
Effects of Potassium Loading over Iron–Silica Interaction, Phase Evolution and Catalytic Behavior of Precipitated Iron-Based Catalysts for Fischer-Tropsch Synthesis

Abstract
:1. Introduction
2. Results and Discussion
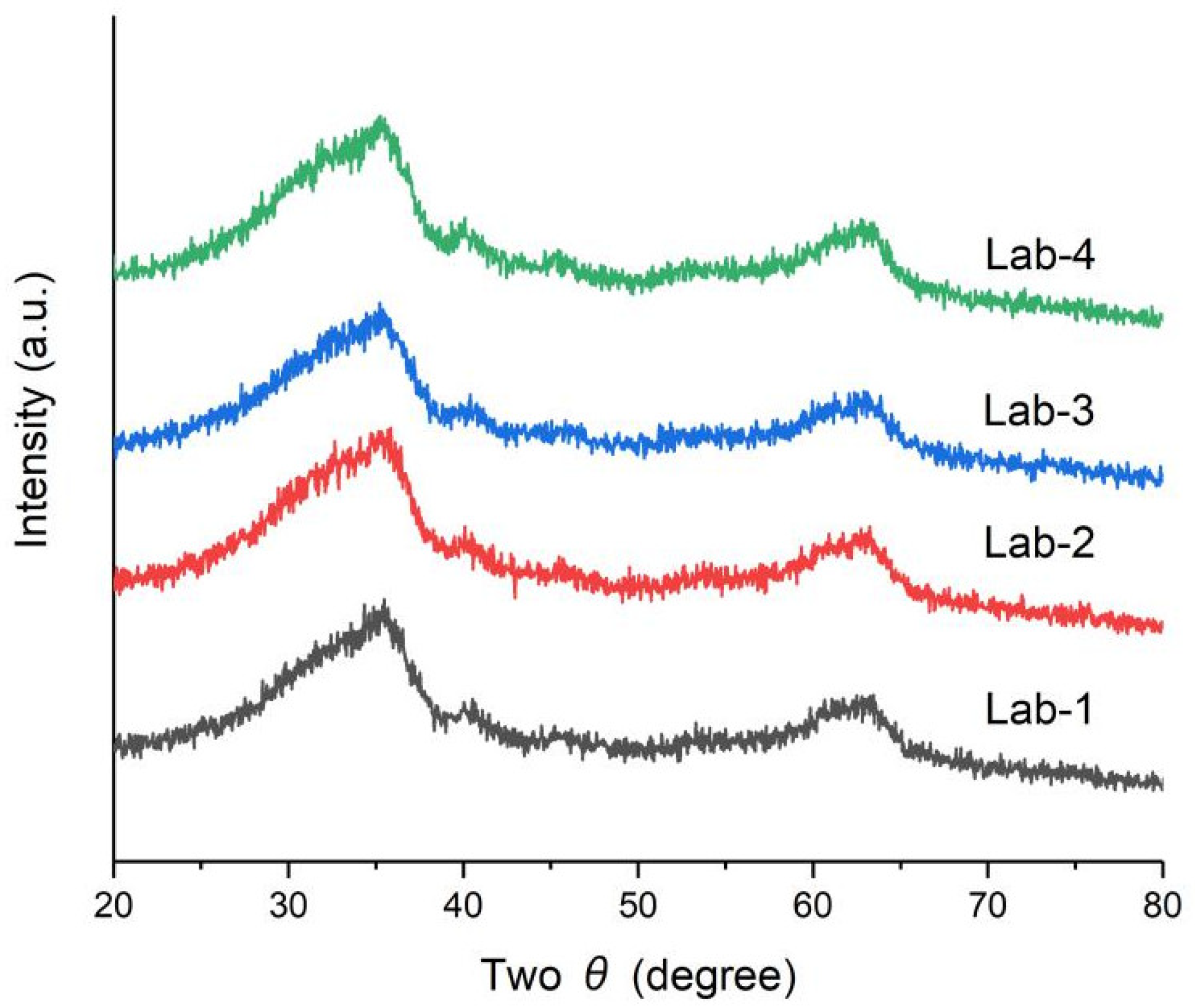
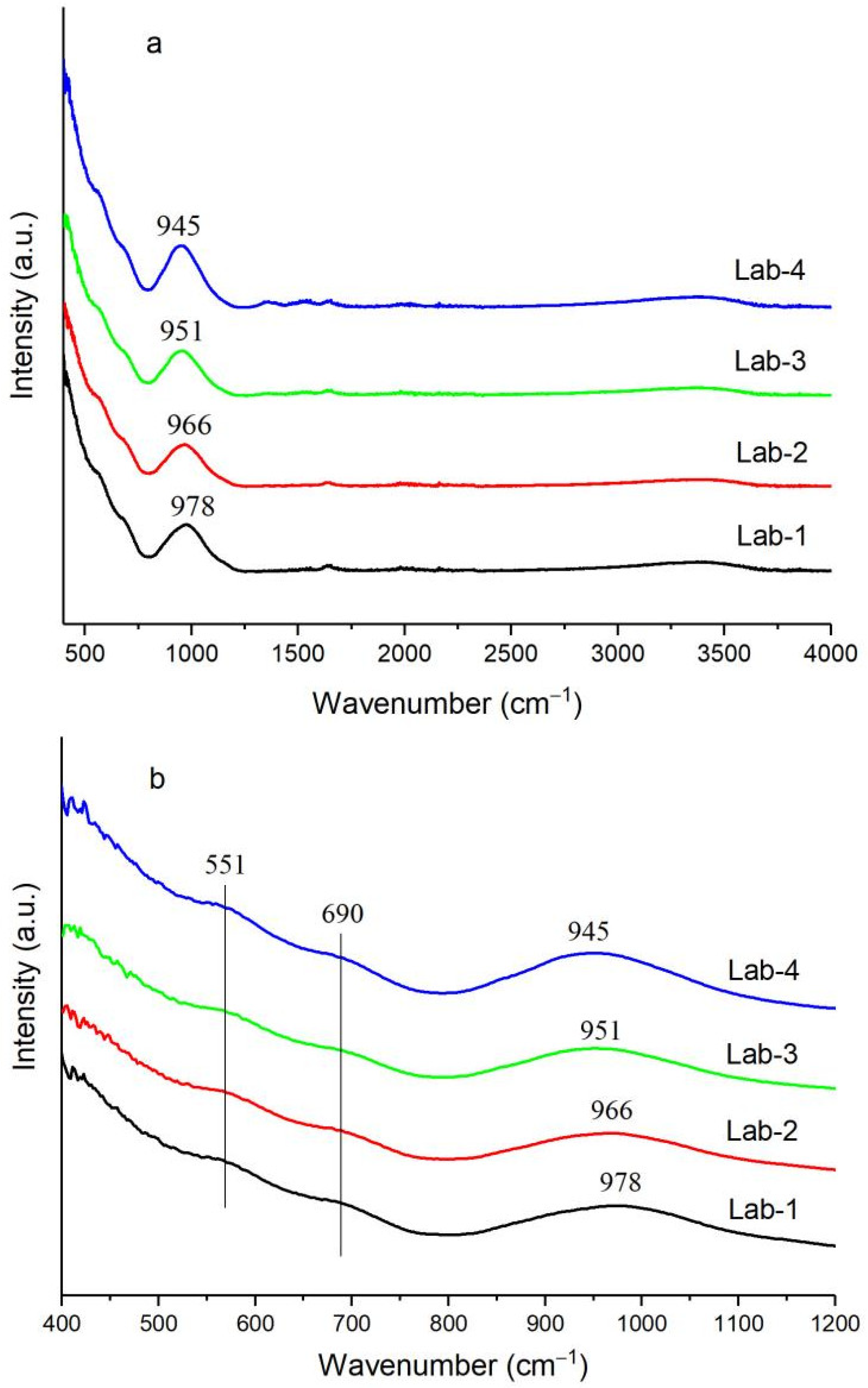
2.1. Effect of K Loading on Fe–O–Si Interaction
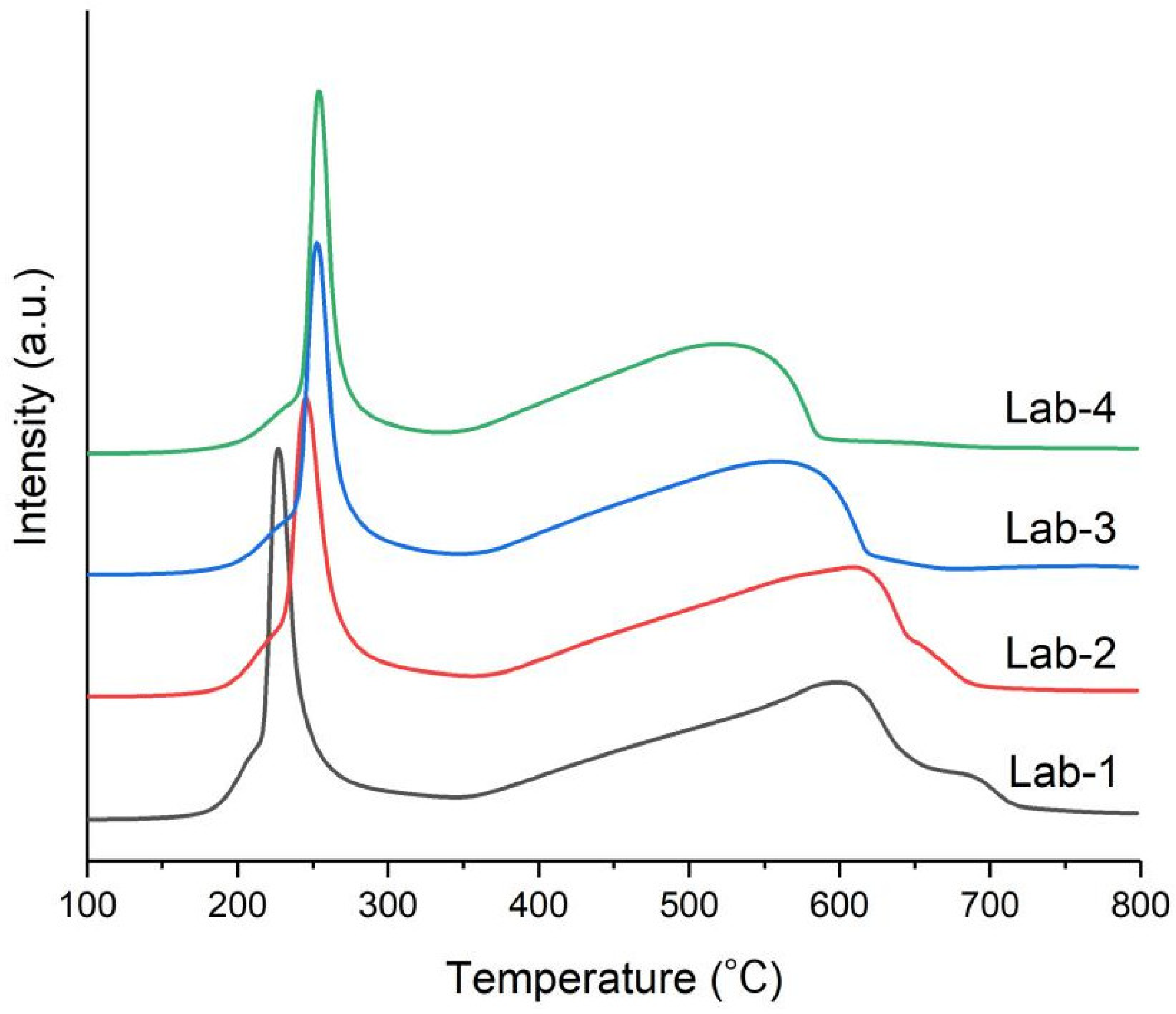
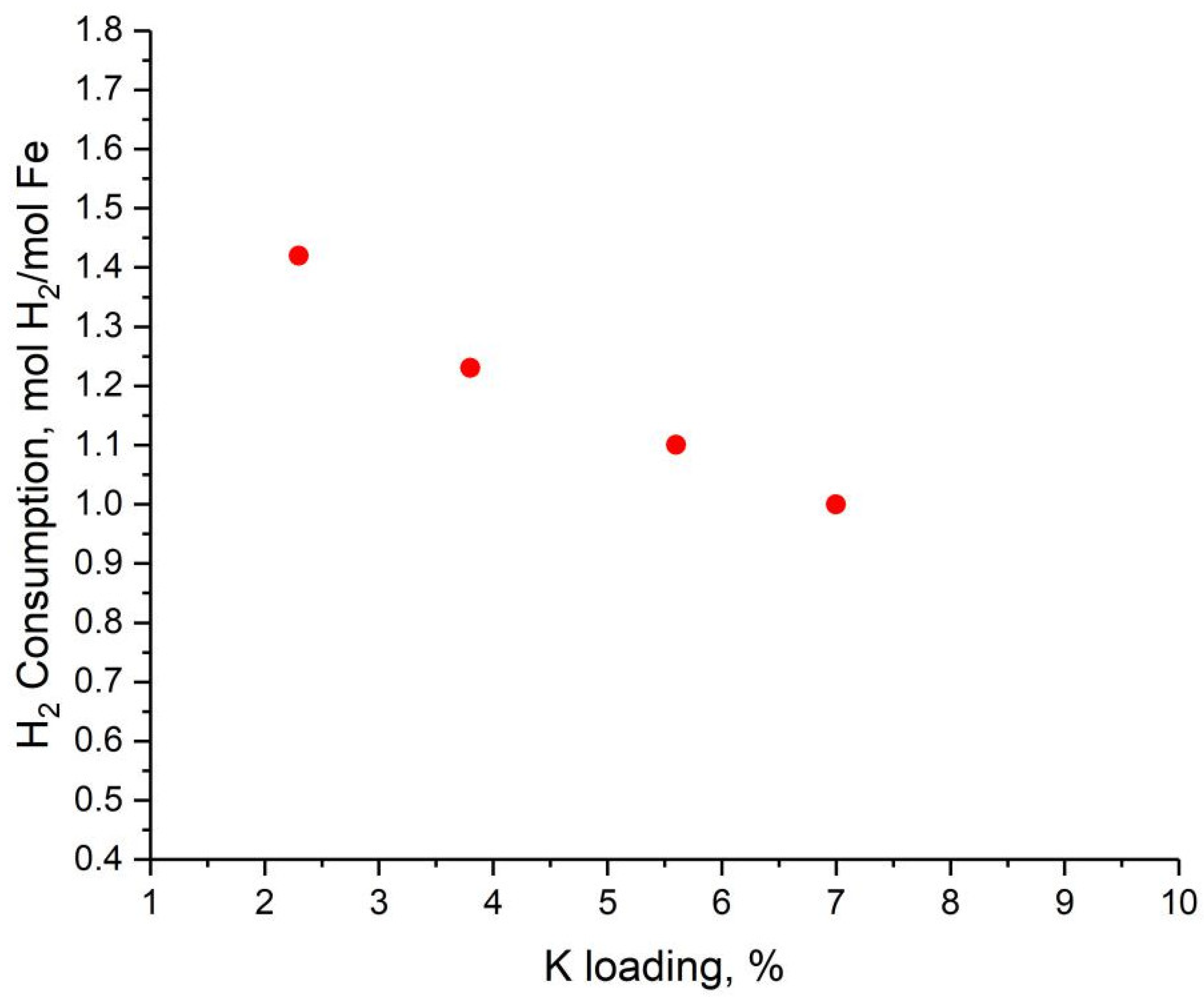
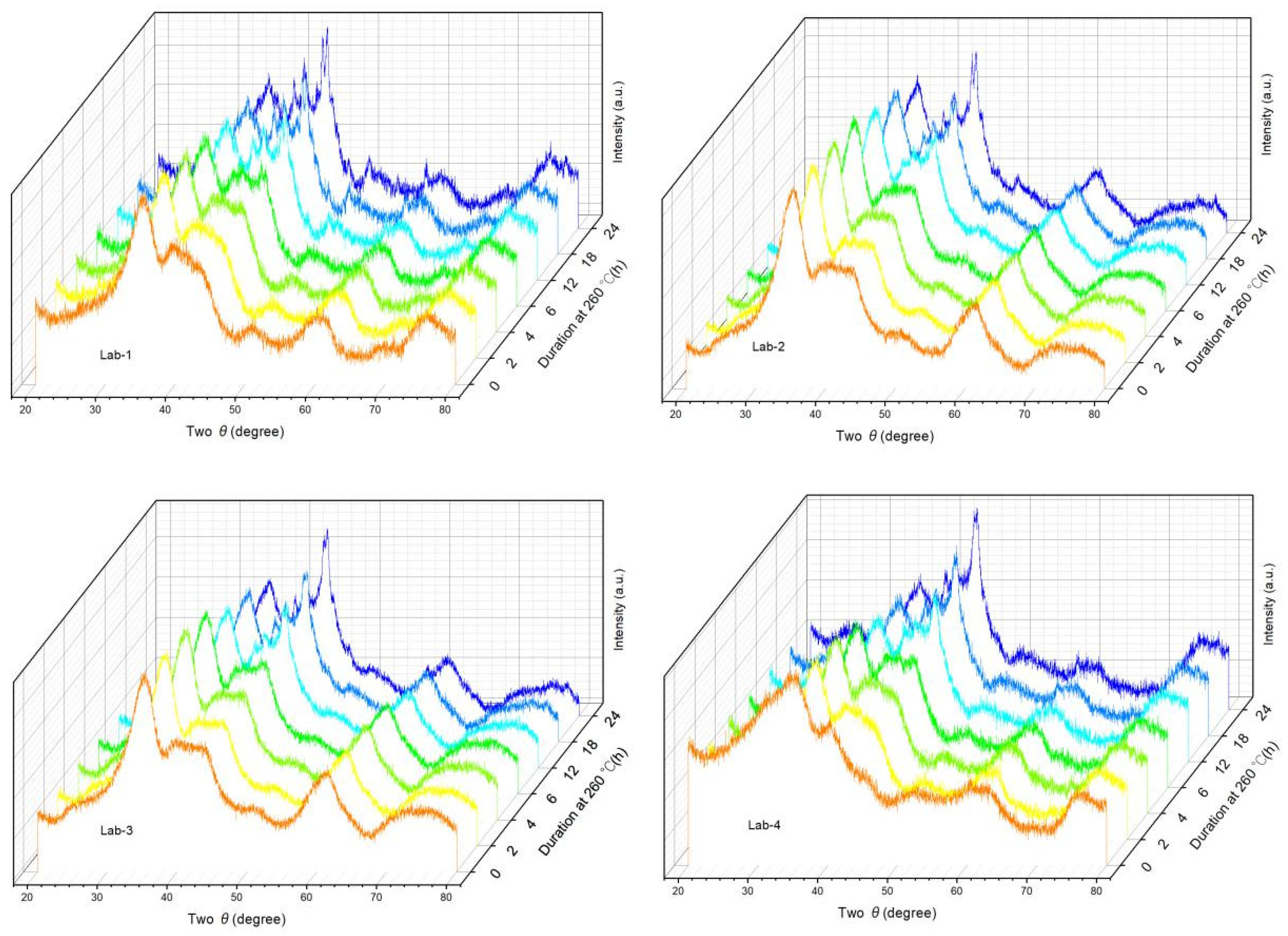
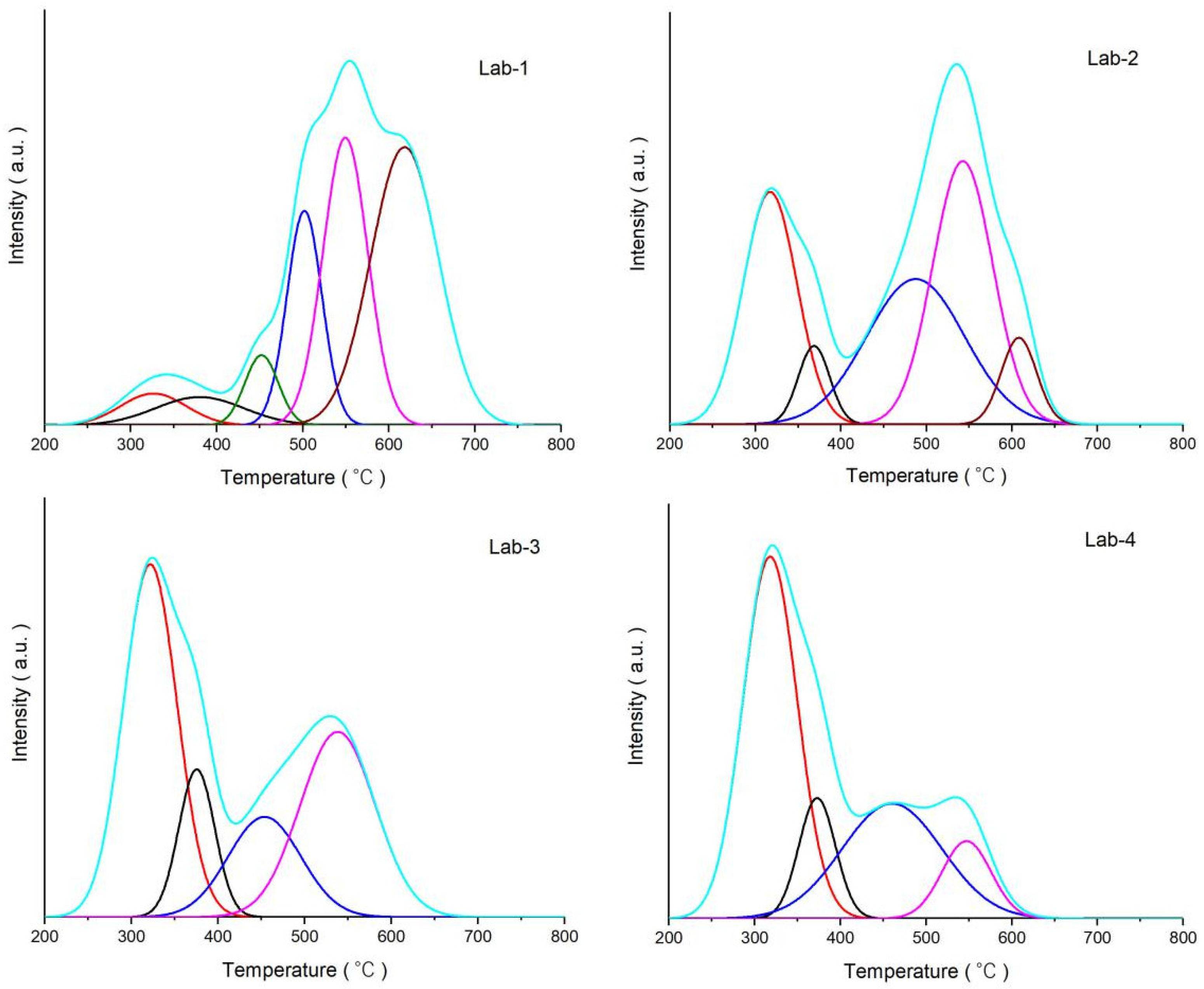
2.2. Effects of K Loading on Carburization of Catalysts
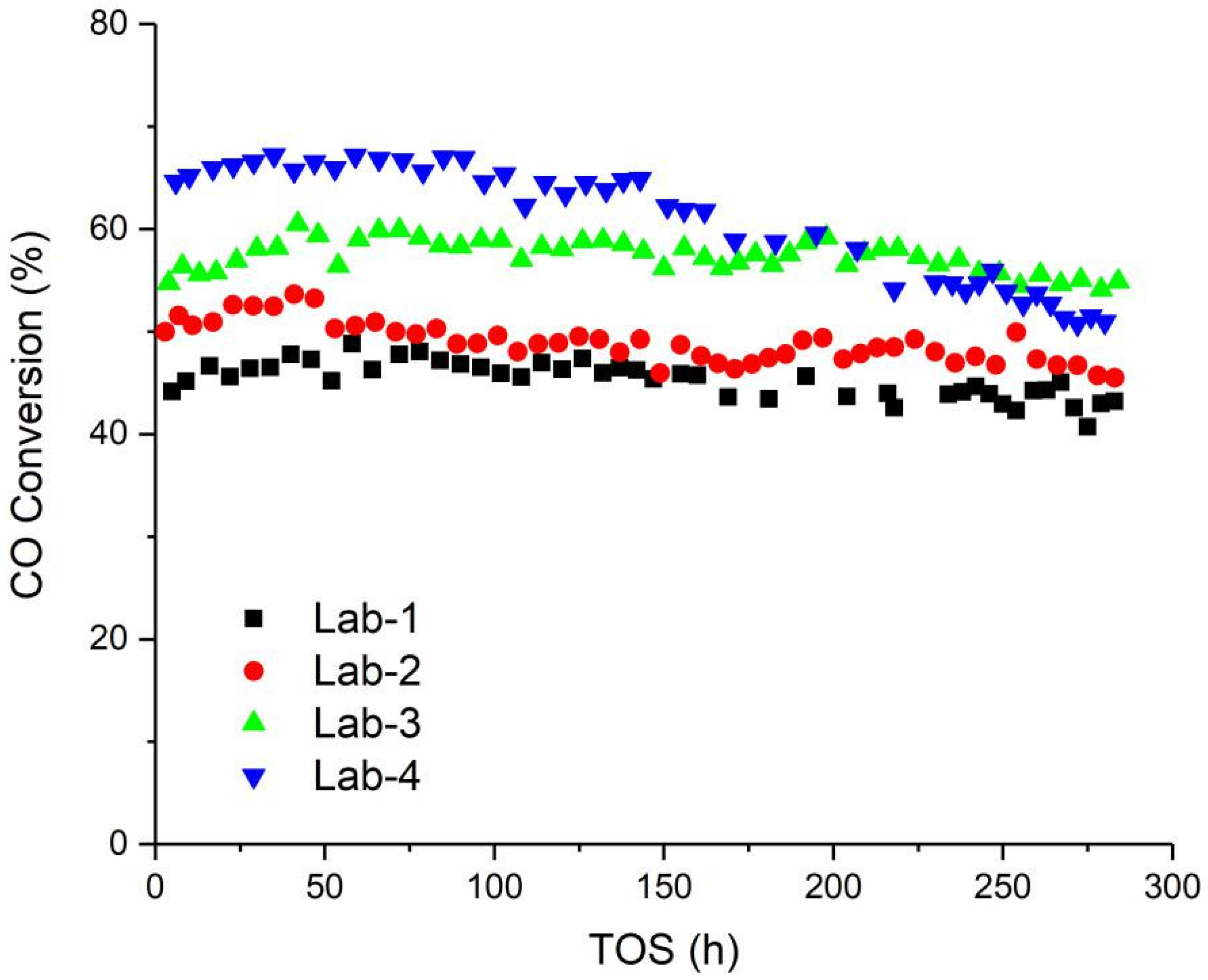
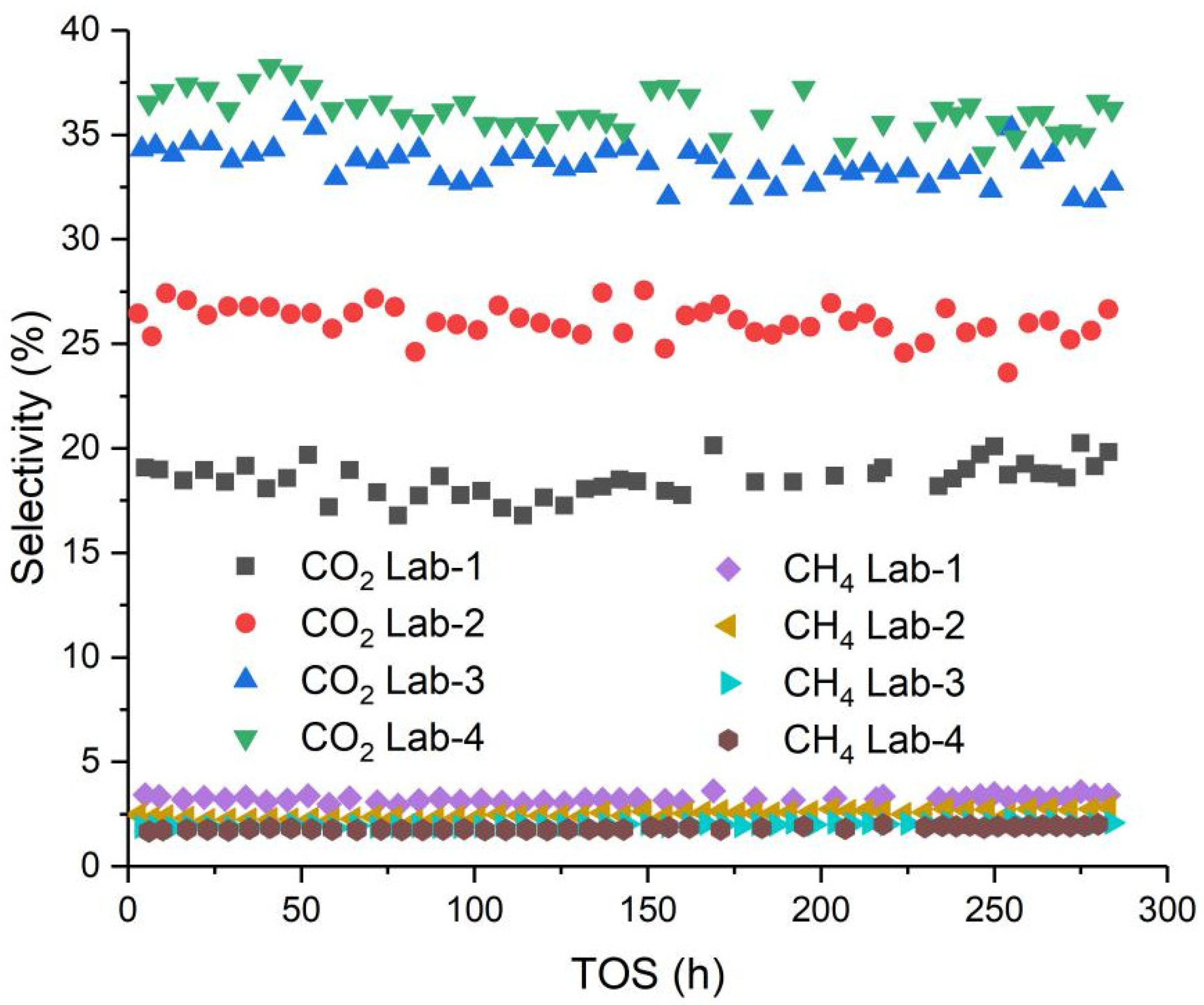
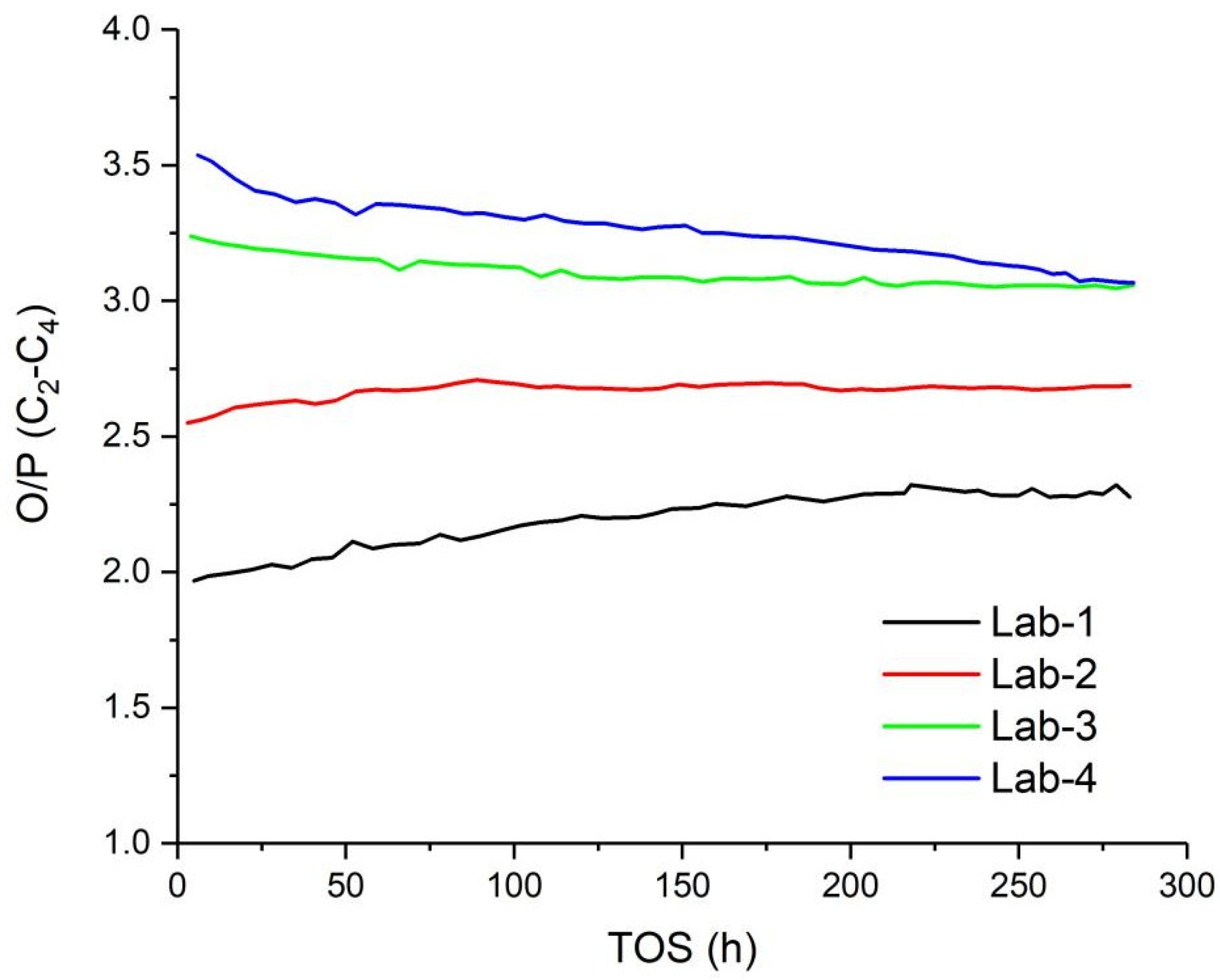
2.3. Fischer-Tropsch Synthesis Performance
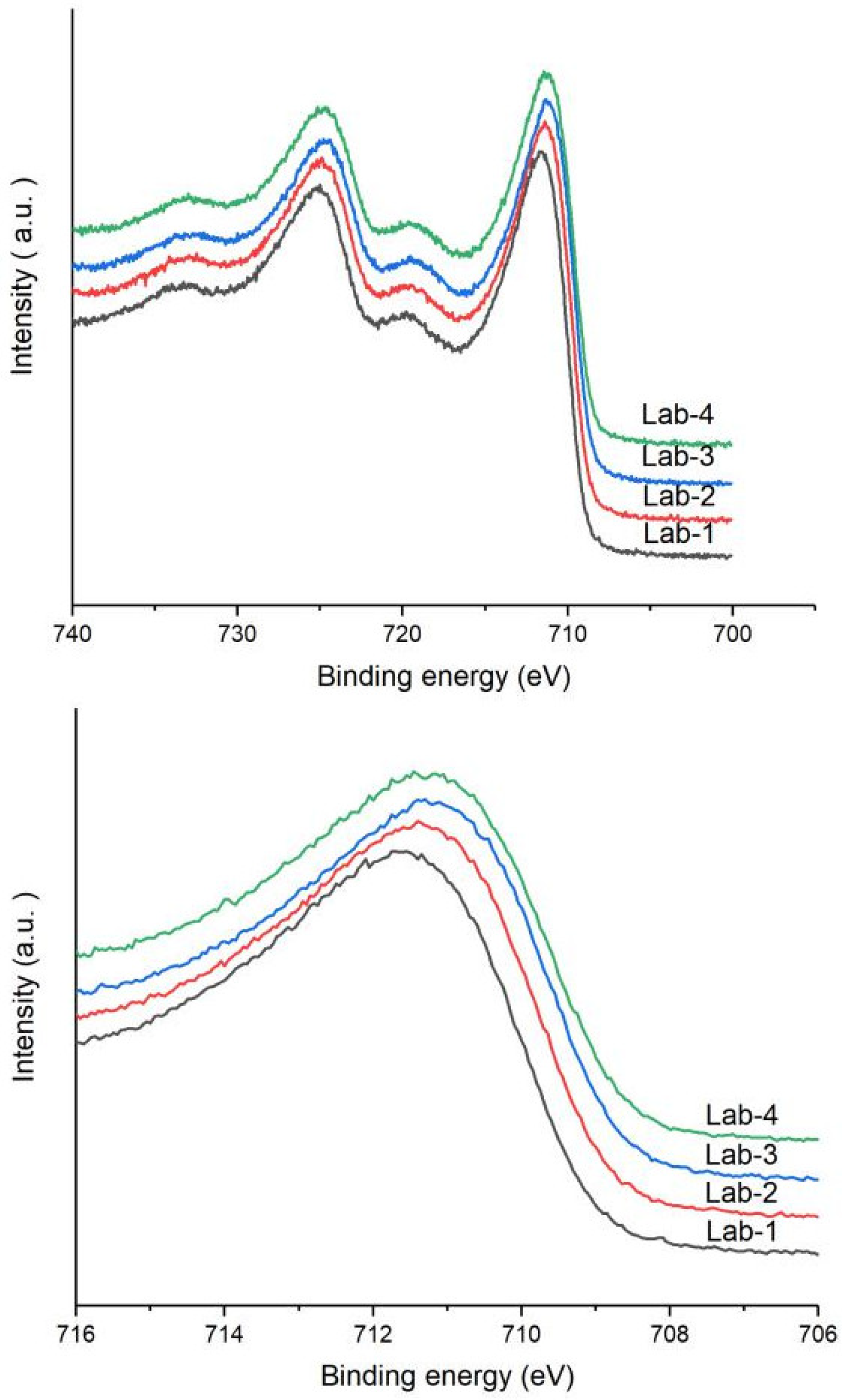
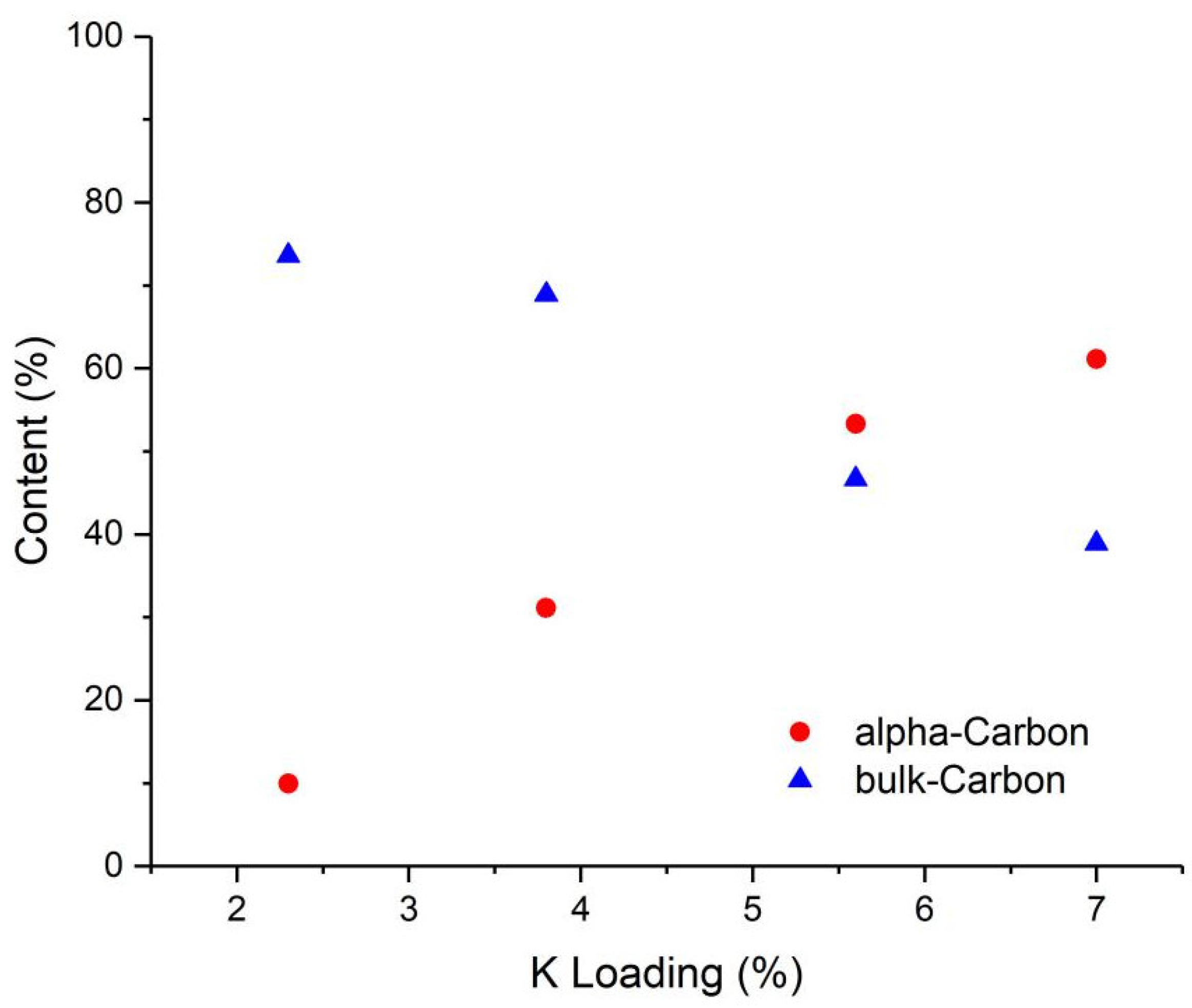
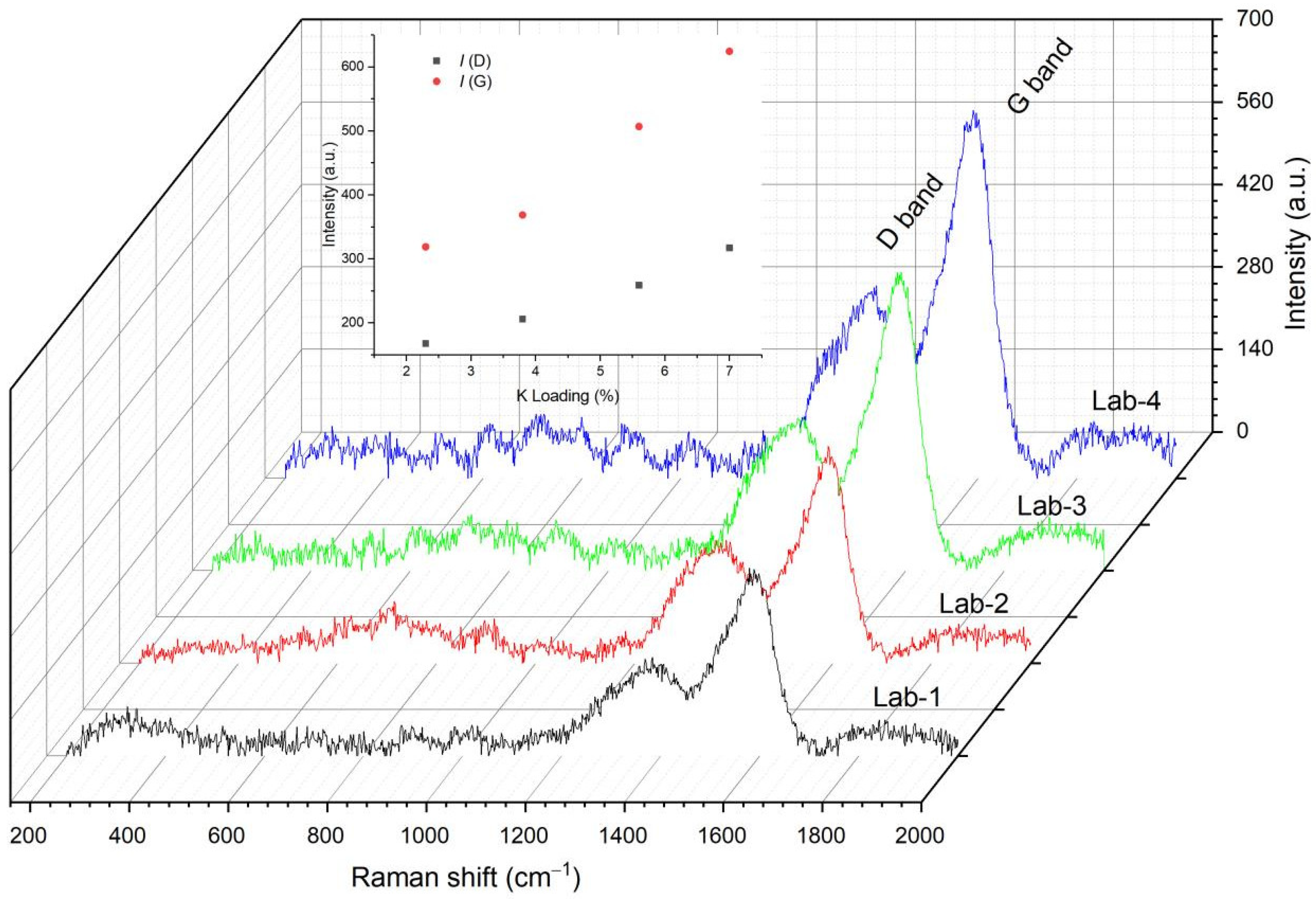
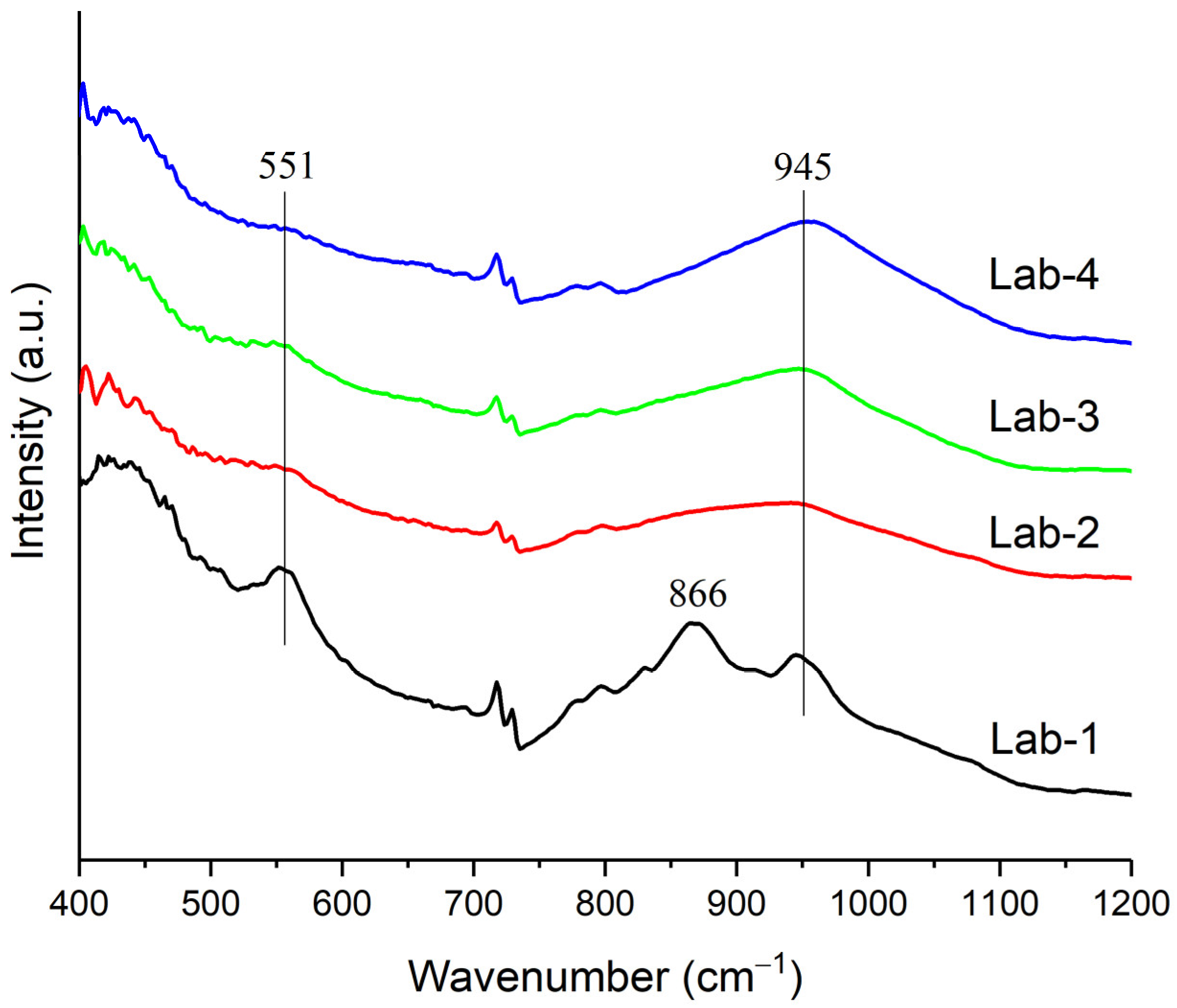
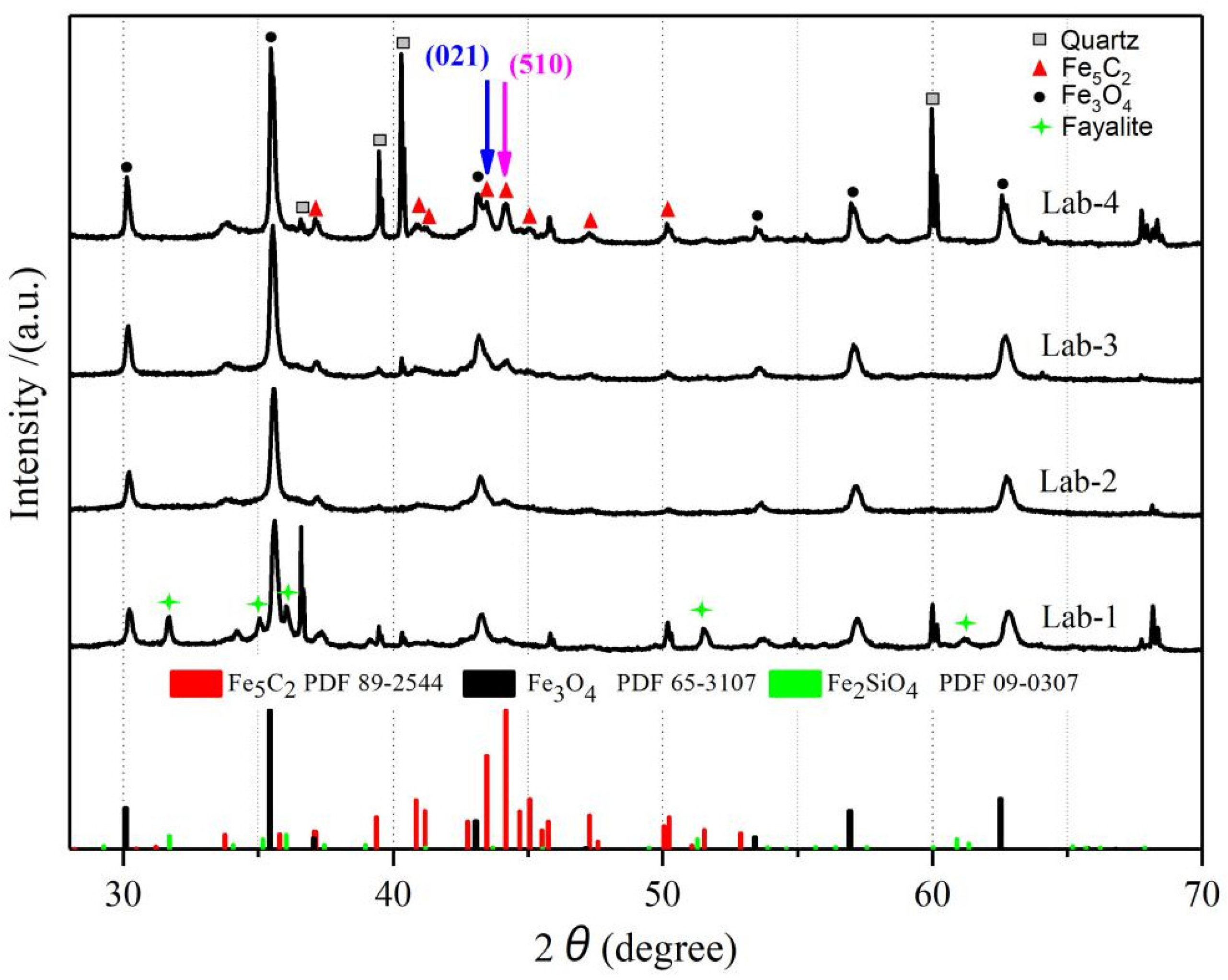
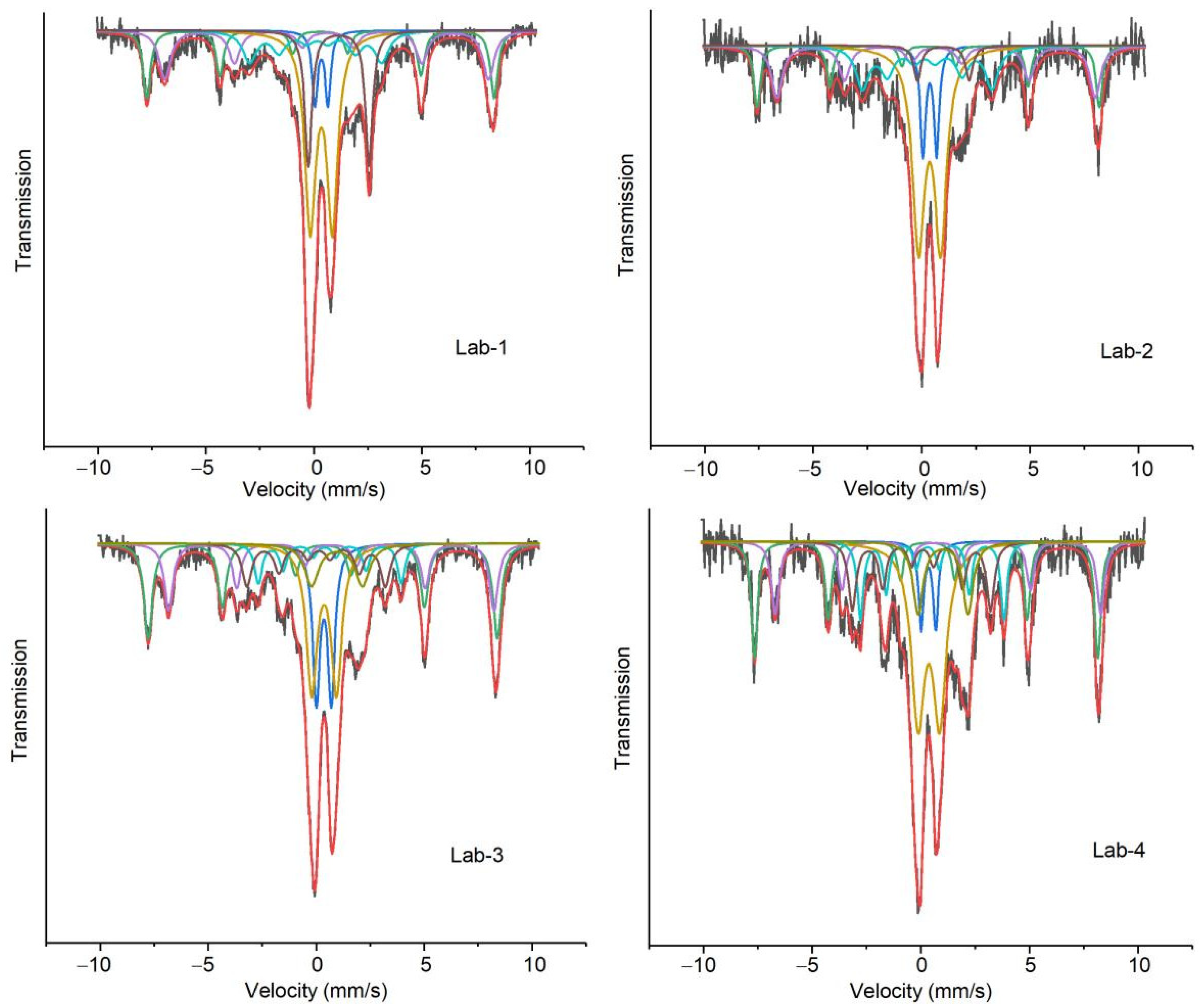
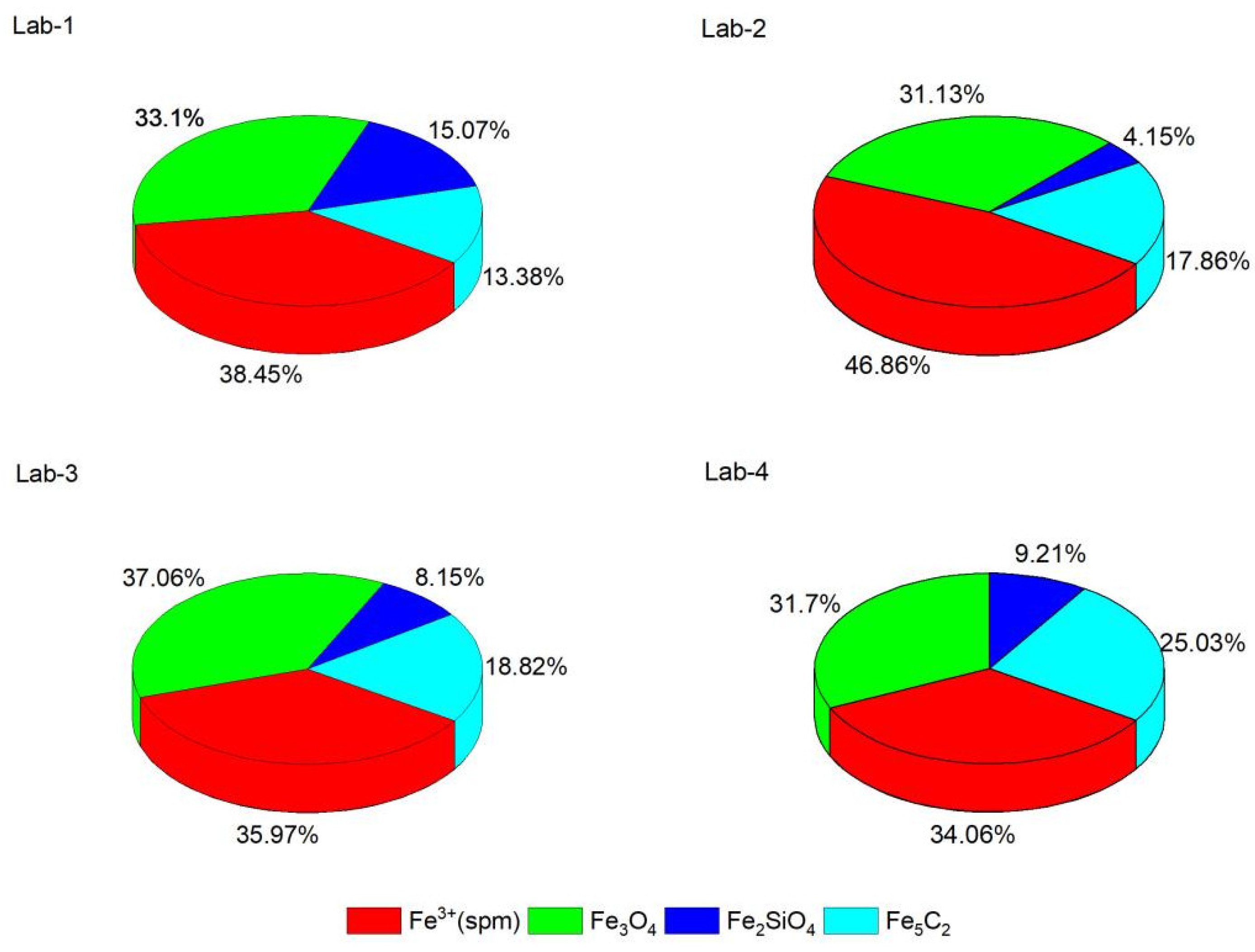
2.4. Characterization of Spent Catalyst Samples
3. Materials and Methods
3.1. Materials
3.2. Catalyst Preparation
3.3. Catalyst Characterization
3.3.1. Nitrogen Adsorption/Desorption
3.3.2. X-ray Diffraction
3.3.3. In Situ XRD
3.3.4. FTIR/ATR Spectra
3.3.5. Temperature-Programmed Reduction (TPR)
3.3.6. Temperature-Programmed Hydrogenation (TPH)
3.3.7. X-ray Photoelectron Spectroscopy (XPS)
3.3.8. Raman Spectrum
3.3.9. Mössbauer Spectra (MES)
3.4. Fischer-Tropsch Synthesis Performance Test
3.4.1. FTS Test in Fixed-Bed Reactor (FBR)
3.4.2. Product Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dry, M.E. The Fischer-Tropsch process: 1950–2000. Catal. Today 2002, 71, 227–241. [Google Scholar] [CrossRef]
- Botes, F.G.; Niemantsverdriet, J.W.; van de Loosdrecht, J. A comparison of cobalt and iron based slurry phase Fischer-Tropsch synthesis. Catal. Today 2013, 215, 112–120. [Google Scholar] [CrossRef]
- Suo, H.Y.; Wang, S.G.; Zhang, C.H.; Xu, J.; Wu, B.S.; Yang, Y.; Xiang, H.W.; Li, Y.W. Chemical and structural effects of silica in iron-based Fischer-Tropsch synthesis catalysts. J. Catal. 2012, 286, 111–123. [Google Scholar] [CrossRef]
