
Gas-Phase Selective Oxidation of Methane into Methane Oxygenates


Abstract
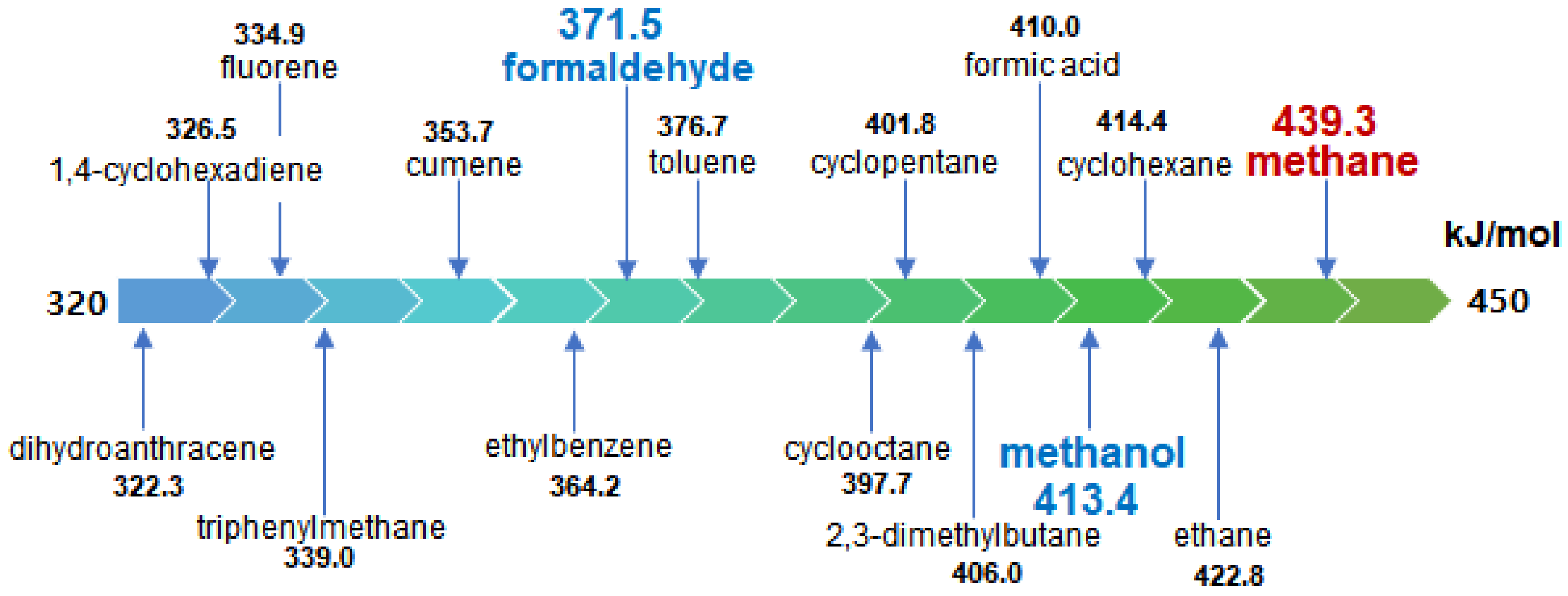
:1. Introduction
2. Catalytic Gas-Phase Partial Oxidation of Methane into Formaldehyde
2.1. Molybdenum-Based Catalyst
2.1.1. Mechanism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | BET Surface (m2/g) | Active Species Loading (wt%) | T (°C) | CH4 Conversion (%) | HCHO Selectivity (%) | CO Selectivity (%) | HCHO Yield (%) | GHSV (mLgcat−1×h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| SiO2 | 475 | / | 590 | 0.10 | 100 | 0 | 0.08 | 60,000 | [52] |
| WO3/SiO2 | 160 | 2.0 | 650 | 1.2 | 17 | 51.3 | 0.02 | 5640 | [53] |
| MoO3 | 3 | / | 600 | 1.0 | 85 | / | 0.85 | 1000-48,000 | [54] |
| MoO3/SiO2 | / | / | 600 | 8.2 | 35 | 17.0 | 2.9 | 2800 b | [55] |
| Li-MoOx/SiO2 | / | 1.0 | 650 | 4.8 | 65 | 0 | 3.1 | 5600 | [56] |
| Mo/KIT-6 | 559 | 4.6 | 675 | 7.3 | 13 | 72.9 | 0.9 | 36,000 | [57] |
| Mo-KIT-6 | 569 | 8.0 | 675 | 7.3 | 29 | 60.1 | 2.1 | 36,000 | [57] |
| eMoOx/SBA-15 | / | 20 | 600 | 1.9 | 70 | 28.0 | 1.4 | 33,000 | [58] |
| PMoV-mesoSiO2 | 526 | 3.4 | 640 | 5.9 | 52 | / | 3.1 | 36,200 | [59] |
| Mo/ZrO2 | 34.3 | 12 | 400 | 8.3 | 48 | 17.5 | 4.0 | 12,000 | [60] |
| Cu-MoOx | / | / | 700 | 1.6 | 62 | 28.0 | 1.0 | 84,000 | [61] |
| Mo-SBA-1 | 1271 | 9.9 | 680 | 8.2 | 20 | / | / | 15,600 | [62] |
| P-MoOx/SBA-15 | 382 | 5.0 | 675 | 5.8 | 90 | / | 5.2 | 35,840 | [63] |
| K2MoO4/SiO2 | / | 2.0 | 650 | 1.3 | 32 | 21.0 | 0.42 | 6000 | [64] |
| V2O5/SiO2 | / | / | 620 | 4.8 | 24 | 65.0 | 1.1 | 3500 | [65] |
| VOx/MCF-17 | 750 | 1.0 | 600 | 20 | 46 | 24.0 | 9.2 | 24,000 | [66] |
| SiO2@V2O5@Al2O3 | 14 | / | 600 | 22 | 58 | 27.0 | 12.8 | 24,000 | [67] |
| V/SBA-15 | 762 | 1.7 | 640 | 4.7 | 42 | / | 2.0 | 480,000 | [68] |
| VOx/α-Al2O3 | / | 0.4 | 450 | 9.0 | 60 | 18.0 | 5.4 | 10,000 | [69] |
| V/MCM-41 | 682 | 2.8 | 600 | 4.7 | 26 | / | 1.2 | 180,000 | [70] |
| V/MCM-48 | 878 | 2.8 | 600 | 4.0 | 26 | / | 1.0 | 180,000 | [70] |
| FePO4 | 22 | / | 500 | 0.51 | 39 | / | 0.2 | 36,000 | [71] |
| FePO4/MCM-41 a | 310 | 40 | 450 | 3.0 | 50 | / | 1.5 | 7200 | [32] |
| FeOx/SBA-15 | 601 | 0.05 | 650 | 5.0 | 37 | 39.0 | 1.9 | 72,000 | [33] |
