
Stability of Pt-Adsorbed CO on Catalysts for Room Temperature-Oxidation of CO †



Abstract
:
1. Introduction
2. Experimental Section
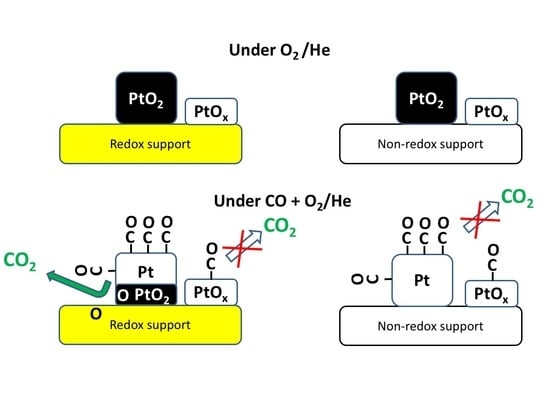
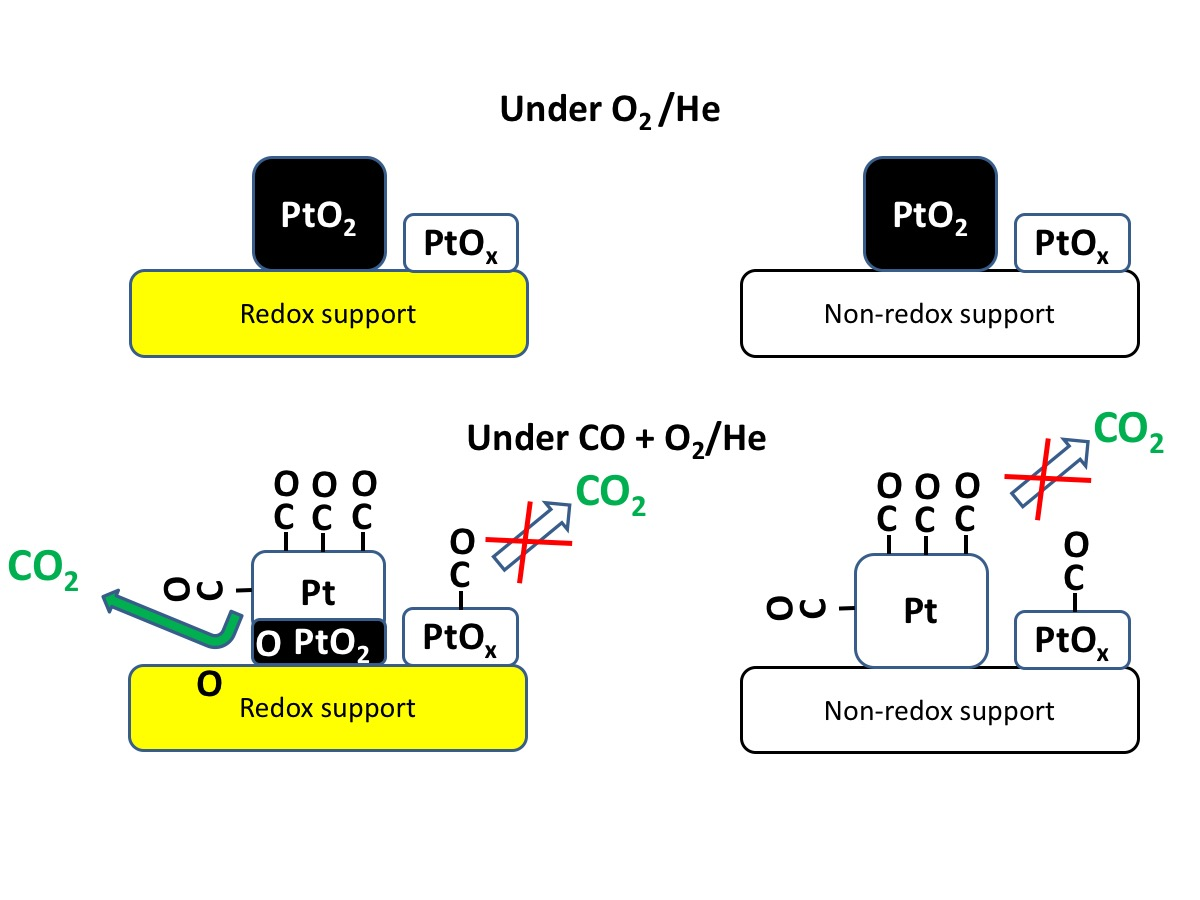
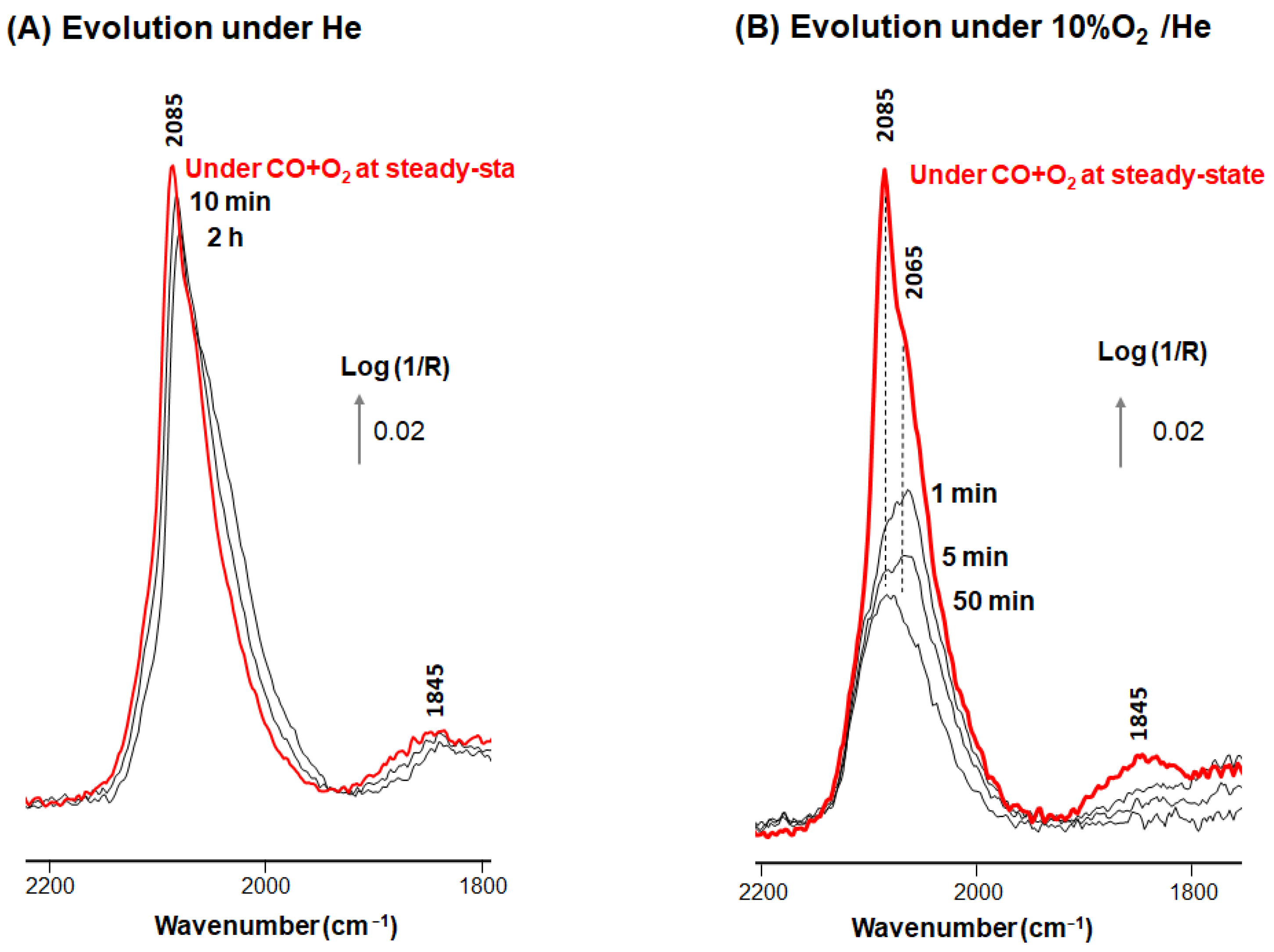
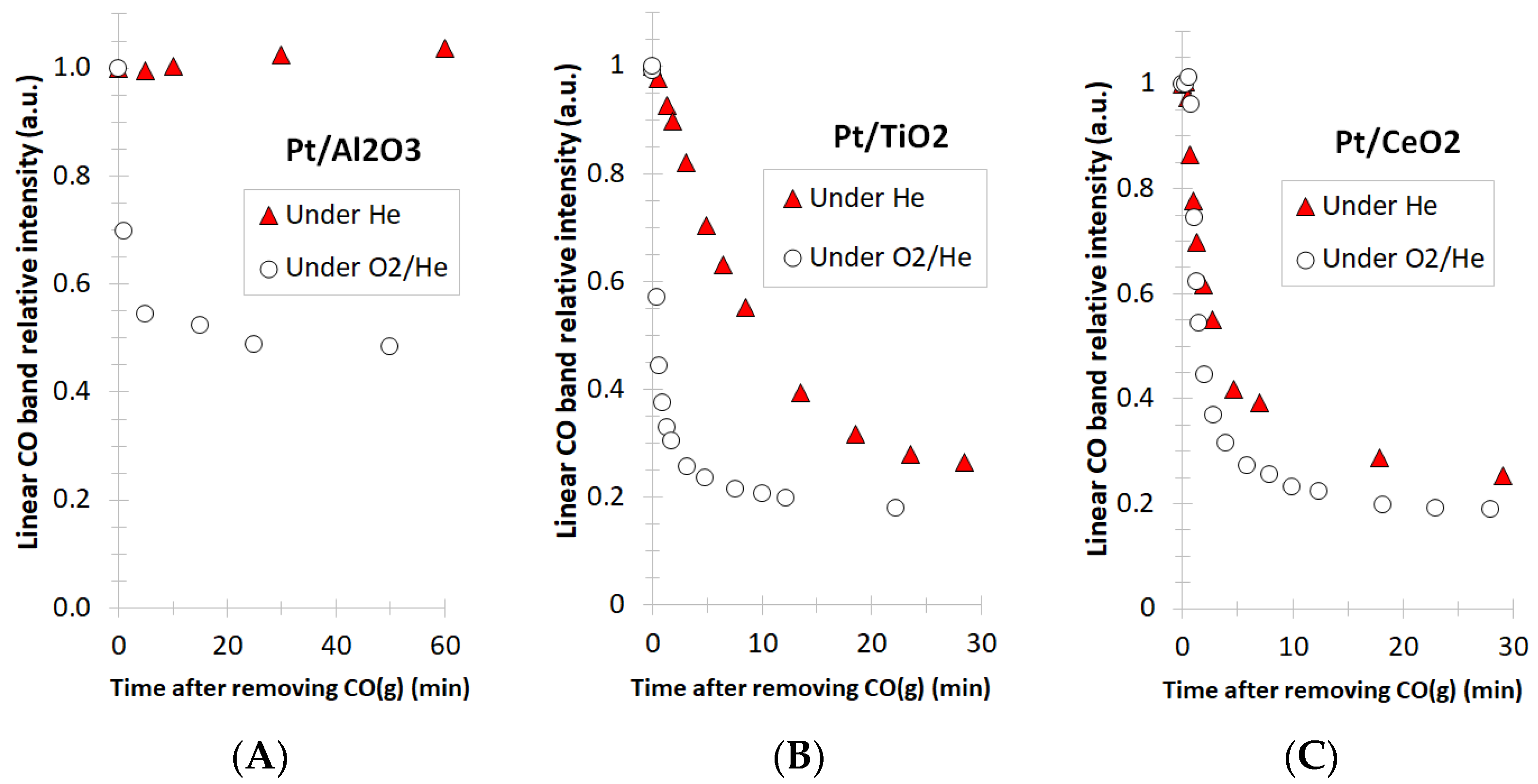
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bi, F.; Zhang, X.; Du, Q.; Yue, K.; Wang, R.; Li, F.; Liu, N.; Huang, Y. Influence of pretreatment conditions on low-temperature CO oxidation over Pd supported UiO-66 catalysts. Mol. Catal. 2021, 509, 111633. [Google Scholar] [CrossRef]
- Di, M.; Simmance, K.; Schaefer, A.; Feng, Y.; Hemmingsson, F.; Skoglundh, M.; Bell, T.; Thompsett, D.; Jensen, L.I.A.; Blomberg, S.; et al. Chasing PtOx species in ceria supported platinum during CO oxidation extinction with correlative operando spectroscopic techniques. J. Catal. 2022, 409, 1–11. [Google Scholar] [CrossRef]
- Rangel, R.; González-A, E.; Solís-García, A.; Zepeda, T.A.; Galván, D.H.; Gómez-Cortés, A.; Díaz, G. Pt and Ir supported on mixed Ce0.97Ru0.03O2 oxide as low-temperature CO oxidation catalysts. Catal. Today 2022, 392–393, 3–12. [Google Scholar] [CrossRef]
- Kale, M.J.; Gidcumb, D.; Gulian, F.J.; Miller, S.P.; Clark, C.H.; Christopher, P. Evaluation of platinum catalysts for naval submarine pollution control. Appl. Catal. B Environ. 2017, 203, 533–540. [Google Scholar] [CrossRef]
- Saavedra, J.; Doan, H.A.; Pursell, C.J.; Grabow, L.C.; Chandler, B.D. The critical role of water at the gold-titania interface in catalytic CO oxidation. Science 2014, 345, 1599–1602. [Google Scholar] [CrossRef] [Green Version]