- Dry, M.E. Fischer-Tropsch synthesis. In Catalysis, Science and Technology, 1st ed.; Anderson, J.R., Boudart, M., Eds.; Springer: New York, NY, USA, 1981; pp. 159–255. [Google Scholar]
- Dry, M.E. Present and future applications of the Fischer-Tropsch process. Appl. Catal. A Gen. 2004, 276, 1–3. [Google Scholar] [CrossRef]
- Yang, Y.; Xiang, H.W.; Xu, Y.Y.; Bai, L.; Li, Y.W. Effect of potassium promoter on precipitated iron-manganese catalyst for Fischer-Tropsch synthesis. Appl. Catal. A Gen. 2004, 266, 181–194. [Google Scholar] [CrossRef]
- Raje, A.P.; O’Brien, R.J.; Davis, B.H. Effect of Potassium Promotion on Iron-Based Catalysts for Fischer-Tropsch Synthesis. J. Catal. 1998, 180, 36–43. [Google Scholar] [CrossRef]
- Bukur, D.B.; Mukesh, D.S.; Patal, A. Promoter Effects on Precipitated Iron Catalysts for Fischer-Tropsch Synthesis. Ind. Eng. Chem. Res. 1990, 29, 194–204. [Google Scholar] [CrossRef]
- Pendyala, V.R.R.; Graham, U.M.; Jacobs, G.; Hamdeh, H.H.; Davis, B.H. Fischer-Tropsch Synthesis: Deactivation as a Function of Potassium Promoter Loading for Precipitated Iron Catalyst. Catal. Lett. 2014, 144, 704–1716. [Google Scholar] [CrossRef]
- Niu, L.W.; Liu, X.W.; Wen, X.D.; Yang, Y.; Xu, J.; Li, Y.W. Effect of potassium promoter on phase transformation during H2 pretreatment of a Fe2O3 Fischer Tropsch synthesis catalyst precursor. Catal. Today 2020, 343, 101–111. [Google Scholar] [CrossRef]
- Eliason, S.A.; Bartholomew, C.H. Reaction and deactivation kinetics for Fischer-Tropsch synthesis on unpromoted and potassium-promoted iron catalysts. Appl. Catal. A Gen. 1999, 186, 229–243. [Google Scholar] [CrossRef]
- Kölbel, H. Kalium als strucktureller und Energetischer Promotor in Eisenkatalysatoren. In Actes du Deuxieme Congress International de Catalyse; Tchnip: Paris, France, 1960; Volume II, p. 2075. [Google Scholar]
- Soled, S.; Iglesia, E.; Miseo, S.; DeRites, B.A.; Fiato, R.A. Selective synthesis of a-olefins on Fe-Zn Fischer-Tropsch catalysts. Top. Catal. 1995, 2, 193. [Google Scholar] [CrossRef]
- Frohning, C.D.; Kölbel, H.; Ralek, M.; Rottig, W.; Schuur, F.; Schulz, H. Fischer-Tropsch-Synthese. In Chemierohstoffe aus Kohle; Falbe, J., Ed.; Georg Thieme Verlag: Stuttgart, Germany, 1977; pp. 219–299. [Google Scholar]