| FeOx/SiO2 | 597 | / | 650 | 37 | 33 | 29.0 | 12.2 | 60,000 | [34] |
| CuOx/SBA-15 | 617 | 0.008 | 625 | 2.3 | 58 | 32.0 | 1.3 | 144,000 | [35] |
| B2O3/Al2O3 | / | 20 | 550 | 6.8 | 46 | 50.4 | 3.1 | 4650 | [36] |
| SbOx/SiO2 | 237 | 20 | 600 | 1.1 | 25 | 52.5 | 0.28 | 7840 | [72] |
| Co/SiO2 | 333 | 0.1 | 500 | / | 38 | / | / | 132,000 | [73] |
2.1.2. Support
2.1.3. Promoter
2.2. Vanadium-Based Catalyst
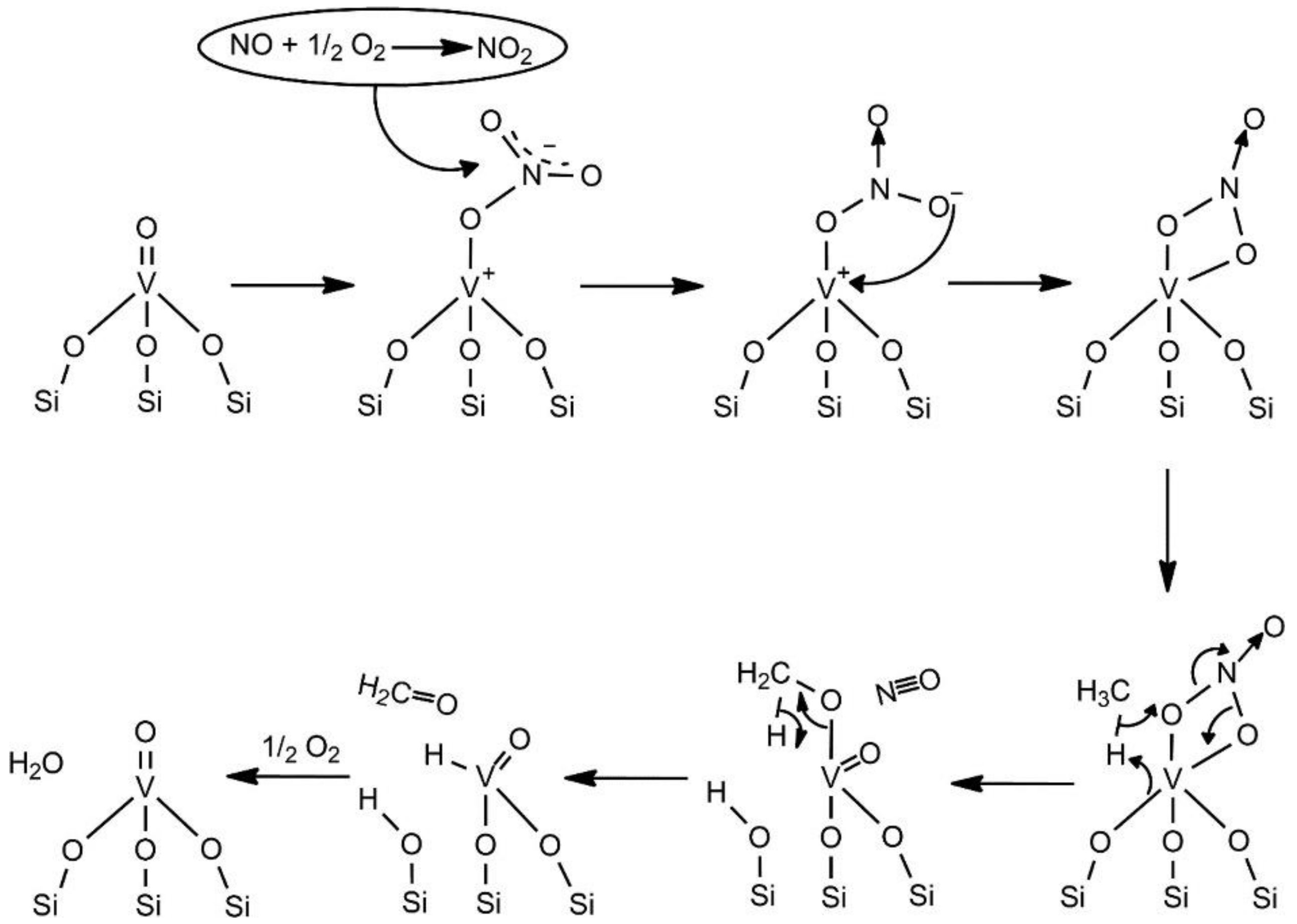
2.2.1. Mechanism
2.2.2. Support
2.2.3. Promoter
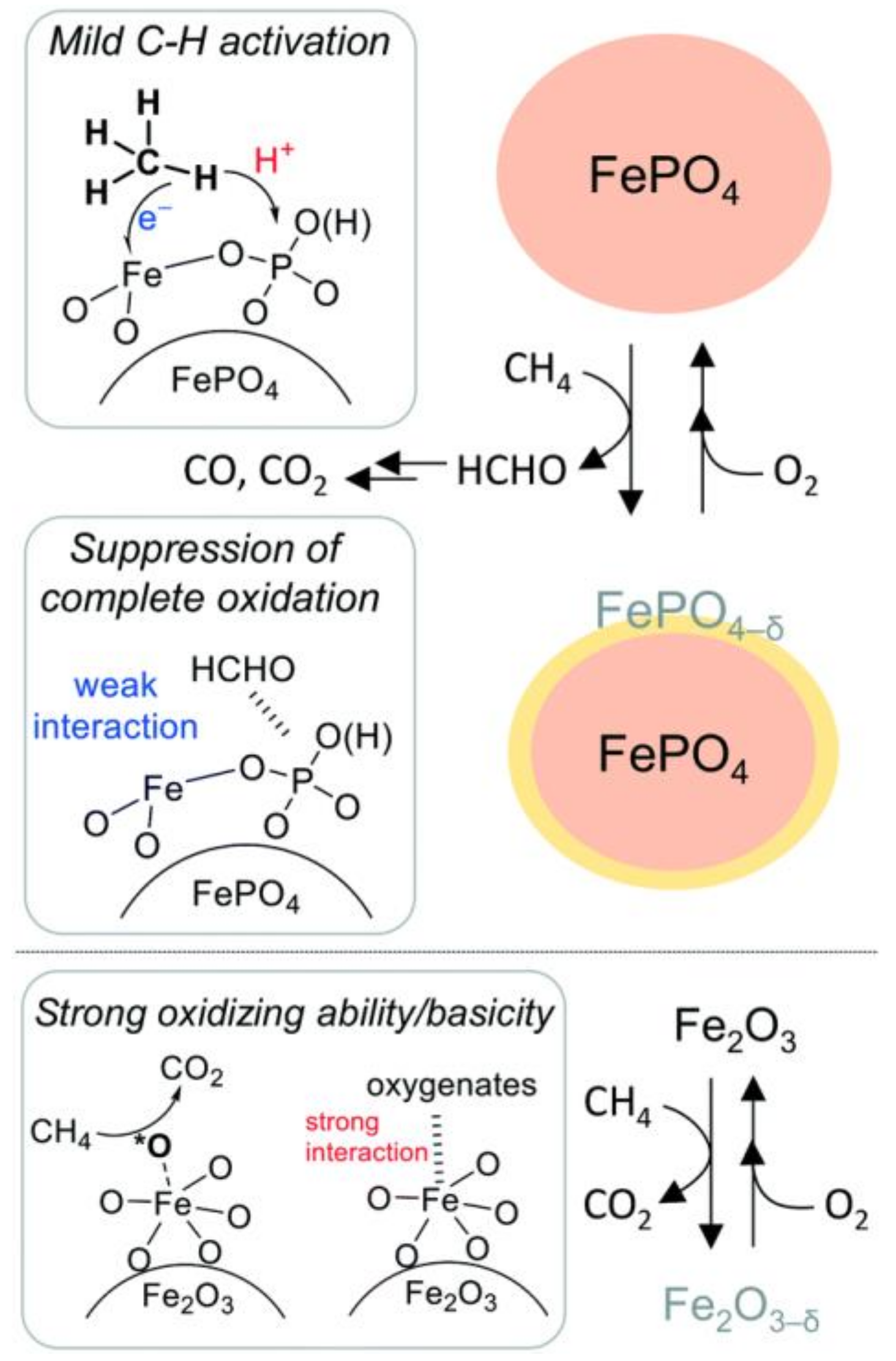
2.3. Iron-Based Catalyst
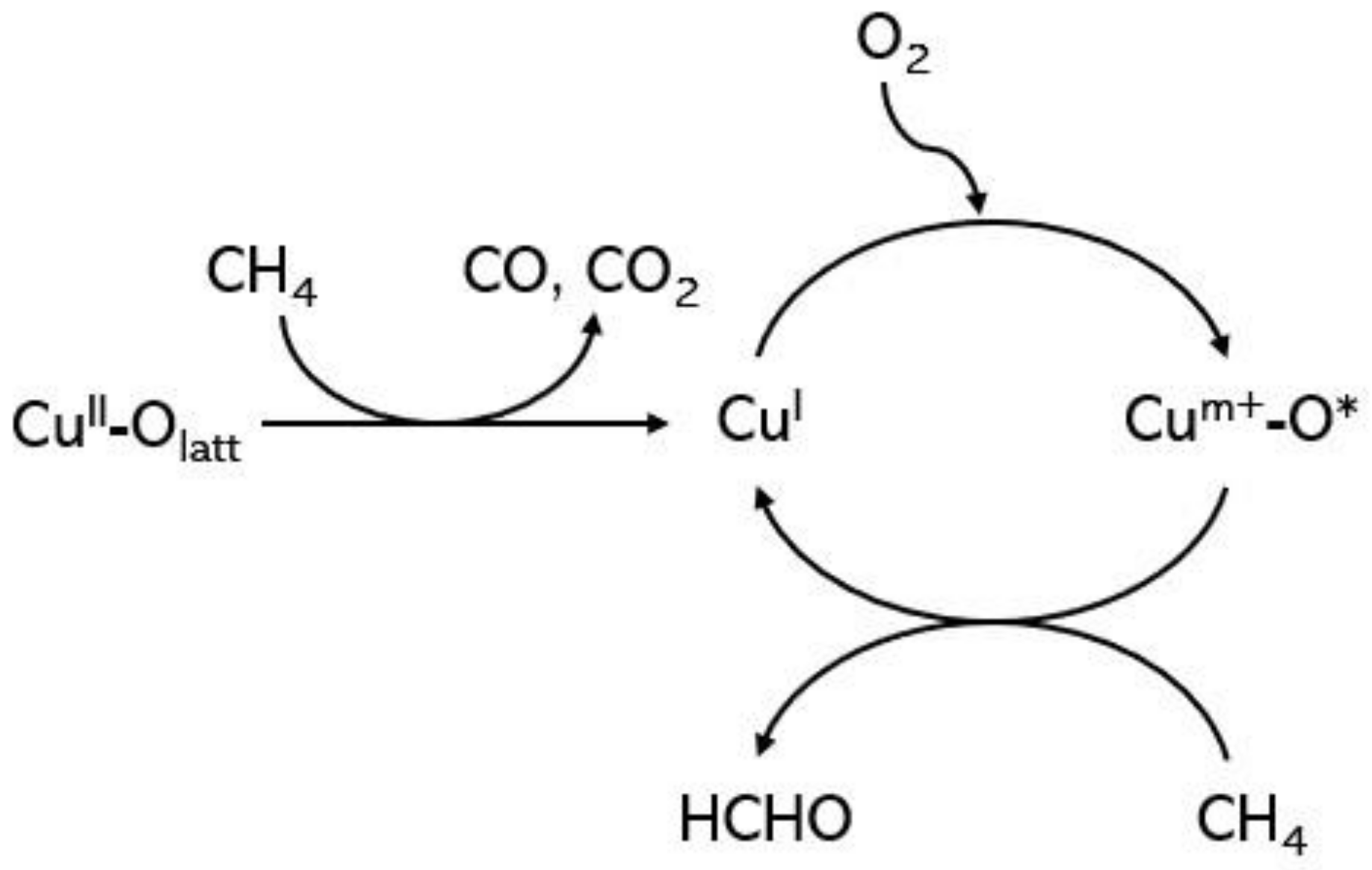
2.4. Copper-Based Catalyst
2.5. Other Catalysts
3. Catalytic Gas-Phase Partial Oxidation of Methane into Methanol
3.1. Molybdenum-Based Catalyst
| Catalysts | T (°C) | Oxidant | CH3OH Productivity (mmol/molmetal/h) | CH3OH Selectivity (%) | Ref. |
|---|---|---|---|---|---|
| Mo/SiO2 | 600 | N2O + H2O | 0.36 a | 60 | [151] |
| MoOx/La-Co-O | 420 | O2 | / | 60 | [149] |
| V2O5/SiO2 | 460 | O2 | 0.55 a | 34 | [152] |
| Cu/CHA | 300 | H2O + O2 | 542 | 91 | [153] |
| Cu/CHA | 270 | H2O + O2 | 26 | 53 | [154] |
| Cu-H-MOR | 400 | H2O + O2 | 143 | 99 | [155] |
| Cu-H-MOR | 350 | H2O | 29 | 100 | [155] |
| Cu-Fe/Al2O3 | 450 | H2O + O2 | 1.3 a | / | [156] |
| Fe-Cu-BEA | 270 | N2O + H2O | 0.26 a | 72 | [157] |
| Rh-dB-ZSM-5 | 150 | O2 + H2O + CO | 0.80 a | 44 | [158] |
| NiFeO/CZ | 250 | H2O | 3.2 × 10−3 a | 1.24 | [159] |
| Cu-SSZ-39 | 325 | N2O + H2O | 1044 | 34 | [160] |
| Cu-MOR | 350 | H2O | 332 | / | [161] |
| ZnO/Cu2O/Cu | 177 | H2O + O2 | 7.02 × 1017 b | 87.5 | [162] |
| Cu-CHA | 300 | H2O + O2 | 0.68 a | 45 | [163] |
3.2. Vanadium-Based Catalyst
3.3. Iron-Based Catalyst
3.4. Copper-Based Catalyst
4. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Olah, G.A. Beyond Oil and Gas: The Methanol Economy. Angew. Chem. Int. Ed. 2005, 44, 2636–2639. [Google Scholar] [CrossRef] [PubMed]
- Tiwary, R.K. Environmental Impact of Coal Mining on Water Regime and Its Management. Water Air Soil Pollut. 2001, 132, 185–199. [Google Scholar] [CrossRef]
- NaturalGas.Org. Available online: http://naturalgas.org/overview/background/ (accessed on 14 February 2022).