- Meunier, F.C.; Cardenas, L.; Kaper, H.; Šmíd, B.; Vorokhta, M.; Grosjean, R.; Aubert, D.; Dembélé, K.; Lunkenbein, T. Synergy between metallic and oxidized Pt sites unravelled during room temperature CO oxidation on Pt/ceria. Angew. Chem. Int. Ed. 2021, 60, 3799–3805. [Google Scholar] [CrossRef]
- Lear, T.; Marshall, R.; Lopez-Sanchez, J.A.; Jackson, S.D.; Klapötke, T.M.; Baümer, M.; Rupprechter, G.; Freund, H.-J.; Lennon, D. The application of infrared spectroscopy to probe the surface morphology of alumina-supported palladium catalysts. J. Chem. Phys. 2005, 123, 174706. [Google Scholar] [CrossRef] [Green Version]
- Aleksandrov, H.A.; Neyman, K.M.; Hadjiivanov, K.I.; Vayssilov, G.N. Can the state of platinum species be unambiguously determined by the stretching frequency of an adsorbed CO probe molecule? Phys. Chem. Chem. Phys. 2016, 18, 22108–22121. [Google Scholar] [CrossRef] [Green Version]
- DeRita, L.; Dai, S.; Lopez–Zepeda, K.; Pham, N.; Graham, G.W.; Pan, X.; Christopher, P. Catalyst architecture for stable single atom dispersion enables site–specific spectroscopic and reactivity measurements of CO adsorbed to Pt atoms, oxidized Pt clusters, and metallic Pt clusters on TiO2. J. Am. Chem. Soc. 2017, 139, 14150–14165. [Google Scholar] [CrossRef]
- Meunier, F.C.; Kdhir, R.; Potrzebowska, N.; Perret, N.; Besson, M. Unravelling platinum-zirconia interfacial sites using CO adsorption. Inorg. Chem. 2019, 58, 8021–8029. [Google Scholar] [CrossRef]