- Jothimurugesan, K.; Spivey, J.J.; Gangwal, S.K.; Goodwin, J.G. Effect of silica on iron-based Fischer-Tropsch catalysts. Natural Gas Conversion V. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1998; Volume 119, pp. 215–220. [Google Scholar]
- Hou, W.J.; Wu, B.S.; Yang, Y.; Hao, Q.L.; Tian, L.; Xiang, H.W.; Li, Y.W. Effect of SiO2 content on iron-based catalysts for slurry Fischer-Tropsch synthesis. Fuel Process. Technol. 2008, 89, 284–291. [Google Scholar] [CrossRef]
- Chang, H.; Cheng, M.; Lin, Q.; Zhu, J.Q.; Lv, Y.J.; Men, Z.W. Effects of binder addition process parameters on physical-chemical and catalytic performance of iron-based Fischer-Tropsch (F-T) synthesis catalyst. J. China Coal Soc. 2021, 46, 3350–3356. [Google Scholar]
- Lin, Q.; Cheng, M.; Zhang, K.; Li, W.Z.; Wu, P.; Chang, H.; Lv, Y.J.; Men, Z.W. Development of an Iron-Based Fischer-Tropsch Catalyst with High Attrition Resistance and Stability for Industrial Application. Catalysts 2021, 11, 908. [Google Scholar] [CrossRef]
- Bukur, D.B.; Carreto-Vazqueza, V.H.; Ma, W.P. Catalytic performance and attrition strength of spray-dried iron catalysts for slurry phase Fischer-Tropsch synthesis. Appl. Catal. A Gen. 2010, 388, 240–247. [Google Scholar] [CrossRef]
- Yang, Y.; Xiang, H.W.; Tian, L.; Wang, H.; Zhang, C.H.; Tao, Z.C.; Xu, Y.Y.; Zhong, B.; Li, Y.W. Structure and Fischer-Tropsch performance of iron-manganese catalyst incorporated with SiO2. Appl. Catal. A Gen. 2005, 284, 105–122. [Google Scholar] [CrossRef]
- Zhang, C.-H.; Wan, H.-J.; Yang, Y.; Xiang, H.-W.; Li, Y.-W. Study on the iron–silica interaction of a co-precipitated Fe/SiO2 Fischer-Tropsch synthesis catalyst. Catal. Commun. 2006, 7, 733–738. [Google Scholar] [CrossRef]
- Mogorosi, R.; Fischer, N.; Claeys, M.; Steen, E.V. Strong-metal–support interaction by molecular design: Fe–silicate interactions in Fischer-Tropsch catalysts. J. Catal. 2012, 289, 140–150. [Google Scholar] [CrossRef]
- Dlamini, H.; Motjope, T.; Joorst, G.; Stege, G.T.; Mdleleni, M. Changes in physico-chemical properties of iron-based Fischer-Tropsch catalyst induced by SiO2 addition. Catal. Lett. 2002, 78, 201–207. [Google Scholar] [CrossRef]