- BP Statistical Review of World Energy Globally Consistent Data on World Energy Markets and Authoritative Publications in the Field of Energy. BP Energy Outlook 2021, 70, 8–20.
- Schwach, P.; Pan, X.; Bao, X. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef] [PubMed]
- Kerr, R.A. Natural Gas From Shale Bursts Onto the Scene. Science 2010, 328, 1624–1626. [Google Scholar] [CrossRef]
- Dry, M.E. The Fischer-Tropsch Process: 1950–2000. Catal. Today 2002, 71, 227–241. [Google Scholar] [CrossRef]
- Schulz, H. Short History and Present Trends of Fischer–Tropsch Synthesis. Appl. Catal. A Gen. 1999, 186, 3–12. [Google Scholar] [CrossRef]
- Cheng, K.; Kang, J.; King, D.L.; Subramanian, V.; Zhou, C.; Zhang, Q.; Wang, Y. Advances in Catalysis for Syngas Conversion to Hydrocarbons; Song, C.B.T.-A., Ed.; Academic Press: Cambridge, MA, USA, 2017; Volume 60, pp. 125–208. [Google Scholar]
- Holmen, A. Direct Conversion of Methane to Fuels and Chemicals. Catal. Today 2009, 142, 2–8. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, B.; Zhang, Q.; Deng, W.; Wang, Y.; Yang, Y. Recent Advances in Heterogeneous Selective Oxidation Catalysis for Sustainable Chemistry. Chem. Soc. Rev. 2014, 43, 3480. [Google Scholar] [CrossRef]
- Otsuka, K.; Wang, Y. Direct Conversion of Methane into Oxygenates. Appl. Catal. A Gen. 2001, 222, 145–161. [Google Scholar] [CrossRef]
- Spivey, J.J.; Hutchings, G. Catalytic Aromatization of Methane. Chem. Soc. Rev. 2014, 43, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Kondratenko, E.v.; Peppel, T.; Seeburg, D.; Kondratenko, V.A.; Kalevaru, N.; Martin, A.; Wohlrab, S. Methane Conversion into Different Hydrocarbons or Oxygenates: Current Status and Future Perspectives in Catalyst Development and Reactor Operation. Catal. Sci. Technol. 2017, 7, 366–381. [Google Scholar] [CrossRef]
- Tang, P.; Zhu, Q.; Wu, Z.; Ma, D. Methane Activation: The Past and Future. Energy Environ. Sci. 2014, 7, 2580–2591. [Google Scholar] [CrossRef]
- Alvarez-Galvan, M.C.; Mota, N.; Ojeda, M.; Rojas, S.; Navarro, R.M.; Fierro, J.L.G. Direct Methane Conversion Routes to Chemicals and Fuels. Catal. Today 2011, 171, 15–23. [Google Scholar] [CrossRef]
- Zavyalova, U.; Holena, M.; Schlögl, R.; Baerns, M. Statistical Analysis of Past Catalytic Data on Oxidative Methane Coupling for New Insights into the Composition of High-Performance Catalysts. ChemCatChem 2011, 3, 1935–1947. [Google Scholar] [CrossRef]
- Gunsalus, N.J.; Koppaka, A.; Park, S.H.; Bischof, S.M.; Hashiguchi, B.G.; Periana, R.A. Homogeneous Functionalization of Methane. Chem. Rev. 2017, 117, 8521–8573. [Google Scholar] [CrossRef]
- Ravi, M.; Ranocchiari, M.; van Bokhoven, J.A. The Direct Catalytic Oxidation of Methane to Methanol-A Critical Assessment. Angew. Chem. Int. Ed. 2017, 56, 16464–16483. [Google Scholar] [CrossRef]
- De Vekki, A.v.; Marakaev, S.T. Catalytic Partial Oxidation of Methane to Formaldehyde. Russ. J. Appl. Chem. 2009, 82, 521–536. [Google Scholar] [CrossRef]
- Olivos-Suarez, A.I.; Szécsényi, À.; Hensen, E.J.M.; Ruiz-Martinez, J.; Pidko, E.A.; Gascon, J. Strategies for the Direct Catalytic Valorization of Methane Using Heterogeneous Catalysis: Challenges and Opportunities. ACS Catal. 2016, 6, 2965–2981. [Google Scholar] [CrossRef]
- Sirajuddin, S.; Rosenzweig, A.C. Enzymatic Oxidation of Methane. Biochemistry 2015, 54, 2283–2294. [Google Scholar] [CrossRef] [Green Version]
- Gol’Dshleger, N.F.; Es’ Kova, V.v.; Shilov, A.E.; Shteinman, A.A. Reactions of Alkanes in Solutions of Platinum Chloride Complexes. Zhurnal Fiz. Khimi 1972, 46, 1353–1354. [Google Scholar]
- Periana, R.A.; Taube, D.J.; Evitt, E.R.; Löffler, D.G.; Wentrcek, P.R.; Voss, G.; Masuda, T. A Mercury-Catalyzed, High-Yield System for the Oxidation of Methane to Methanol. Science 1993, 259, 340–343. [Google Scholar] [CrossRef]
- Periana, R.A.; Taube, D.J.; Gamble, S.; Taube, H.; Satoh, T.; Fujii, H. Platinum Catalysts for the High-Yield Oxidation of Methane to a Methanol Derivative. Science 1998, 280, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Zerella, M.; Bell, A.T. A High-Yield, Liquid-Phase Approach for the Partial Oxidation of Methane to Methanol Using SO3 as the Oxidant. Adv. Synth. Catal. 2005, 347, 1203–1206. [Google Scholar] [CrossRef]
- Newton, M.A.; Knorpp, A.J.; Sushkevich, V.L.; Palagin, D.; van Bokhoven, J.A. Active Sites and Mechanisms in the Direct Conversion of Methane to Methanol Using Cu in Zeolitic Hosts: A Critical Examination. Chem. Soc. Rev. 2020, 49, 1449–1486. [Google Scholar] [CrossRef]
- Sen, A.; Benvenuto, M.A.; Lin, M.; Hutson, A.C.; Basickes, N. Activation of Methane and Ethane and Their Selective Oxidation to the Alcohols in Protic Media. J. Am. Chem. Soc. 1994, 116, 998–1003. [Google Scholar] [CrossRef]
- Hashiguchi, B.G.; Bischof, S.M.; Konnick, M.M.; Periana, R.A. Designing Catalysts for Functionalization of Unactivated C–H Bonds Based on the CH Activation Reaction. Acc. Chem. Res. 2012, 45, 885–898. [Google Scholar] [CrossRef]
- Ohyama, J.; Hirayama, A.; Kondou, N.; Yoshida, H.; Machida, M.; Nishimura, S.; Hirai, K.; Miyazato, I.; Takahashi, K. Data Science Assisted Investigation of Catalytically Active Copper Hydrate in Zeolites for Direct Oxidation of Methane to Methanol Using H2O2. Sci. Rep. 2021, 11, 2067. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Y.; Guan, N.; Li, L. Methane Activation and Utilization: Current Status and Future Challenges. Energy Technol. 2020, 8, 1900826. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Tang, Q.; Guo, Q.; Zhang, Q.; Wan, H. MCM-41-Supported Iron Phosphate Catalyst for Partial Oxidation of Methane to Oxygenates with Oxygen and Nitrous Oxide. J. Catal. 2003, 217, 457–467. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, Y.; An, D.; Wang, Y. Catalytic Behavior and Kinetic Features of FeOx/SBA-15 Catalyst for Selective Oxidation of Methane by Oxygen. Appl. Catal. A Gen. 2009, 356, 103–111. [Google Scholar] [CrossRef]
- Arena, F.; Gatti, G.; Martra, G.; Coluccia, S.; Stievano, L.; Spadaro, L.; Famulari, P.; Parmaliana, A. Structure and Reactivity in the Selective Oxidation of Methane to Formaldehyde of Low-Loaded FeOx/SiO2 Catalysts. J. Catal. 2005, 231, 365–380. [Google Scholar] [CrossRef]
- Li, Y.; An, D.; Zhang, Q.; Wang, Y. Copper-Catalyzed Selective Oxidation of Methane by Oxygen: Studies on Catalytic Behavior and Functioning Mechanism of CuOx/SBA-15. J. Phys. Chem. C 2008, 112, 13700–13708. [Google Scholar] [CrossRef]
- Tian, J.; Tan, J.; Zhang, Z.; Han, P.; Yin, M.; Wan, S.; Lin, J.; Wang, S.; Wang, Y. Direct Conversion of Methane to Formaldehyde and CO on B2O3 Catalysts. Nat. Commun. 2020, 11, 5693. [Google Scholar] [CrossRef]
- Moro-Oka, Y.; Ueda, W. Multicomponent Bismuth Molybdate Catalyst: A Highly Functionalized Catalyst System for the Selective Oxidation of Olefin. Adv. Catal. 1994, 40, 233–273. [Google Scholar] [CrossRef]
- Mars, P.; van Krevelen, D.W. Oxidations Carried out by Means of Vanadium Oxide Catalysts. Chem. Eng. Sci. 1954, 3, 41–59. [Google Scholar] [CrossRef]
- Schlögl, R.; Knop-Gericke, A.; Hävecker, M.; Wild, U.; Frickel, D.; Ressler, T.; Jentoft, R.E.; Wienold, J.; Mestl, G.; Blume, A.; et al. In Situ Analysis of Metal-Oxide Systems Used for Selective Oxidation Catalysis: How Essential Is Chemical Complexity? Top. Catal. 2001, 15, 219–228. [Google Scholar] [CrossRef]
- Grasselli, R.K. Fundamental Principles of Selective Heterogeneous Oxidation Catalysis. Top. Catal. 2002, 21, 79–88. [Google Scholar] [CrossRef]
- Grasselli, R.K. Advances and Future Trends in Selective Oxidation and Ammoxidation Catalysis. Catal. Today 1999, 49, 141–153. [Google Scholar] [CrossRef]
- Haber, J.; Turek, W. Kinetic Studies as a Method to Differentiate between Oxygen Species Involved in the Oxidation of Propene. J. Catal. 2000, 190, 320–326. [Google Scholar] [CrossRef]
- Ohler, N.; Bell, A.T. Study of the Elementary Processes Involved in the Selective Oxidation of Methane over MoOx/SiO2. J. Phys. Chem. B 2006, 110, 2700–2709. [Google Scholar] [CrossRef] [PubMed]
