K.; Stangl, J.; Daum, D.; Steinhart, M.; Schulz-Kölbel, C.; Han, W.; Wollschläger, J.; et al. Steel-based electrocatalysts for efficient and durable oxygen evolution in acidic media. Catal. Sci. Technol. 2018, 8, 2104–2116. [Google Scholar] [CrossRef]
- Liu, H.; Peng, X.; Liu, X.; Qi, G.; Luo, J. Porous Mn-doped FeP/Co3(PO4)2 nanosheets as efficient electrocatalysts for overall water splitting in a wide pH range. ChemSusChem 2019, 12, 1334–1341. [Google Scholar] [CrossRef]
- Blasco-Ahicart, M.; Soriano-López, J.; Carbó, J.J.; Poblet, J.M.; Galan-Mascaros, J.R. Polyoxometalate electrocatalysts based on earth-abundant metals for efficient water oxidation in acidic media. Nat. Chem. 2018, 10, 24–30. [Google Scholar] [CrossRef]
- Chang, S.-Q.; Cheng, C.-C.; Cheng, P.-Y.; Huang, C.-L.; Lu, S.-Y. Pulse electrodeposited feconimnw high entropy alloys as efficient and stable bifunctional electrocatalysts for acidic water splitting. Chem. Eng. J. 2022, 446, 137452. [Google Scholar] [CrossRef]
- Cai, Z.-X.; Goou, H.; Ito, Y.; Tokunaga, T.; Miyauchi, M.; Abe, H.; Fujita, T. Nanoporous ultra-high-entropy alloys containing fourteen elements for water splitting electrocatalysis. Chem. Sci. 2021, 12, 11306–11315. [Google Scholar] [CrossRef]
- Zhu, H.; Zhu, Z.; Hao, J.; Sun, S.; Lu, S.; Wang, C.; Ma, P.; Dong, W.; Du, M. High-entropy alloy stabilized active Ir for highly efficient acidic oxygen evolution. Chem. Eng. J. 2022, 431, 133251. [Google Scholar] [CrossRef]













| Electrolyte | Half-Cell Reaction | |
|---|---|---|
| Hydrogen evolution reaction (HER) | ||
| Acid | (1) | |
| Alkaline | (2) | |
| Oxygen evolution reaction (OER) | ||
| Acid | (3) | |
| Alkaline | (4) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Higareda, A.; Hernández-Arellano, D.L.; Ordoñez, L.C.; Barbosa, R.; Alonso-Vante, N. Advanced Electrocatalysts for the Oxygen Evolution Reaction: From Single- to Multielement Materials. Catalysts 2023, 13, 1346. https://doi.org/10.3390/catal13101346
Higareda A, Hernández-Arellano DL, Ordoñez LC, Barbosa R, Alonso-Vante N. Advanced Electrocatalysts for the Oxygen Evolution Reaction: From Single- to Multielement Materials. Catalysts. 2023; 13(10):1346. https://doi.org/10.3390/catal13101346
Chicago/Turabian StyleHigareda, América, Diana Laura Hernández-Arellano, Luis Carlos Ordoñez, Romeli Barbosa, and Nicolas Alonso-Vante. 2023. "Advanced Electrocatalysts for the Oxygen Evolution Reaction: From Single- to Multielement Materials" Catalysts 13, no. 10: 1346. https://doi.org/10.3390/catal13101346