- Maurer, F.; Jelic, J.; Wang, J.; Gänzler, A.; Dolcet, P.; Wöll, C.; Wang, Y.; Studt, F.; Casapu, M.; Grunwaldt, J.D. Tracking the formation, fate and consequence for catalytic activity of Pt single sites on CeO2. Nat. Catal. 2020, 3, 824–833. [Google Scholar] [CrossRef]
- Resasco, J.; DeRita, L.; Dai, S.; Chada, J.P.; Xu, M.; Yan, X.; Finzel, J.; Hanukovich, S.; Hoffman, A.S.; Graham, G.W.; et al. Uniformity is key in defining structure–function relationships for atomically dispersed metal catalysts: The case of Pt/CeO2. J. Am. Chem. Soc. 2020, 142, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Meira, D.M.; Arenal, R.; Concepcion, P.; Puga, A.V.; Corma, A. Determination of the evolution of heterogeneous single metal atoms and nanoclusters under reaction conditions: Which are the working catalytic sites? ACS Catal. 2019, 9, 10626–10639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Thrimurthulu, G.; Delannoy, L.; Louis, C.; Mottet, C.; Creuze, J.; Legrand, B.; Guesmi, H. Evidence of Pd segregation and stabilization at edges of AuPd nano-clusters in the presence of CO: A combined DFT and DRIFTS study. J. Catal. 2013, 308, 272–281. [Google Scholar] [CrossRef]
- Jbir, I.; Couble, J.; Khaddar-Zine, S.; Ksibi, Z.; Meunier, F.; Bianchi, D. Individual heat of adsorption of adsorbed CO species on palladium and Pd–Sn nanoparticles supported on Al2O3by using temperature-programmed adsorption equilibrium methods. ACS Catal. 2016, 6, 2545. [Google Scholar] [CrossRef]