- Ohler, N.; Bell, A.T. Selective Oxidation of Methane over MoOx/SiO2: Isolation of the Kinetics of Reactions Occurring in the Gas Phase and on the Surfaces of SiO2 and MoOx. J. Catal. 2005, 231, 115–130. [Google Scholar] [CrossRef]
- Baldwin, T.R.; Burch, R.; Squire, G.D.; Tsang, S.C. Influence of Homogeneous Gas Phase Reactions in the Partial Oxidation of Methane to Methanol and Formaldehyde in the Presence of Oxide Catalysts. Appl. Catal. 1991, 74, 137–152. [Google Scholar] [CrossRef]
- Ohler, N.; Bell, A.T. A Study of the Redox Properties of MoOx/SiO2. J. Phys. Chem. B 2005, 109, 23419–23429. [Google Scholar] [CrossRef] [PubMed]
- Chempath, S.; Bell, A.T. A DFT Study of the Mechanism and Kinetics of Methane Oxidation to Formaldehyde Occurring on Silica-Supported Molybdena. J. Catal. 2007, 247, 119–126. [Google Scholar] [CrossRef]
- Lee, E.L.; Wachs, I.E. In Situ Raman Spectroscopy of SiO2-Supported Transition Metal Oxide Catalysts: An Isotopic 18O-16O Exchange Study. J. Phys. Chem. C 2008, 112, 6487–6498. [Google Scholar] [CrossRef]
- Lee, E.L.; Wachs, I.E. In Situ Spectroscopic Investigation of the Molecular and Electronic Structures of SiO2 Supported Surface Metal Oxides. J. Phys. Chem. C 2007, 111, 14410–14425. [Google Scholar] [CrossRef]
- Handzlik, J.; Ogonowski, J. Structure of Isolated Molybdenum(VI) and Molybdenum(IV) Oxide Species on Silica: Periodic and Cluster DFT Studies. J. Phys. Chem. C 2012, 116, 5571–5584. [Google Scholar] [CrossRef]
- Tian, H.; Roberts, C.A.; Wachs, I.E. Molecular Structural Determination of Molybdena in Different Environments: Aqueous Solutions, Bulk Mixed Oxides, and Supported MoO3 Catalysts. J. Phys. Chem. C 2010, 114, 14110–14120. [Google Scholar] [CrossRef]
- Shimura, K.; Fujitani, T. Effects of Promoters on the Performance of a VO/SiO2 Catalyst for the Oxidation of Methane to Formaldehyde. Appl. Catal. A Gen. 2019, 577, 44–51. [Google Scholar] [CrossRef]
- Erdőhelyi, A.; Németh, R.; Hancz, A.; Oszkó, A. Partial Oxidation of Methane on Potassium-Promoted WO3/SiO2 and on K2WO4/SiO2 Catalysts. Appl. Catal. A Gen. 2001, 211, 109–121. [Google Scholar] [CrossRef]
- Hargreaves, J.S.J.; Hutchings, G.J.; Joyner, R.W. Control of Product Selectivity in the Partial Oxidation of Methane. Nature 1990, 348, 428–429. [Google Scholar] [CrossRef]
- Aoki, K.; Ohmae, M.; Nanba, T.; Takeishi, K.; Azuma, N.; Ueno, A.; Ohfune, H.; Hayashi, H.; Udagawa, Y. Direct Conversion of Methane into Methanol over MoO3/SiO2 Catalyst in an Excess Amount of Water Vapor. Catal. Today 1998, 45, 29–33. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, T.Y.; Song, C.K.; Lee, K.R.; Bae, S.; Park, H.; Yun, D.; Yun, Y.S.; Nam, I.; Park, J.; et al. Redox-Driven Restructuring of Lithium Molybdenum Oxide Nanoclusters Boosts the Selective Oxidation of Methane. Nano Energy 2021, 82, 105704. [Google Scholar] [CrossRef]
- Chen, P.; Xie, Z.; Zhao, Z.; Li, J.; Liu, B.; Liu, B.; Fan, X.; Kong, L.; Xiao, X. Study on the Selective Oxidation of Methane over Highly Dispersed Molybdenum-Incorporated KIT-6 Catalysts. Catal. Sci. Technol. 2021, 11, 4083–4097. [Google Scholar] [CrossRef]
- Lou, Y.; Tang, Q.; Wang, H.; Chia, B.; Wang, Y.; Yang, Y. Selective Oxidation of Methane to Formaldehyde by Oxygen over SBA-15-Supported Molybdenum Oxides. Appl. Catal. A Gen. 2008, 350, 118–125. [Google Scholar] [CrossRef]
- Pei, S.; Yue, B.; Qian, L.; Yan, S.; Cheng, J.; Zhou, Y.; Xie, S.; He, H. Preparation and Characterization of P–Mo–V Mixed Oxide-Incorporating Mesoporous Silica Catalysts for Selective Oxidation of Methane to Formaldehyde. Appl. Catal. A Gen. 2007, 329, 148–155. [Google Scholar] [CrossRef]
- Zhang, X.; He, D.H.; Zhang, Q.J.; Ye, Q.; Xu, B.Q.; Zhu, Q.M. Selective Oxidation of Methane to Formaldehyde over Mo/ZrO2 Catalysts. Appl. Catal. A Gen. 2003, 249, 107–117. [Google Scholar] [CrossRef]
- Akiyama, T.; Sei, R.; Takenaka, S. Partial Oxidation of Methane to Formaldehyde over Copper–Molybdenum Complex Oxide Catalysts. Catal. Sci. Technol. 2021, 11, 5273–5281. [Google Scholar] [CrossRef]
- Dai, L.X.; Teng, Y.H.; Tabata, K.; Suzuki, E.; Tatsumi, T. Catalytic Application of Mo-Incorporated SBA-1 Mesoporous Molecular Sieves to Partial Oxidation of Methane. Microporous Mesoporous Mater. 2001, 44–45, 573–580. [Google Scholar] [CrossRef]
- Yang, W.; Wang, X.; Guo, Q.; Zhang, Q.; Wang, Y. Superior Catalytic Performance of Phosphorus-Modified Molybdenum Oxide Clusters Encapsulated inside SBA-15 in the Partial Oxidation of Methane. New J. Chem. 2003, 27, 1301. [Google Scholar] [CrossRef] [Green Version]
- Erdohelyi, A.; Fodor, K.; Solymosi, F. Partial Oxidation of Methane on Supported Potassium Molybdate. J. Catal. 1997, 166, 244–253. [Google Scholar] [CrossRef]
- Kastanas, G.N.; Tsigdinos, G.A.; Schwank, J. Selective Oxidation of Methane over Vycor Glass, Quartz Glass and Various Silica, Magnesia and Alumina Surfaces. Appl. Catal. 1988, 44, 33–51. [Google Scholar] [CrossRef]
- Yang, E.; Lee, J.G.; Park, E.D.; An, K. Methane Oxidation to Formaldehyde over Vanadium Oxide Supported on Various Mesoporous Silicas. Korean J. Chem. Eng. 2021, 38, 1224–1230. [Google Scholar] [CrossRef]
- Yang, E.; Lee, J.G.; Kim, D.H.; Jung, Y.S.; Kwak, J.H.; Park, E.D.; An, K. SiO2@V2O5@Al2O3 Core–Shell Catalysts with High Activity and Stability for Methane Oxidation to Formaldehyde. J. Catal. 2018, 368, 134–144. [Google Scholar] [CrossRef]
- Kunkel, B.; Wohlrab, S. Enhancement and Limits of the Selective Oxidation of Methane to Formaldehyde over V-SBA-15: Influence of Water Cofeed and Product Decomposition. Catal. Commun. 2021, 155, 106317. [Google Scholar] [CrossRef]
- Volpe, M.A. Partial Oxidation of Methane over VOx/α-Al2O3 Catalysts. Appl. Catal. A Gen. 2001, 210, 355–361. [Google Scholar] [CrossRef]
- Berndt, H.; Martin, A.; Brückner, A.; Schreier, E.; Müller, D.; Kosslick, H.; Wolf, G.U.; Lücke, B. Structure and Catalytic Properties of VOx/MCM Materials for the Partial Oxidation of Methane to Formaldehyde. J. Catal. 2000, 191, 384–400. [Google Scholar] [CrossRef]
- Matsuda, A.; Tateno, H.; Kamata, K.; Hara, M. Iron Phosphate Nanoparticle Catalyst for Direct Oxidation of Methane into Formaldehyde: Effect of Surface Redox and Acid–Base Properties. Catal. Sci. Technol. 2021, 11, 6987–6998. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, K.; Feng, Z.; Ying, P.; Li, C. Studies on the SbOx Species of SbOx/SiO2 Catalysts for Methane-Selective Oxidation to Formaldehyde. Appl. Catal. A Gen. 2006, 305, 110–119. [Google Scholar] [CrossRef]
- Ohyama, J.; Abe, D.; Hirayama, A.; Iwai, H.; Tsuchimura, Y.; Sakamoto, K.; Irikura, M.; Nakamura, Y.; Yoshida, H.; Machida, M.; et al. Selective Oxidation of Methane to Formaldehyde over a Silica-Supported Cobalt Single-Atom Catalyst. J. Phys. Chem. C 2022, 126, 1785–1792. [Google Scholar] [CrossRef]
- Foster, N.R. Direct Catalytic Oxidation of Methane to Methanol—A Review. Appl. Catal. 1985, 19, 1–11. [Google Scholar] [CrossRef]
- Banares, M.A.; Fierro, J.L.G.; Moffat, J.B. The Partial Oxidation of Methane on MoO3/SiO2 Catalysts: Influence of the Molybdenum Content and Type of Oxidant. J. Catal. 1993, 142, 406–417. [Google Scholar] [CrossRef]
- Spencer, N. Partial Oxidation of Methane to Formaldehyde by Means of Molecular Oxygen. J. Catal. 1988, 109, 187–197. [Google Scholar] [CrossRef]
- Barbaux, Y.; Elamrani, A.R.; Payen, E.; Gengembre, L.; Bonnelle, J.P.; Grzybowska, B. Silica Supported Molybdena Catalysts. Appl. Catal. 1988, 44, 117–132. [Google Scholar] [CrossRef]
- Liu, H.F.; Liu, R.S.; Liew, K.Y.; Johnson, R.E.; Lunsford, J.H. Partial Oxidation of Methane by Nitrous Oxide over Molybdenum on Silica. J. Am. Chem. Soc. 1984, 106, 4117–4121. [Google Scholar] [CrossRef]
- Plyuto, Y.V.; Babich, I.V.; Plyuto, I.V.; van Langeveld, A.D.; Moulijn, J.A. XPS Studies of MoO3/Al2O3 and MoO3/SiO2 Systems. Appl. Surf. Sci. 1997, 119, 11–18. [Google Scholar] [CrossRef]
- Thomas, D.J.; Willi, R.; Baiker, A. Partial Oxidation of Methane: The Role of Surface Reactions. Ind. Eng. Chem. Res. 1992, 31, 2272–2278. [Google Scholar] [CrossRef]
- Liu, Y.M.; Cao, Y.; Yi, N.; Feng, W.L.; Dai, W.L.; Yan, S.R.; He, H.Y.; Fan, K.N. Vanadium Oxide Supported on Mesoporous SBA-15 as Highly Selective Catalysts in the Oxidative Dehydrogenation of Propane. J. Catal. 2004, 224, 417–428. [Google Scholar] [CrossRef]