- Bukur, D.B.; Lang, X.S.; Mukesh, D.; Zimmerman, W.H.; Rosynek, M.P.; Li, C.P. Binder/support effects on the activity and selectivity of iron catalysts in the Fischer-Tropsch synthesis. Ind. Eng. Chem. Res. 1990, 29, 1588–1599. [Google Scholar] [CrossRef]
- Lohitharn, N.; Goodwin, J.G. Effect of K promotion of Fe and FeMn Fischer-Tropsch synthesis catalysts: Analysis at the site level using SSITKA. J. Catal. 2008, 260, 7–16. [Google Scholar] [CrossRef]
- Liu, H.; Li, P.; Lu, B.; Wei, Y.; Sun, Y.H. Transformation of ferrihydrite in the presence or absence of trace Fe(Ⅱ): The effect of preparation procedures of ferrihydrite. J. Solid State Chem. 2009, 182, 1767–1771. [Google Scholar] [CrossRef]
- Seehra, M.; Roy, P.; Raman, A.; Manivannan, A. Structural investigations of synthetic ferrihydrite nanoparticles doped with Si. Solid State Commun. 2004, 130, 597–601. [Google Scholar] [CrossRef]
- Stolyar, S.V.; Yaroslavtsev, R.N.; Bayukov, C.A.; Balaev, D.A.; Krasikov, A.A.; Iskhakov, R.S.; Vorotynov, A.M.; Pladygina, V.P.; Purtov, K.V.; Volochaev, M.N. Preparation, structure and magnetic properties of synthetic ferrihydrite nanoparticles. J. Phys. Conf. Ser. 2018, 994, 0120023. [Google Scholar] [CrossRef]
- Krehula, S.; Music, S. Influence of aging in an alkaline medium on the microstructural properties of a-FeOOH. J. Cryst. Growth 2008, 310, 513–520. [Google Scholar] [CrossRef]
- Rzepa1, G.; Pieczara1, G.; Gaweł1, A.; Tomczyk, A.; Zalecki, R. The inflfluence of silicate on transformation pathways of synthetic 2-line ferrihydrite. J. Therm. Anal. Calorim. 2016, 125, 407–421. [Google Scholar] [CrossRef]
- Chernyshova, I.V.; Hochella, M.F., Jr.; Madden, A.S. Size-dependent structural transformations of hematite nanoparticles. 1. Phase transition. Phys. Chem. Chem. Phys. 2007, 9, 1736–1750. [Google Scholar] [CrossRef] [PubMed]
- Swedlund, P.J.; Gordon, M.; Miskelly, G.M.; McQuillan, A.J. An attenuated total reflflectance IR study of silicic acid adsorbed onto a ferric oxyhydroxide surface. Geochim. Cosmochim. Acta 2009, 73, 4199–4214. [Google Scholar] [CrossRef]
- Das, S.K.; Majhi, S.; Mohanty, P.; Pant, K.K. CO-hydrogenation of syngas to fuel using silica supported Fe-Cu-K catalysts: Effects of active components. Fuel Process. Technol. 2014, 118, 82–89. [Google Scholar] [CrossRef]