- Meunier, F.; Maffre, M.; Schuurman, Y.; Colussi, S.; Trovarelli, A. Acetylene semi-hydrogenation over Pd-Zn/CeO2: Relevance of CO adsorption and methanation as descriptors of selectivity. Catal. Commun. 2018, 105, 52–55. [Google Scholar] [CrossRef]
- Meunier, F.C. Pitfalls and benefits of in situ and operando diffuse reflectance FT-IR spectroscopy (DRIFTS) applied to catalytic reactions. React. Chem. Eng. 2016, 1, 134–141. [Google Scholar] [CrossRef]
- Paredes-Nunez, A.; Jbir, I.; Bianchi, D.; Meunier, F.C. Spectrum baseline artefacts and correction of gas-phase species signal during diffuse reflectance FT-IR analyses of catalysts at variable temperatures. Appl. Catal. A Gen. 2015, 495, 17–22. [Google Scholar] [CrossRef]
- Meunier, F.C. On the contamination with nickel and nickel tetracarbonyl during FT-IR investigation of catalysts under CO-containing gases. J. Catal. 2019, 372, 388. [Google Scholar] [CrossRef]
- Meunier, F.C. Comment on “Stabilizing platinum atoms on CeO2 oxygen vacancies by metal-support interaction induced interface distortion: Mechanism and application” by Jiang et al. Appl. Catal. B 2020, 278, 119304. Appl. Catal. B Environ. 2022, 302, 120841. [Google Scholar] [CrossRef]
- Li, H.; Rivallan, M.; Thibault-Starzyk, F.; Travert, A.; Meunier, F.C. Effective bulk and surface temperatures of the catalyst bed of FT-IR cells used for in situ and operando studies. Phys. Chem. Chem. Phys. 2013, 15, 7321–7327. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Mei, D.; Xiong, H.; Peng, B.; Ren, Z.; Hernandez, X.I.P.; DeLaRiva, A.; Wang, M.; Engelhard, M.H.; Kovarik, L.; et al. Activation of surface lattice oxygen in single–atom Pt/CeO2 for low–temperature CO oxidation. Science 2017, 358, 1419–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Guo, Y.; Huang, Y.; Xi, W.; Xu, J.; Luo, J.; Qi, H.; Ren, Y.; Liu, X.; Qiao, B.; et al. Strong metal–support interactions between Pt single atoms and TiO2. Angew. Chem. Int. Ed. 2020, 59, 11824–11829. [Google Scholar] [CrossRef]
- Kottwitz, M.; Li, Y.; Palomino, R.M.; Liu, Z.; Wang, G.; Wu, Q.; Huang, J.; Timoshenko, J.; Senanayake, S.D.; Balasubramanian, M.; et al. Local structure and electronic state of atomically dispersed Pt supported on nanosized CeO2. ACS Catal. 2019, 9, 8738–8748. [Google Scholar] [CrossRef]
- Lu, Y.; Zhou, S.; Kuo, C.-T.; Kunwar, D.; Thompson, C.; Hoffman, A.S.; Boubnov, A.; Lin, S.; Datye, A.K.; Guo, H.; et al. Unraveling the intermediate reaction complexes and critical role of support-derived oxygen atoms in CO oxidation on single-atom Pt/CeO2. ACS Catal. 2021, 11, 8701–8715. [Google Scholar] [CrossRef]
- Jeong, H.; Shin, D.; Kim, B.; Bae, J.; Shin, S.; Choe, C.; Han, J.W.; Lee, H. Controlling the oxidation state of Pt single atoms for maximizing catalytic activity. Angew. Chem. Int. Ed. 2020, 59, 20691–20696. [Google Scholar] [CrossRef] [PubMed]
- Thang, H.V.; Pacchioni, G.; DeRita, L.; Christopher, P. Nature of stable single atom Pt catalysts dispersed on anatase TiO2. J. Catal. 2018, 367, 104–114. [Google Scholar] [CrossRef]
- Ertl, G.; Neumann, M.; Streit, K.M. Chemisorption of CO on the Pt(111) Surface. Surf. Sci. 1977, 64, 393–410. [Google Scholar] [CrossRef]