- Nieminen, V.; Kumar, N.; Salmi, T.; Murzin, D.Y. N-Butane Isomerization over Pt–H–MCM-41. Catal. Commun. 2004, 5, 15–19. [Google Scholar] [CrossRef]
- Solsona, B.; Blasco, T.; López Nieto, J.M.; Peña, M.L.; Rey, F.; Vidal-Moya, A. Vanadium Oxide Supported on Mesoporous MCM-41 as Selective Catalysts in the Oxidative Dehydrogenation of Alkanes. J. Catal. 2001, 203, 443–452. [Google Scholar] [CrossRef]
- Fornés, V.; López, C.; López, H.H.; Martínez, A. Catalytic Performance of Mesoporous VOx/SBA-15 Catalysts for the Partial Oxidation of Methane to Formaldehyde. Appl. Catal. A Gen. 2003, 249, 345–354. [Google Scholar] [CrossRef]
- Chen, K.; Xie, S.; Bell, A.T.; Iglesia, E. Alkali Effects on Molybdenum Oxide Catalysts for the Oxidative Dehydrogenation of Propane. J. Catal. 2000, 195, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Liu, J.; Duan, A.; Xu, C.; Kobayashi, T.; Wachs, I.E. Effects of Alkali Metal Cations on the Structures, Physico-Chemical Properties and Catalytic Behaviors of Silica-Supported Vanadium Oxide Catalysts for the Selective Oxidation of Ethane and the Complete Oxidation of Diesel Soot. Top. Catal. 2006, 38, 309–325. [Google Scholar] [CrossRef]
- Ivars, F.; Solsona, B.; Botella, P.; Soriano, M.D.; López Nieto, J.M. Selective Oxidation of Propane over Alkali-Doped Mo-V-Sb-O Catalysts. Catal. Today 2009, 141, 294–299. [Google Scholar] [CrossRef]
- Grant, J.T.; Carrero, C.A.; Love, A.M.; Verel, R.; Hermans, I. Enhanced Two-Dimensional Dispersion of Group V Metal Oxides on Silica. ACS Catal. 2015, 5, 5787–5793. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.; Kobayashi, T. Reaction Pathways for the Oxygenates Formation from Propane and Oxygen over Potassium-modified Fe/SiO2 Catalysts. Catal. Lett. 1998, 55, 33–38. [Google Scholar] [CrossRef]
- Wallis, P.; Schönborn, E.; Kalevaru, V.N.; Martin, A.; Wohlrab, S. Enhanced Formaldehyde Selectivity in Catalytic Methane Oxidation by Vanadia on Ti-Doped SBA-15. RSC Adv. 2015, 5, 69509–69513. [Google Scholar] [CrossRef]
- Grzybowska, B.; Mekšs, P.; Grabowski, R.; Wcisto, K.; Barbaux, Y.; Gengembre, L. Effect of Potassium Addition to V2O5/TiO2 and MoO3/TiO2 Catalysts on Their Physicochemical and Catalytic Properties in Oxidative Dehydrogenation of Propane. In New Developments in Selective Oxidation II; Corberán, V.C., Bellón, S.V.B.T.-S., Eds.; Elsevier: Amsterdam, The Netherlands, 1994; Volume 82, pp. 151–158. [Google Scholar]
- Grzybowska-Świerkosz, B. Effect of Additives on the Physicochemical and Catalytic Properties of Oxide Catalysts in Selective Oxidation Reactions. Top. Catal. 2002, 21, 35–46. [Google Scholar] [CrossRef]
- Deo, G.; Wachs, I.E. Reactivity of Supported Vanadium Oxide Catalysts: The Partial Oxidation of Methanol. J. Catal. 1994, 146, 323–334. [Google Scholar] [CrossRef]
- Liu, Q.; Li, J.; Zhao, Z.; Gao, M.; Kong, L.; Liu, J.; Wei, Y. Synthesis, Characterization, and Catalytic Performances of Potassium-Modified Molybdenum-Incorporated KIT-6 Mesoporous Silica Catalysts for the Selective Oxidation of Propane to Acrolein. J. Catal. 2016, 344, 38–52. [Google Scholar] [CrossRef]
- Verbruggen, N.F.D.; von Hippel, L.M.J.; Mestl, G.; Lengeler, B.; Knözinger, H. Structure of K-Doped Molybdena-on-Silica Catalysts As Studied by X-ray Absorption and Raman Spectroscopy. Langmuir 2002, 10, 3073–3080. [Google Scholar] [CrossRef]
- Liu, J.; Yu, L.; Zhao, Z.; Chen, Y.; Zhu, P.; Wang, C.; Luo, Y.; Xu, C.; Duan, A.; Jiang, G. Potassium-Modified Molybdenum-Containing SBA-15 Catalysts for Highly Efficient Production of Acetaldehyde and Ethylene by the Selective Oxidation of Ethane. J. Catal. 2012, 285, 134–144. [Google Scholar] [CrossRef]
- Millner, T.; Neugebauer, J. Volatility of the Oxides of Tungsten and Molybdenum in the Presence of Water Vapour. Nature 1949, 163, 601–602. [Google Scholar] [CrossRef]
- Blasco, T.; Nieto, J.M.L. Oxidative Dyhydrogenation of Short Chain Alkanes on Supported Vanadium Oxide Catalysts. Appl. Catal. A Gen. 1997, 157, 117–142. [Google Scholar] [CrossRef]
- Faraldos, M.; Bañares, M.A.; Anderson, J.A.; Hu, H.; Wachs, I.E.; Fierro, J.L.G. Comparison of Silica-Supported MoO3 and V2O5 Catalysts in the Selective Partial Oxidation of Methane. J. Catal. 1996, 160, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Jehng, J.M.; Wachs, I.E. In Situ UV-Vis-NIR Diffuse Reflectance and Raman Spectroscopic Studies of Propane Oxidation over ZrO2-Supported Vanadium Oxide Catalysts. J. Catal. 2002, 209, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Bell, A.T.; Iglesia, E. Kinetics and Mechanism of Oxidative Dehydrogenation of Propane on Vanadium, Molybdenum, and Tungsten Oxides. J. Phys. Chem. B 2000, 104, 1292–1299. [Google Scholar] [CrossRef] [Green Version]
- Argyle, M.D.; Chen, K.; Resini, C.; Krebs, C.; Bell, A.T.; Iglesia, E. Extent of Reduction of Vanadium Oxides during Catalytic Oxidation of Alkanes Measured by In-Situ UV−Visible Spectroscopy. J. Phys. Chem. B 2004, 108, 2345–2353. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.; Loridant, S.; Launay, H.; Pigamo, A.; Dubois, J.; Millet, J. Study of New Catalysts Based on Vanadium Oxide Supported on Mesoporous Silica for the Partial Oxidation of Methane to Formaldehyde: Catalytic Properties and Reaction Mechanism. J. Catal. 2006, 237, 38–48. [Google Scholar] [CrossRef]
- Ding, X.L.; Zhao, Y.X.; Wu, X.N.; Wang, Z.C.; Ma, J.B.; He, S.G. Hydrogen-Atom Abstraction from Methane by Stoichiometric Vanadium-Silicon Heteronuclear Oxide Cluster Cations. Chem.-A Eur. J. 2010, 16, 11463–11470. [Google Scholar] [CrossRef] [PubMed]
- Datka, J.; Turek, A.M.; Jehng, J.M.; Wachs, I.E. Acidic Properties of Supported Niobium Oxide Catalysts: An Infrared Spectroscopy Investigation. J. Catal. 1992, 135, 186–199. [Google Scholar] [CrossRef]
- Busca, G.; Ramis, G.; Lorenzelli, V. FT-IR Study of the Surface Properties of Polycrystalline Vanadia. J. Mol. Catal. 1989, 50, 231–240. [Google Scholar] [CrossRef]
- Hu, P.; Lang, W.Z.; Yan, X.; Chen, X.F.; Guo, Y.J. Vanadium-Doped Porous Silica Materials with High Catalytic Activity and Stability for Propane Dehydrogenation Reaction. Appl. Catal. A Gen. 2018, 553, 65–73. [Google Scholar] [CrossRef]
- Xue, X.-L.; Lang, W.-Z.; Yan, X.; Guo, Y.-J. Dispersed Vanadium in Three-Dimensional Dendritic Mesoporous Silica Nanospheres: Active and Stable Catalysts for the Oxidative Dehydrogenation of Propane in the Presence of CO2. ACS Appl. Mater. Interfaces 2017, 9, 15408–15423. [Google Scholar] [CrossRef]
- Ghampson, I.T.; Lundin, S.-T.B.; Vargheese, V.; Kobayashi, Y.; Huff, G.S.; Schlögl, R.; Trunschke, A.; Oyama, S.T. Methane Selective Oxidation on Metal Oxide Catalysts at Low Temperatures with O2 Using an NO/NO2 Oxygen Atom Shuttle. J. Catal. 2021. [Google Scholar] [CrossRef]
- Barbero, J.A.; Alvarez, M.C.; Bañares, M.A.; Peña, M.A.; Fierro, J.L.G. Breakthrough in the Direct Conversion of Methane into C1-Oxygenates. Chem. Commun. 2002, 11, 1184–1185. [Google Scholar] [CrossRef]
- Bañares, M.A.; Cardoso, J.H.; Hutchings, G.J.; Bueno, J.M.C.; Fierro, J.L.G. Selective Oxidation of Methane to Methanol and Formaldehyde over V2O5/SiO2 Catalysts. Role of NO in the Gas Phase. Catal. Lett. 1998, 56, 149–153. [Google Scholar] [CrossRef]
- Arena, F.; Giordano, N.; Parmaliana, A. Working Mechanism of Oxide Catalysts in the Partial Oxidation of Methane to Formaldehyde. II. Redox Properties and Reactivity of SiO2, MoO3/SiO2, V2O5/SiO2, TiO2, and V2O5/TiO2 Systems. J. Catal. 1997, 167, 66–76. [Google Scholar] [CrossRef]
- Wallis, P.; Wohlrab, S.; Kalevaru, V.N.; Frank, M.; Martin, A. Impact of Support Pore Structure and Morphology on Catalyst Performance of VOx/SBA-15 for Selective Methane Oxidation. Catal. Today 2016, 278, 120–126. [Google Scholar] [CrossRef]
- Du, G.; Yang, Y.; Qiu, W.; Lim, S.; Pfefferle, L.; Haller, G. Statistical Design and Modeling of the Process of Methane Partial Oxidation Using V-MCM-41 Catalysts and the Prediction of the Formaldehyde Production. Appl. Catal. A Gen. 2006, 313, 1–13. [Google Scholar] [CrossRef]