- Choi, J.-S.; Yoon, S.-S.; Jang, S.-H.; Ahn, W.-S. Phenol hydroxylation using Fe-MCM-41 catalysts. Catal. Today 2006, 111, 280–287. [Google Scholar] [CrossRef]
- Swedlund, P.J.; Miskelly, G.M.; McQuillan, A.J. Silicic acid adsorption and oligomerization at the ferrihydrite-water interface: Interpretation of ATR-IR spectra based on a model surface structure. Langmuir 2010, 26, 3394–3401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qing, M.; Wang, H.; Liu, X.; Liu, S.; Wan, H.; Li, L.; Gao, X.; Yang, Y.; Wen, X.; et al. Comprehensive Understanding of SiO2-promoted Fe Fischer-Tropsch Synthesis Catalysts: Fe-SiO2 Interaction and Beyond. Catal. Today 2021, 368, 96–105. [Google Scholar] [CrossRef]
- Qing, M.; Yang, Y.; Wu, B.; Xu, J.; Zhang, C.; Gao, P.; Li, Y. Modification of Fe-SiO2 interaction with zirconia for iron-based Fischer-Tropsch catalysts. J. Catal. 2011, 279, 111–122. [Google Scholar] [CrossRef]
- Bukur, D.B.; Sivaraj, C. Supported iron catalyst for slurry phase Fischer-Tropsch synthesis. Appl. Catal. A Gen. 2002, 231, 201–214. [Google Scholar] [CrossRef]
- Suo, H.; Zhang, C.; Wu, B.; Xu, J.; Yang, Y.; Xiang, H.; Li, Y. A comparative study of Fe/SiO2 Fischer-Tropsch synthesis catalysts using tetraethoxysilane and acidic silica sol as silica sources. Catal. Today 2012, 183, 88–95. [Google Scholar] [CrossRef]
- Wan, H.-J.; Wu, B.-S.; Tao, Z.-C.; Li, T.-Z.; An, X.; Xiang, H.-W.; Li, Y.-W. Study of an iron-based Fischer-Tropsch synthesis catalyst incorporated with SiO2. J. Mol. Catal. A Chem. 2006, 260, 255–263. [Google Scholar] [CrossRef]
- Qiu, T.; Wang, L.; Lv, S.; Sun, B.; Zhang, Y.; Liu, Z.; Yang, W.; Li, J. SAPO-34 zeolite encapsulated Fe3C nanoparticles as highly selective Fischer-Tropsch catalysts for the production of light olefins. Fuel 2017, 203, 811–816. [Google Scholar] [CrossRef]
- Xu, J.; Bartholomew, C.H. Temperature-programmed hydrogenation (TPH) and in situ Mössbauer spectroscopy studies of carbonaceous species on silica-supported iron Fischer-Tropsch catalysts. Phys. Chem. B 2005, 109, 2392–2403. [Google Scholar] [CrossRef]
- Nakhaei Pour, A.; Housaindokht, M.R.; Tayyari, S.F.; Zarkesh, J.; Alaei, M.R. Deactivation studies of Fischer-Tropsch synthesis on nano-structured iron catalyst. J. Mol. Catal. A Chem. 2010, 330, 112–120. [Google Scholar] [CrossRef]