- Altman, E.I.; Gorte, R.J. The Desorption of CO from Small Pt Particles on Al2O3. Surf. Sci. 1986, 172, 71–80. [Google Scholar] [CrossRef]
- Kopelent, R.; van Bokhoven, J.A.; Szlachetko, J.; Edebeli, J.; Paun, C.; Nachtegaal, M.; Safonova, O.V. Catalytically active and spectator Ce3+ in ceria-supported metal catalysts. Angew. Chem. Int. Ed. 2015, 54, 8728–8731. [Google Scholar] [CrossRef]
- Meunier, F.C. Relevance of IR Spectroscopy of Adsorbed CO for the Characterization of Heterogeneous Catalysts Containing Isolated Atoms. J. Phys. Chem. C 2021, 125, 21810–21823. [Google Scholar] [CrossRef]
- Gatla, S.; Aubert, D.; Flaud, V.; Grosjean, R.; Lunkenbein, T.; Mathon, O.; Pascarelli, S.; Kaper, H. Facile synthesis of high-surface area platinum-doped ceria for low temperature CO oxidation. Catal. Today 2019, 333, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Meunier, F.C.; Goguet, A.; Shekhtman, S.; Rooney, D.; Daly, H. A modified commercial DRIFTS cell for kinetically relevant operando studies of heterogeneous catalytic reactions. Appl. Catal. A Gen. 2008, 340, 196–202. [Google Scholar] [CrossRef]
- Scalbert, J.; Meunier, F.C.; Daniel, C.; Schuurman, Y. An operando DRIFTS investigation into the resistance against CO2 poisoning of a Rh/alumina catalyst during toluene hydrogenation. Phys. Chem. Chem. Phys. 2012, 14, 2159–2163. [Google Scholar] [CrossRef] [PubMed]
- Sirita, J.; Phanichphant, S.; Meunier, F.C. Quantitative analysis of adsorbate concentrations by diffuse reflectance FT-IR. Anal. Chem. 2007, 79, 3912–3918. [Google Scholar] [CrossRef] [Green Version]