- Dang, T.T.H.; Seeburg, D.; Radnik, J.; Kreyenschulte, C.; Atia, H.; Vu, T.T.H.; Wohlrab, S. Influence of V-Sources on the Catalytic Performance of VMCM-41 in the Selective Oxidation of Methane to Formaldehyde. Catal. Commun. 2018, 103, 56–59. [Google Scholar] [CrossRef]
- Kunkel, B.; Kabelitz, A.; Buzanich, A.G.; Wohlrab, S. Increasing the Efficiency of Optimized V-SBA-15 Catalysts in the Selective Oxidation of Methane to Formaldehyde by Artificial Neural Network Modelling. Catalysts 2020, 10, 1411. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, J.; Sun, K.; Feng, Z.; Ying, P.; Li, C. Catalytic Performance of the Sb–V Mixed Oxide on Sb–V–O/SiO2 Catalysts in Methane Selective Oxidation to Formaldehyde. Catal. Lett. 2006, 106, 89–93. [Google Scholar] [CrossRef]
- Battiston, A.A.; Bitter, J.H.; de Groot, F.M.F.; Overweg, A.R.; Stephan, O.; van Bokhoven, J.A.; Kooyman, P.J.; van der Spek, C.; Vankó, G.; Koningsberger, D.C. Evolution of Fe Species during the Synthesis of Over-Exchanged Fe/ZSM-5 Obtained by Chemical Vapor Deposition of FeCl3. J. Catal. 2003, 213, 251–271. [Google Scholar] [CrossRef] [Green Version]
- Marturano, P.; Drozdová, L.; Kogelbauer, A.; Prins, R. Fe/ZSM-5 Prepared by Sublimation of FeCl3: The Structure of the Fe Species as Determined by IR, 27Al MAS NMR, and EXAFS Spectroscopy. J. Catal. 2000, 192, 236–247. [Google Scholar] [CrossRef]
- Kobayashi, T.; Guilhaume, N.; Miki, J.; Kitamura, N.; Haruta, M. Oxidation of Methane to Formaldehyde over Fe/SiO2 and Sn-W Mixed Oxides. Catal. Today 1996, 32, 171–175. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, W.; Wang, X.; Wang, Y.; Shishido, T.; Takehira, K. Coordination Structures of Vanadium and Iron in MCM-41 and the Catalytic Properties in Partial Oxidation of Methane. Microporous Mesoporous Mater. 2005, 77, 223–234. [Google Scholar] [CrossRef]
- Ertl, G.; Knözinger, H.; Weitkamp, J. Handbook of Heterogeneous Catalysis; Wiley-VCH Verlag GmbH: Hoboken, NJ, USA, 2008; Volumes 1–5, ISBN 9783527619474. [Google Scholar]
- He, J.; Li, Y.; An, D.; Zhang, Q.; Wang, Y. Selective Oxidation of Methane to Formaldehyde by Oxygen over Silica-Supported Iron Catalysts. J. Nat. Gas Chem. 2009, 18, 288–294. [Google Scholar] [CrossRef]
- McCormick, R.L.; Alptekin, G.O.; Williamson, D.L.; Ohno, T.R. Methane Partial Oxidation by Silica-supported Iron Phosphate Catalysts. Influence of Iron Phosphate Content on Selectivity and Catalyst Structure. Top. Catal. 2000, 10, 115–122. [Google Scholar] [CrossRef]
- Alptekin, G.O.; Herring, A.M.; Williamson, D.L.; Ohno, T.R.; McCormick, R.L. Methane Partial Oxidation by Unsupported and Silica Supported Iron Phosphate Catalysts. J. Catal. 1999, 181, 104–112. [Google Scholar] [CrossRef]
- Cortés Ortiz, W.G.; Delgado, D.; Guerrero Fajardo, C.A.; Agouram, S.; Sanchís, R.; Solsona, B.; López Nieto, J.M. Partial Oxidation of Methane and Methanol on FeOx-, MoOx- and FeMoOx-SiO2 Catalysts Prepared by Sol-Gel Method: A Comparative Study. Mol. Catal. 2020, 491, 110982. [Google Scholar] [CrossRef]
- Kanai, S.; Nagahara, I.; Kita, Y.; Kamata, K.; Hara, M. A Bifunctional Cerium Phosphate Catalyst for Chemoselective Acetalization. Chem. Sci. 2017, 8, 3146–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, A.; Ogo, S.; Kamata, K.; Takeno, Y.; Yabe, T.; Yamamoto, T.; Matsumura, S.; Hara, M.; Sekine, Y. Ambient-Temperature Oxidative Coupling of Methane in an Electric Field by a Cerium Phosphate Nanorod Catalyst. Chem. Commun. 2019, 55, 4019–4022. [Google Scholar] [CrossRef] [PubMed]
- Krisnandi, Y.K.; Nurani, D.A.; Alfian, D.V.; Sofyani, U.; Faisal, M.; Saragi, I.R.; Pamungkas, A.Z.; Pratama, A.P. The New Challenge of Partial Oxidation of Methane over Fe2O3/NaY and Fe3O4/NaY Heterogeneous Catalysts. Heliyon 2021, 7, e08305. [Google Scholar] [CrossRef]
- Gomonaj, V.; Toulhoat, H. Selective Oxidation of Methane to Formaldehyde Catalyzed by Phosphates: Kinetic Description by Bond Strengths and Specific Total Acidities. ACS Catal. 2018, 8, 8263–8272. [Google Scholar] [CrossRef]
- Dasireddy, V.D.B.C.; Hanzel, D.; Bharuth-Ram, K.; Likozar, B. The Effect of Oxidant Species on Direct, Non-Syngas Conversion of Methane to Methanol over an FePO4 Catalyst Material. RSC Adv. 2019, 9, 30989–31003. [Google Scholar] [CrossRef] [Green Version]
- Paunović, V.; Zichittella, G.; Moser, M.; Amrute, A.P.; Pérez-Ramírez, J. Catalyst Design for Natural-Gas Upgrading through Oxybromination Chemistry. Nat. Chem. 2016, 8, 803–809. [Google Scholar] [CrossRef]
- Zichittella, G.; Paunović, V.; Amrute, A.P.; Pérez-Ramírez, J. Catalytic Oxychlorination versus Oxybromination for Methane Functionalization. ACS Catal. 2017, 7, 1805–1817. [Google Scholar] [CrossRef]
- Bozbag, S.E.; Alayon, E.M.C.; Pecháček, J.; Nachtegaal, M.; Ranocchiari, M.; van Bokhoven, J.A. Methane to Methanol over Copper Mordenite: Yield Improvement through Multiple Cycles and Different Synthesis Techniques. Catal. Sci. Technol. 2016, 6, 5011–5022. [Google Scholar] [CrossRef]
- Le, H.v.; Parishan, S.; Sagaltchik, A.; Göbel, C.; Schlesiger, C.; Malzer, W.; Trunschke, A.; Schomäcker, R.; Thomas, A. Solid-State Ion-Exchanged Cu/Mordenite Catalysts for the Direct Conversion of Methane to Methanol. ACS Catal. 2017, 7, 1403–1412. [Google Scholar] [CrossRef]
- Ipek, B.; Wulfers, M.J.; Kim, H.; Göltl, F.; Hermans, I.; Smith, J.P.; Booksh, K.S.; Brown, C.M.; Lobo, R.F. Formation of [Cu2O2]2+ and [Cu2O]2+ toward C–H Bond Activation in Cu-SSZ-13 and Cu-SSZ-39. ACS Catal. 2017, 7, 4291–4303. [Google Scholar] [CrossRef]
- Wulfers, M.J.; Teketel, S.; Ipek, B.; Lobo, R.F. Conversion of Methane to Methanol on Copper-Containing Small-Pore Zeolites and Zeotypes. Chem. Commun. 2015, 51, 4447–4450. [Google Scholar] [CrossRef] [PubMed]
- Beznis, N.v.; Weckhuysen, B.M.; Bitter, J.H. Cu-ZSM-5 Zeolites for the Formation of Methanol from Methane and Oxygen: Probing the Active Sites and Spectator Species. Catal. Lett. 2010, 138, 14–22. [Google Scholar] [CrossRef] [Green Version]
- An, D.; Zhang, Q.; Wang, Y. Copper Grafted on SBA-15 as Efficient Catalyst for the Selective Oxidation of Methane by Oxygen. Catal. Today 2010, 157, 143–148. [Google Scholar] [CrossRef]
- Kim, D.S.; Ostromecki, M.; Wachs, I.E.; Kohler, S.D.; Ekerdt, J.G. Preparation and Characterization of WO3/SiO2 Catalysts. Catal. Lett. 1995, 33, 209–215. [Google Scholar] [CrossRef]
- Kohler, S.D.; Ekerdt, J.G.; Kim, D.S.; Wachs, I.E. Relationship between Structure and Point of Zero Surface Charge for Molybdenum and Tungsten Oxides Supported on Alumina. Catal. Lett. 1992, 16, 231–239. [Google Scholar] [CrossRef]
- De Lucas, A.; Valverde, J.L.; Cañizares, P.; Rodriguez, L. Partial Oxidation of Methane to Formaldehyde over W/SiO2 Catalysts. Appl. Catal. A Gen. 1999, 184, 143–152. [Google Scholar] [CrossRef]
- Krisnandi, Y.K.; Putra, B.A.P.; Bahtiar, M.; Zahara; Abdullah, I.; Howe, R.F. Partial Oxidation of Methane to Methanol over Heterogeneous Catalyst Co/ZSM-5. Procedia Chem. 2015, 14, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Beznis, N.v.; van Laak, A.N.C.; Weckhuysen, B.M.; Bitter, J.H. Oxidation of Methane to Methanol and Formaldehyde over Co–ZSM-5 Molecular Sieves: Tuning the Reactivity and Selectivity by Alkaline and Acid Treatments of the Zeolite ZSM-5 Agglomerates. Microporous Mesoporous Mater. 2011, 138, 176–183. [Google Scholar] [CrossRef]
- Shi, L.; Wang, Y.; Yan, B.; Song, W.; Shao, D.; Lu, A.H. Progress in Selective Oxidative Dehydrogenation of Light Alkanes to Olefins Promoted by Boron Nitride Catalysts. Chem. Commun. 2018, 54, 10936–10946. [Google Scholar] [CrossRef] [PubMed]
- Venegas, J.M.; McDermott, W.P.; Hermans, I. Serendipity in Catalysis Research: Boron-Based Materials for Alkane Oxidative Dehydrogenation. Acc. Chem. Res. 2018, 51, 2556–2564. [Google Scholar] [CrossRef]
- Atroshchenko, V.I.; Shchedrinskaya, Z.M.; Gavrya, N.A. Catalysts for the Oxidation of Natural Gas to Formaldehyde and Methanol. Zhurnal Prikl. Khimii 1965, 38, 643–649. [Google Scholar]
- Dowden, D.A.; Walker, G.T. Oxygenated Hydrocarbons Production. British Patent 1,244,001, 25 August 1971. Available online: https://patents.google.com/patent/GB1244001A/en (accessed on 14 February 2022).