- Mo, W.L.; Wang, X.X.; Zou, M.; Huang, X.J.; Ma, F.Y.; Zhao, J.Z.; Zhao, T.S. Influence of Ni Precursors on the Structure, Performance, and Carbon Deposition of Ni-Al2O3 Catalysts for CO Methanation. ACS Omega 2021, 6, 16373–16380. [Google Scholar] [CrossRef]
- Paalanen, P.P.; van Vreeswijk, S.H.; Weckhuysen, B.M. Combined in situ x-ray diffractrometry/raman spectroscopy of iron carbide and carbon species evolution in Fe(-Na-S)/a-Al2O3 catalysts during Fisch-Tropsch synthesis. ACS Catal. 2020, 10, 9837–9855. [Google Scholar] [CrossRef]
- Brunetto, R.; Lantz, C.; Ledu, D.; Baklouti, D.; Barucci, M.A.; Beck, P.; Delauche, L.; Dionnet, Z.; Dumas, P.; Duprat, J.; et al. Iron irradiation of allende meteorite probed by visible, IR, and Raman spectroscopy. Icarus 2014, 237, 278–292. [Google Scholar] [CrossRef]
- Wielers, A.F.H.; Kock, A.J.H.M.; Hop, C.E.C.A.; Geus, J.W.; van der Kran, A.M. The Reduction Behavior of Silica-Supported and Alumina-Supported Iron Catalysts: A Mossbauer and Infrared Spectroscopic Study. J. Catal. 1989, 117, 1–18. [Google Scholar] [CrossRef]
- Hou, W.J.; Wu, B.S.; An, X.; Li, T.Z.; Tao, Z.C.; Zheng, H.Y.; Xiang, H.W.; Li, Y.W. Effect of the ratio of precipitated SiO2 to binder SiO2 on iron-based catalysts for Fischer-Tropsch synthesis. Catal. Lett. 2007, 119, 353–360. [Google Scholar] [CrossRef]
- Pariona, N.; Camacho-Aguilar, K.I.; Ramos-González, R.; Martinez, A.I.; Herrera-Trejo, M.; Baggio-Saitovitch, E. Magnetic and structural properties of ferrihydrite/hematite nanocomposites. J. Magn. Magn. Mater. 2016, 406, 221–227. [Google Scholar] [CrossRef]
- Huggins, F.E.; Bali, S.; Huffman, G.P.; Eyring, E.M. Iron-oxide aerogel and xerogel catalyst formulations: Characterization by 57Fe Mössbauer and XAFS spectroscopies. Spectrochim. Acta Part A 2010, 76, 74–83. [Google Scholar] [CrossRef]
- Hafner, S.S.; Stanek, J.; Stanek, M. 57Fe Hyperfine interactions in the magnetic phase of fayalite, Fe2SiO4. J. Phys. Chem. Solids 1990, 51, 203–208. [Google Scholar] [CrossRef]
- Yuen, S.; Chen, Y.; Kubsh, J.E.; Dumesic, J.A.; TopsØe, N.; TopsØe, H. Metal Oxide-Support Interactions in Silica-Supported Iron Oxide Catalysts Probed by Nitric Oxide Adsorption. J. Phys. Chem. 1982, 86, 3022–3032. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Peak | Peak Centre, °C | H2 Consumption, mol H2/mol Fe | Fe% in First Peak | |