- Gänzler, A.M.; Casapu, M.; Vernoux, P.; Loridant, S.; Cadete Santos Aires, F.J.; Epicier, T.; Betz, B.; Hoyer, R.; Grunwaldt, J.-D. Tuning the Structure of Platinum Particles on Ceria In Situ for Enhancing the Catalytic Performance of Exhaust Gas Catalysts. Angew. Chem. Int. Ed. 2017, 56, 13078. [Google Scholar] [CrossRef]
- Alayon, E.M.C.; Singh, J.; Nachtegaal, M.; Harfouche, M.; van Bokhoven, J.A. On highly active partially oxidized platinum in carbon monoxide oxidation over supported platinum catalysts. J. Catal. 2009, 263, 228–238. [Google Scholar] [CrossRef]
- Kunwar, D.; Zhou, S.; De La Riva, A.; Peterson, E.J.; Xiong, H.; Pereira-Hernández, X.I.; Purdy, S.C.; ter Veen, R.; Brongersma, H.H.; Miller, J.T.; et al. Stabilizing High Metal Loadings of Thermally Stable Platinum Single Atoms on an Industrial Catalyst Support. ACS Catal. 2019, 9, 3978–3990. [Google Scholar] [CrossRef]
- Kim, G.J.; Kwon, D.W.; Hong, S.C. Effect of Pt Particle Size and Valence State on the Performance of Pt/TiO2 Catalysts for CO Oxidation at Room Temperature. J. Phys. Chem. C 2016, 120, 17996–18004. [Google Scholar] [CrossRef]
- Bazin, P.; Saur, O.; Lavalley, J.C.; Daturi, M.; Blanchard, G. Effect of Pt Particle Size and Valence State on the Performance of Pt/TiO2 Catalysts for CO Oxidation at Room Temperature. Phys. Chem. Chem. Phys. 2005, 7, 187–194. [Google Scholar] [CrossRef]