- Zhang, X.; He, D.; Zhang, Q.; Xu, B.; Zhu, Q. Comparative Studies on Direct Conversion of Methane to Methanol/Formaldehyde over La-Co-O and ZrO2 Supported Molybdenum Oxide Catalysts. Top. Catal. 2005, 32, 215–223. [Google Scholar] [CrossRef]
- Sugino, T.; Kido, A.; Azuma, N.; Ueno, A.; Udagawa, Y. Partial Oxidation of Methane on Silica-Supported Silicomolybdic Acid Catalysts in an Excess Amount of Water Vapor. J. Catal. 2000, 190, 118–127. [Google Scholar] [CrossRef]
- Liu, R.-S.; Iwamoto, M.; Lunsford, J.H. Partial Oxidation of Methane by Nitrous Oxide over Molybdenum Oxide Supported on Silica. J. Chem. Soc. Chem. Commun. 1982, 78. [Google Scholar] [CrossRef]
- Chen, S.Y.; Willcox, D. Effect of Vanadium Oxide Loading on the Selective Oxidation of Methane over Vanadium Oxide (V2O5)/Silica. Ind. Eng. Chem. Res. 1993, 32, 584–587. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Y.; Wang, C.; Xie, Z.; Guan, N.; Li, L. Water-Involved Methane-Selective Catalytic Oxidation by Dioxygen over Copper Zeolites. Chem 2021, 7, 1557–1568. [Google Scholar] [CrossRef]
- Dinh, K.T.; Sullivan, M.M.; Narsimhan, K.; Serna, P.; Meyer, R.J.; Dincă, M.; Román-Leshkov, Y. Continuous Partial Oxidation of Methane to Methanol Catalyzed by Diffusion-Paired Copper Dimers in Copper-Exchanged Zeolites. J. Am. Chem. Soc. 2019, 141, 11641–11650. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.R.; Jung, H.; Kang, J.; Han, J.W.; Park, E.D. Continuous Synthesis of Methanol from Methane and Steam over Copper-Mordenite. ACS Catal. 2021, 11, 1065–1070. [Google Scholar] [CrossRef]
- Dasireddy, V.D.B.C.; Likozar, B. Direct Methanol Production from Mixed Methane/H2O/N2O Feedstocks over Cu–Fe/Al2O3 Catalysts. Fuel 2021, 301, 121084. [Google Scholar] [CrossRef]
- Li, Y.; Liu, N.; Dai, C.; Xu, R.; Yu, G.; Wang, N.; Zhang, J.; Chen, B. Synergistic Effect of Neighboring Fe and Cu Cation Sites Boosts FenCum-BEA Activity for the Continuous Direct Oxidation of Methane to Methanol. Catalysts 2021, 11, 1444. [Google Scholar] [CrossRef]
- Wang, H.; Xin, W.; Wang, Q.; Zheng, X.; Lu, Z.; Pei, R.; He, P.; Dong, X. Direct Methane Conversion with Oxygen and CO over Hydrophobic DB-ZSM-5 Supported Rh Single-Atom Catalyst. Catal. Commun. 2022, 162, 106374. [Google Scholar] [CrossRef]
- Lyu, Y.; Jocz, J.N.; Xu, R.; Williams, O.C.; Sievers, C. Selective Oxidation of Methane to Methanol over Ceria-Zirconia Supported Mono and Bimetallic Transition Metal Oxide Catalysts. ChemCatChem 2021, 13, 2832–2842. [Google Scholar] [CrossRef]
- Memioglu, O.; Ipek, B. A Potential Catalyst for Continuous Methane Partial Oxidation to Methanol Using N2O: Cu-SSZ-39. Chem. Commun. 2021, 57, 1364–1367. [Google Scholar] [CrossRef]
- Lee, S.H.; Kang, J.K.; Park, E.D. Continuous Methanol Synthesis Directly from Methane and Steam over Cu(II)-Exchanged Mordenite. Korean J. Chem. Eng. 2018, 35, 2145–2149. [Google Scholar] [CrossRef]
- Huang, E.; Orozco, I.; Ramírez, P.J.; Liu, Z.; Zhang, F.; Mahapatra, M.; Nemšák, S.; Senanayake, S.D.; Rodriguez, J.A.; Liu, P. Selective Methane Oxidation to Methanol on ZnO/Cu2O/Cu(111) Catalysts: Multiple Site-Dependent Behaviors. J. Am. Chem. Soc. 2021, 143, 19018–19032. [Google Scholar] [CrossRef]
- Hirayama, A.; Tsuchimura, Y.; Yoshida, H.; Machida, M.; Nishimura, S.; Kato, K.; Takahashi, K.; Ohyama, J. Catalytic Oxidation of Methane to Methanol over Cu-CHA with Molecular Oxygen. Catal. Sci. Technol. 2021, 11, 6217–6224. [Google Scholar] [CrossRef]
- Arutyunov, V.S.; Basevich, V.Y.; Vedeneev, V.I. Direct High-Pressure Gas-Phase Oxidation of Natural Gas to Methanol and Other Oxygenates. Russ. Chem. Rev. 1996, 65, 197–224. [Google Scholar] [CrossRef]
- Panov, G.I.; Sobolev, V.I.; Dubkov, K.A.; Parmon, V.N.; Ovanesyan, N.S.; Shilov, A.E.; Shteinman, A.A. Iron Complexes in Zeolites as a New Model of Methane Monooxygenase. React. Kinet. Catal. Lett. 1997, 61, 251–258. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; van Bokhoven, J.A. Methane-to-Methanol: Activity Descriptors in Copper-Exchanged Zeolites for the Rational Design of Materials. ACS Catal. 2019, 9, 6293–6304. [Google Scholar] [CrossRef]
- Sobolev, V.I.; Dubkov, K.A.; Panna, O.v.; Panov, G.I. Selective Oxidation of Methane to Methanol on a FeZSM-5 Surface. Catal. Today 1995, 24, 251–252. [Google Scholar] [CrossRef]
- Zhu, J.; Liang, W.; Erik, Z.; Kartick, M.; Zhang, M.; Zhang, J.; Wang, C.; Meng, X.; Yang, H.; Carl, M.; et al. Hydrophobic Zeolite Modification for in Situ Peroxide Formation in Methane Oxidation to Methanol. Science 2020, 367, 193–197. [Google Scholar] [CrossRef]
- Narsimhan, K.; Iyoki, K.; Dinh, K.; Román-Leshkov, Y. Catalytic Oxidation of Methane into Methanol over Copper-Exchanged Zeolites with Oxygen at Low Temperature. ACS Cent. Sci. 2016, 2, 424–429. [Google Scholar] [CrossRef] [Green Version]
- Mahyuddin, M.H.; Tanaka, T.; Shiota, Y.; Staykov, A.; Yoshizawa, K. Methane Partial Oxidation over [Cu2(μ-O)]2+ and [Cu3(μ-O)3]2+ Active Species in Large-Pore Zeolites. ACS Catal. 2018, 8, 1500–1509. [Google Scholar] [CrossRef]
- Dyballa, M.; Pappas, D.K.; Kvande, K.; Borfecchia, E.; Arstad, B.; Beato, P.; Olsbye, U.; Svelle, S. On How Copper Mordenite Properties Govern the Framework Stability and Activity in the Methane-to-Methanol Conversion. ACS Catal. 2019, 9, 365–375. [Google Scholar] [CrossRef]
- Xie, J.; Jin, R.; Li, A.; Bi, Y.; Ruan, Q.; Deng, Y.; Zhang, Y.; Yao, S.; Sankar, G.; Ma, D.; et al. Highly Selective Oxidation of Methane to Methanol at Ambient Conditions by Titanium Dioxide-Supported Iron Species. Nat. Catal. 2018, 1, 889–896. [Google Scholar] [CrossRef]
- Cui, X.; Li, H.; Wang, Y.; Hu, Y.; Hua, L.; Li, H.; Han, X.; Liu, Q.; Yang, F.; He, L.; et al. Room-Temperature Methane Conversion by Graphene-Confined Single Iron Atoms. Chem 2018, 4, 1902–1910. [Google Scholar] [CrossRef] [Green Version]
- Wood, B.R.; Reimer, J.A.; Bell, A.T.; Janicke, M.T.; Ott, K.C. Methanol Formation on Fe/Al-MFI via the Oxidation of Methane by Nitrous Oxide. J. Catal. 2004, 225, 300–306. [Google Scholar] [CrossRef]
- Pannov, G.I.; Sobolev, V.I.; Kharitonov, A.S. The Role of Iron in N2O Decomposition on ZSM-5 Zeolite and Reactivity of the Surface Oxygen Formed. J. Mol. Catal. 1990, 61, 85–97. [Google Scholar] [CrossRef]
- Starokon, E.v.; Parfenov, M.v.; Arzumanov, S.S.; Pirutko, L.v.; Stepanov, A.G.; Panov, G.I. Oxidation of Methane to Methanol on the Surface of FeZSM-5 Zeolite. J. Catal. 2013, 300, 47–54. [Google Scholar] [CrossRef]
- Starokon, E.v.; Parfenov, M.v.; Pirutko, L.v.; Abornev, S.I.; Panov, G.I. Room-Temperature Oxidation of Methane by α-Oxygen and Extraction of Products from the FeZSM-5 Surface. J. Phys. Chem. C 2011, 115, 2155–2161. [Google Scholar] [CrossRef]
- Snyder, B.E.R.; Vanelderen, P.; Bols, M.L.; Hallaert, S.D.; Böttger, L.H.; Ungur, L.; Pierloot, K.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. The Active Site of Low-Temperature Methane Hydroxylation in Iron-Containing Zeolites. Nature 2016, 536, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Bols, M.L.; Devos, J.; Rhoda, H.M.; Plessers, D.; Solomon, E.I.; Schoonheydt, R.A.; Sels, B.F.; Dusselier, M. Selective Formation of α-Fe(II) Sites on Fe-Zeolites through One-Pot Synthesis. J. Am. Chem. Soc. 2021, 143, 16243–16255. [Google Scholar] [CrossRef] [PubMed]
- Bols, M.L.; Snyder, B.E.R.; Rhoda, H.M.; Cnudde, P.; Fayad, G.; Schoonheydt, R.A.; van Speybroeck, V.; Solomon, E.I.; Sels, B.F. Coordination and Activation of Nitrous Oxide by Iron Zeolites. Nat. Catal. 2021, 4, 332–340. [Google Scholar] [CrossRef]
- Snyder, B.E.R.; Bol, M.L.; Rhoda, H.M.; Plessers, D.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Cage Effects Control the Mechanism of Methane Hydroxylation in Zeolites. Science 2021, 373, 327–331. [Google Scholar] [CrossRef]
- Zhao, G.; Benhelal, E.; Adesina, A.; Kennedy, E.; Stockenhuber, M. Comparison of Direct, Selective Oxidation of Methane by N2O over Fe-ZSM-5, Fe-Beta, and Fe-FER Catalysts. J. Phys. Chem. C 2019, 123, 27436–27447. [Google Scholar] [CrossRef]