|---|---|---|---|---|---|
| Single | Total | ||||
| Lab-1 | I | 226.8 | 0.27 | 1.42 | 19.11 |
| II | 589.9 | 0.6 | / | ||
| III | 629.4 | 0.55 | / | ||
| Lab-2 | I | 245.3 | 0.24 | 1.23 | 19.83 |
| II | 579.1 | 0.858 | / | ||
| III | 635.3 | 0.125 | / | ||
| Lab-3 | I | 252.3 | 0.237 | 1.1 | 21.42 |
| II | 558.3 | 0.868 | / | ||
| Lab-4 | I | 253.8 | 0.218 | 1.01 | 21.54 |
| II | 526.0 | 0.792 | / | ||
| Sample | TPH Peak Temperature, °C | |||||
|---|---|---|---|---|---|---|
| Carbidic C | Amorphous C | β-C | Bulk C1 | Bulk C2 | Bulk C3 | |
| Lab-1 | 324 | 385 | 446 | 504 | 552 | 625 |
| Lab-2 | 312 | 365 | / | 523 | 562 | 624 |
| Lab-3 | 319 | 385 | / | 473 | 544 | / |
| Lab-4 | 319 | 373 | / | 470 | 540 | / |
| Sample | Carbon Species Assignment and Relative Percentage, % | ||||||
|---|---|---|---|---|---|---|---|
| Carbidic C | Amorphous C | β-C | Bulk C1 | Bulk C2 | Bulk C3 | Total Bulk C | |
| Lab-1 | 4.53 | 5.44 | 16.33 | 5.07 | 27.89 | 40.74 | 73.63 |
| Lab-2 | 25.99 | 5.12 | / | 29.61 | 32.94 | 6.34 | 68.89 |
| Lab-3 | 41.67 | 11.67 | / | 16.02 | 30.64 | / | 46.66 |
| Lab-4 | 49.91 | 11.2 | / | 29.25 | 9.64 | / | 38.89 |
| Crystallite Size, nm | ||||
|---|---|---|---|---|
| Lab-1 | Lab-2 | Lab-3 | Lab-4 | |
| a Fresh samples | <8 | <8 | <8 | <8 |
| b χ-Fe5C2 | 12 | 15 | 20 | 41 |
| Cat. | Phases | Mossbauer Parameters | Phase Percentage (%) | ||||
|---|---|---|---|---|---|---|---|
| IS (mm·s−1) | QS (mm·s−1) | Hhf (T) | Area (%) | ||||
| Lab-1 | Doublet1 | Fe3+ (spm) | 0.3378 | 0.602 | / | 6.16 | FH: 38.45 |
| Doublet2 | Fe3+ (spm) | 0.3356 | 1.025 | / | 32.29 | / | |
| Doublet3 | Fe2SiO4 | 1.138 | 2.806 | / | 15.07 | Fe2SiO4: 15.07 | |
| Mixed M+Q (1) | Fe3O4 (A) | 0.294 | 0.0075 | 49.98 | 15.78 | Fe3O4: 33.1 | |
| Mixed M+Q (2) | Fe3O4 (B) | 0.617 | −0.064 | 46.61 | 17.32 | / | |
| Mixed M+Q (3) | χ-Fe5C2 | 0.109 | −0.0656 | 19.009 | 13.38 | χ-Fe5C2: 13.38 | |
| Lab-2 | Doublet1 | Fe3+ (spm) | 0.363 | 0.628 | / | 9.19 | FH: 46.86 |
| Doublet2 | Fe3+ (spm) | 0.3565 | 1.014 | / | 37.67 | / | |
| Doublet3 | Fe2SiO4 | 0.988 | 2.392 | / | 4.15 | Fe2SiO4: 4.15 | |