- Happel, M.; Myslivecek, J.; Joha’nek, V.; Dvorak, F.; Stetsovych, O.; Lykhach, Y.; Matolin, V.; Libuda, J. Adsorption sites, metal-support interactions, and oxygen spillover identified by vibrational spectroscopy of adsorbed CO: A model study on Pt/ceria catalysts. J. Catal. 2012, 289, 118–126. [Google Scholar] [CrossRef]
- Ivanova, E.; Mihaylov, M.; Thibault-Starzyk, F.; Daturi, M.; Hadjiivanov, K. FTIR spectroscopy study of CO and NO adsorption and co-adsorption on Pt/TiO2. J. Mol. Catal. A Chem. 2007, 274, 179–184. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
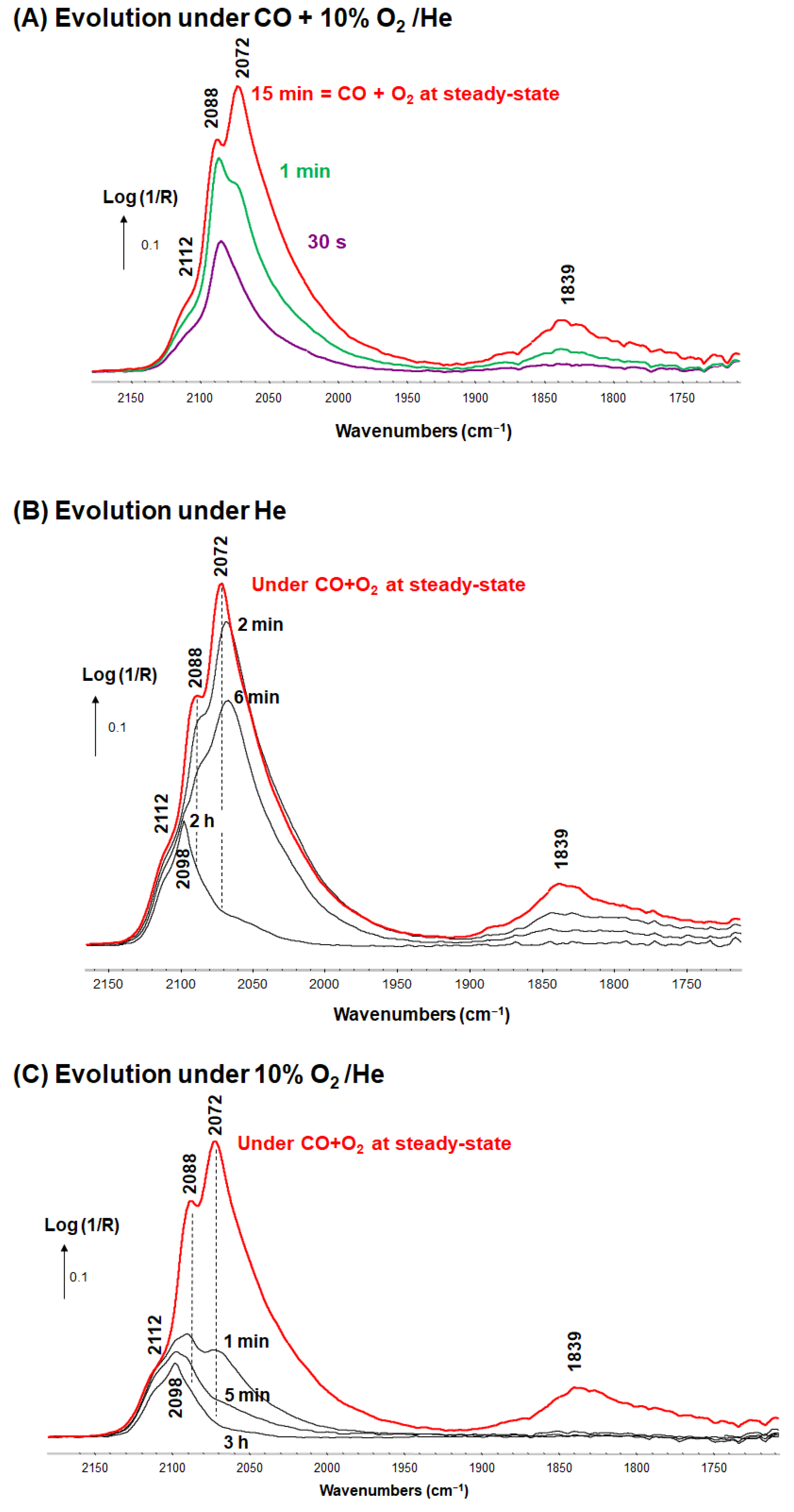{kind=link}
{kind=link}
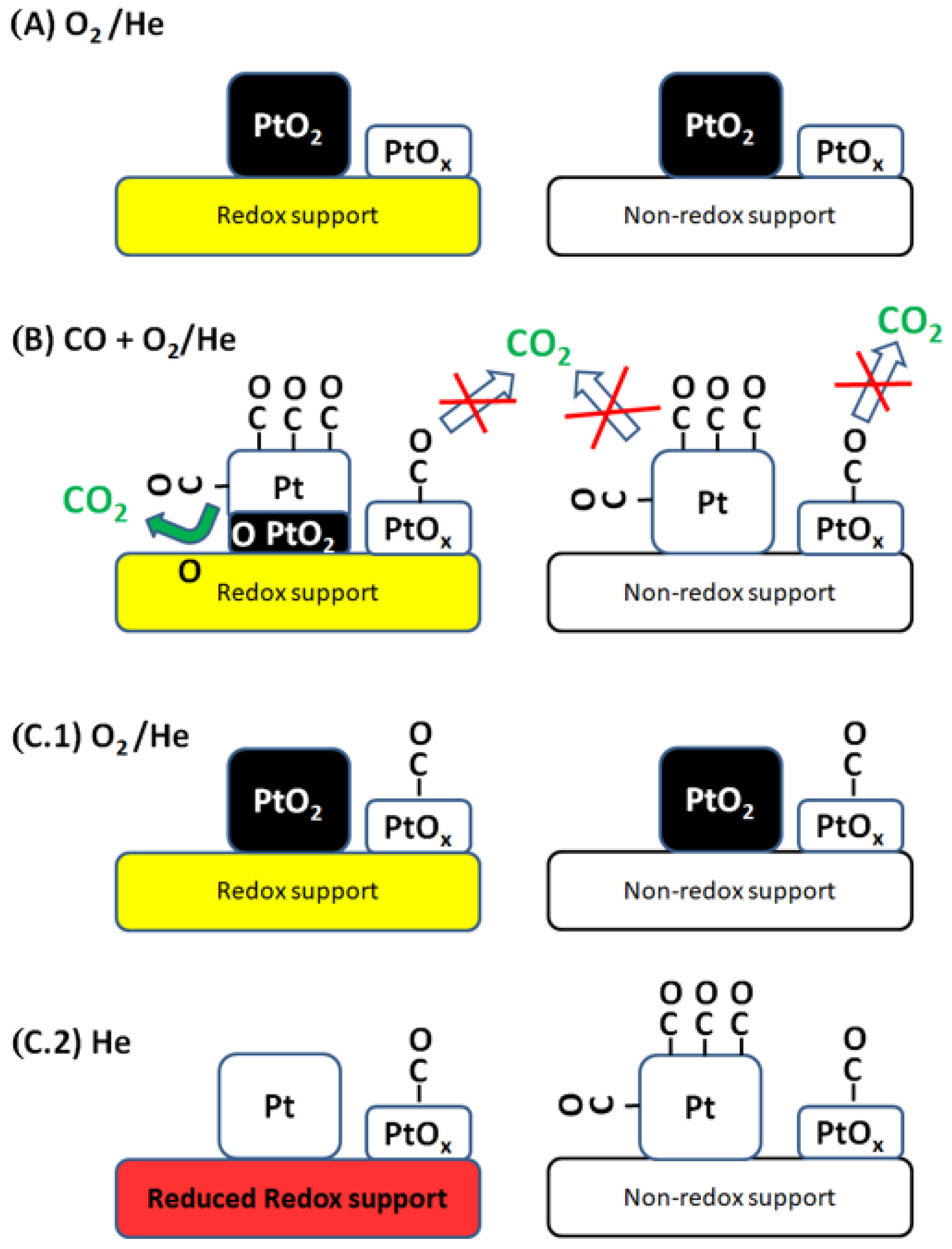{kind=link}
| Wavenumber (cm−1) | References | |
|---|---|---|
| - | PtO2: does not adsorb CO at RT | [42] |
| 2112–2098 | L-CO on PtOx | [8,9,27,38] |
| 2088 | L-CO on O-covered metallic Pt | [40,41] |
| 2075–2060 | L-CO on Pt0 | [8,10] |
| 1850–1830 | B-CO on Pt0 | [8,10] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meunier, F.C.; Elgayyar, T.; Dembélé, K.; Kaper, H. Stability of Pt-Adsorbed CO on Catalysts for Room Temperature-Oxidation of CO. Catalysts 2022, 12, 532. https://doi.org/10.3390/catal12050532
Meunier FC, Elgayyar T, Dembélé K, Kaper H. Stability of Pt-Adsorbed CO on Catalysts for Room Temperature-Oxidation of CO. Catalysts. 2022; 12(5):532. https://doi.org/10.3390/catal12050532
Chicago/Turabian StyleMeunier, Frédéric C., Taha Elgayyar, Kassiogé Dembélé, and Helena Kaper. 2022. "Stability of Pt-Adsorbed CO on Catalysts for Room Temperature-Oxidation of CO" Catalysts 12, no. 5: 532. https://doi.org/10.3390/catal12050532