| Mixed M+Q (1) | Fe3O4 (A) | 0.314 | −0.0368 | 49.015 | 12.73 | Fe3O4: 31.13 | |
| Mixed M+Q (2) | Fe3O4 (B) | 0.671 | −0.0274 | 45.599 | 18.39 | / | |
| Mixed M+Q (3) | χ-Fe5C2 | 0.1936 | 0.1155 | 18.54 | 17.86 | χ-Fe5C2: 17.86 | |
| Lab-3 | Doublet1 | Fe3+ (spm) | 0.345 | 0.695 | / | 14.88 | FH: 35.97 |
| Doublet2 | Fe3+ (spm) | 0.3598 | 1.147 | / | 21.09 | / | |
| Doublet3 | Fe2SiO4 | 0.9594 | 2.354 | / | 8.16 | Fe2SiO4: 8.15 | |
| Mixed M+Q (1) | Fe3O4 (A) | 0.3089 | −0.0238 | 50.235 | 21.49 | Fe3O4: 37.06 | |
| Mixed M+Q (2) | Fe3O4 (B) | 0.6825 | 0.01489 | 46.83 | 15.57 | / | |
| Mixed M+Q (3) | χ-Fe5C2 (I) | 0.5039 | 0.241 | 20.579 | 7.87 | χ-Fe5C2: 18.82 | |
| Mixed M+Q (4) | χ-Fe5C2 (II) | 0.0586 | −0.1324 | 19.926 | 10.95 | / | |
| Lab-4 | Doublet1 | Fe3+ (spm) | 0.339 | 0.6685 | / | 5.49 | FH: 34.06 |
| Doublet2 | Fe3+ (spm) | 0.3594 | 0.9918 | / | 28.57 | / | |
| Doublet3 | Fe2SiO4 | 1.0144 | 2.2969 | / | 9.21 | Fe2SiO4: 9.21 | |
| Mixed M+Q (1) | Fe3O4 (A) | 0.271 | −0.06778 | 49.089 | 17.98 | Fe3O4: 31.7 | |
| Mixed M+Q (2) | Fe3O4 (B) | 0.738 | 0.0879 | 46.539 | 13.72 | / | |
| Mixed M+Q (3) | χ-Fe5C2 (I) | 0.411 | 0.2115 | 20.525 | 11.91 | χ-Fe5C2: 25.03 | |
| Mixed M+Q (4) | χ-Fe5C2 (II) | 0.04487 | −0.05465 | 19.749 | 13.12 | / | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, H.; Lin, Q.; Cheng, M.; Zhang, K.; Feng, B.; Chai, J.; Lv, Y.; Men, Z. Effects of Potassium Loading over Iron–Silica Interaction, Phase Evolution and Catalytic Behavior of Precipitated Iron-Based Catalysts for Fischer-Tropsch Synthesis. Catalysts 2022, 12, 916. https://doi.org/10.3390/catal12080916
Chang H, Lin Q, Cheng M, Zhang K, Feng B, Chai J, Lv Y, Men Z. Effects of Potassium Loading over Iron–Silica Interaction, Phase Evolution and Catalytic Behavior of Precipitated Iron-Based Catalysts for Fischer-Tropsch Synthesis. Catalysts. 2022; 12(8):916. https://doi.org/10.3390/catal12080916
Chicago/Turabian StyleChang, Hai, Quan Lin, Meng Cheng, Kui Zhang, Bo Feng, Jiachun Chai, Yijun Lv, and Zhuowu Men. 2022. "Effects of Potassium Loading over Iron–Silica Interaction, Phase Evolution and Catalytic Behavior of Precipitated Iron-Based Catalysts for Fischer-Tropsch Synthesis" Catalysts 12, no. 8: 916. https://doi.org/10.3390/catal12080916