- Parfenov, M.v.; Starokon, E.v.; Pirutko, L.v.; Panov, G.I. Quasicatalytic and Catalytic Oxidation of Methane to Methanol by Nitrous Oxide over FeZSM-5 Zeolite. J. Catal. 2014, 318, 14–21. [Google Scholar] [CrossRef]
- Li, S.; Fan, L.; Song, L.; Cheng, D.; Chen, F. Influence of Extra-Framework Al in Fe-MOR Catalysts for the Direct Conversion of Methane to Oxygenates by Nitrous Oxide. Chin. J. Chem. Eng. 2021, 33, 132–138. [Google Scholar] [CrossRef]
- Zhao, G.; Adesina, A.; Kennedy, E.; Stockenhuber, M. Formation of Surface Oxygen Species and the Conversion of Methane to Value-Added Products with N2O as Oxidant over Fe-Ferrierite Catalysts. ACS Catal. 2020, 10, 1406–1416. [Google Scholar] [CrossRef]
- Simons, M.C.; Prinslow, S.D.; Babucci, M.; Hoffman, A.S.; Hong, J.; Vitillo, J.G.; Bare, S.R.; Gates, B.C.; Lu, C.C.; Gagliardi, L.; et al. Beyond Radical Rebound: Methane Oxidation to Methanol Catalyzed by Iron Species in Metal–Organic Framework Nodes. J. Am. Chem. Soc. 2021, 143, 12165–12174. [Google Scholar] [CrossRef] [PubMed]
- Park, M.B.; Park, E.D.; Ahn, W.-S. Recent Progress in Direct Conversion of Methane to Methanol Over Copper-Exchanged Zeolites. Front. Chem. 2019, 7, 514. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Shiota, Y.; Staykov, A.; Yoshizawa, K. Theoretical Overview of Methane Hydroxylation by Copper–Oxygen Species in Enzymatic and Zeolitic Catalysts. Acc. Chem. Res. 2018, 51, 2382–2390. [Google Scholar] [CrossRef] [PubMed]
- Sushkevich, V.L.; Artsiusheuski, M.; Klose, D.; Jeschke, G.; van Bokhoven, J.A. Identification of Kinetic and Spectroscopic Signatures of Copper Sites for Direct Oxidation of Methane to Methanol. Angew. Chem. Int. Ed. 2021, 60, 15944–15953. [Google Scholar] [CrossRef] [PubMed]
- Tomkins, P.; Ranocchiari, M.; van Bokhoven, J.A. Direct Conversion of Methane to Methanol under Mild Conditions over Cu-Zeolites and Beyond. Acc. Chem. Res. 2017, 50, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Snyder, B.E.R.; Bols, M.L.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Iron and Copper Active Sites in Zeolites and Their Correlation to Metalloenzymes. Chem. Rev. 2018, 118, 2718–2768. [Google Scholar] [CrossRef] [PubMed]
- Groothaert, M.H.; Smeets, P.J.; Sels, B.F.; Jacobs, P.A.; Schoonheydt, R.A. Selective Oxidation of Methane by the Bis(μ-Oxo)Dicopper Core Stabilized on ZSM-5 and Mordenite Zeolites. J. Am. Chem. Soc. 2005, 127, 1394–1395. [Google Scholar] [CrossRef] [PubMed]
- Woertink, J.S.; Smeets, P.J.; Groothaert, M.H.; Vance, M.A.; Sels, B.F.; Schoonheydt, R.A.; Solomon, E.I. A [Cu2 O]2+ Core in Cu-ZSM-5, the Active Site in the Oxidation of Methane to Methanol. Proc. Natl. Acad. Sci. USA 2009, 106, 18908–18913. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Vassilev, P.; Sanchez-Sanchez, M.; Lercher, J.A.; Hensen, E.J.M.; Pidko, E.A. Stability and Reactivity of Copper Oxo-Clusters in ZSM-5 Zeolite for Selective Methane Oxidation to Methanol. J. Catal. 2016, 338, 305–312. [Google Scholar] [CrossRef]
- Grundner, S.; Markovits, M.A.C.; Li, G.; Tromp, M.; Pidko, E.A.; Hensen, E.J.M.; Jentys, A.; Sanchez-Sanchez, M.; Lercher, J.A. Single-Site Trinuclear Copper Oxygen Clusters in Mordenite for Selective Conversion of Methane to Methanol. Nat. Commun. 2015, 6, 7546. [Google Scholar] [CrossRef]
- Vanelderen, P.; Snyder, B.E.R.; Tsai, M.L.; Hadt, R.G.; Vancauwenbergh, J.; Coussens, O.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Spectroscopic Definition of the Copper Active Sites in Mordenite: Selective Methane Oxidation. J. Am. Chem. Soc. 2015, 137, 6383–6392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Kim, T.Y.; Lee, H.; Yi, J. Distinct Activation of Cu-MOR for Direct Oxidation of Methane to Methanol. Chem. Commun. 2017, 53, 4116–4119. [Google Scholar] [CrossRef] [PubMed]
- Ipek, B.; Lobo, R.F. Catalytic Conversion of Methane to Methanol on Cu-SSZ-13 Using N2O as Oxidant. Chem. Commun. 2016, 52, 13401–13404. [Google Scholar] [CrossRef] [PubMed]
- Tomkins, P.; Mansouri, A.; Sushkevich, V.L.; van der Wal, L.I.; Bozbag, S.E.; Krumeich, F.; Ranocchiari, M.; van Bokhoven, J.A. Increasing the Activity of Copper Exchanged Mordenite in the Direct Isothermal Conversion of Methane to Methanol by Pt and Pd Doping. Chem. Sci. 2019, 10, 167–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozbag, S.E.; Sot, P.; Nachtegaal, M.; Ranocchiari, M.; van Bokhoven, J.A.; Mesters, C. Direct Stepwise Oxidation of Methane to Methanol over Cu–SiO2. ACS Catal. 2018, 8, 5721–5731. [Google Scholar] [CrossRef] [Green Version]
- Elwell, C.E.; Gagnon, N.L.; Neisen, B.D.; Dhar, D.; Spaeth, A.D.; Yee, G.M.; Tolman, W.B. Copper–Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity. Chem. Rev. 2017, 117, 2059–2107. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.R.; Zhao, Z.-J.; Siahrostami, S.; Nørskov, J.K.; Studt, F. Cation-Exchanged Zeolites for the Selective Oxidation of Methane to Methanol. Catal. Sci. Technol. 2018, 8, 114–123. [Google Scholar] [CrossRef]
- Lange, J.-P.; Sushkevich, V.L.; Knorpp, A.J.; van Bokhoven, J.A. Methane-to-Methanol via Chemical Looping: Economic Potential and Guidance for Future Research. Ind. Eng. Chem. Res. 2019, 58, 8674–8680. [Google Scholar] [CrossRef]
- Alayon, E.M.; Nachtegaal, M.; Ranocchiari, M.; van Bokhoven, J.A. Catalytic Conversion of Methane to Methanol over Cu–Mordenite. Chem. Commun. 2012, 48, 404–406. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; van Bokhoven, J.A. Selective Anaerobic Oxidation of Methane Enables Direct Synthesis of Methanol. Science 2017, 356, 523–527. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Palagin, D.; van Bokhoven, J.A. The Effect of the Active-Site Structure on the Activity of Copper Mordenite in the Aerobic and Anaerobic Conversion of Methane into Methanol. Angew. Chem. 2018, 130, 9044–9048. [Google Scholar] [CrossRef]
- Pappas, D.K.; Martini, A.; Dyballa, M.; Kvande, K.; Teketel, S.; Lomachenko, K.A.; Baran, R.; Glatzel, P.; Arstad, B.; Berlier, G.; et al. The Nuclearity of the Active Site for Methane to Methanol Conversion in Cu-Mordenite: A Quantitative Assessment. J. Am. Chem. Soc. 2018, 140, 15270–15278. [Google Scholar] [CrossRef] [PubMed]
- Pappas, D.K.; Borfecchia, E.; Dyballa, M.; Pankin, I.A.; Lomachenko, K.A.; Martini, A.; Signorile, M.; Teketel, S.; Arstad, B.; Berlier, G.; et al. Methane to Methanol: Structure–Activity Relationships for Cu-CHA. J. Am. Chem. Soc. 2017, 139, 14961–14975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markovits, M.A.C.; Jentys, A.; Tromp, M.; Sanchez-Sanchez, M.; Lercher, J.A. Effect of Location and Distribution of Al Sites in ZSM-5 on the Formation of Cu-Oxo Clusters Active for Direct Conversion of Methane to Methanol. Top. Catal. 2016, 59, 1554–1563. [Google Scholar] [CrossRef]
- Brezicki, G.; Zheng, J.; Paolucci, C.; Schlögl, R.; Davis, R.J. Effect of the Co-Cation on Cu Speciation in Cu-Exchanged Mordenite and ZSM-5 Catalysts for the Oxidation of Methane to Methanol. ACS Catal. 2021, 11, 4973–4987. [Google Scholar] [CrossRef]
- Zhou, C.; Zhang, H.; Yang, L.; Wu, D.; He, S.; Yang, H.; Xiong, H. Methane-Selective Oxidation to Methanol and Ammonia Selective Catalytic Reduction of NOx over Monolithic Cu/SSZ-13 Catalysts: Are Hydrothermal Stability and Active Sites Same? Fuel 2022, 309, 122178. [Google Scholar] [CrossRef]
- Xiong, H.; Zhang, H.; Lv, J.; Zhang, Z.; Du, C.; Wang, S.; Lin, J.; Wan, S.; Wang, Y. Oxidation of Methane to Methanol by Water over Cu/SSZ-13: Impact of Cu Loading and Formation of Active Sites. ChemCatChem 2022, 14, e202101609. [Google Scholar] [CrossRef]
- Park, M.B.; Ahn, S.H.; Mansouri, A.; Ranocchiari, M.; van Bokhoven, J.A. Comparative Study of Diverse Copper Zeolites for the Conversion of Methane into Methanol. ChemCatChem 2017, 9, 3705–3713. [Google Scholar] [CrossRef]
- Knorpp, A.J.; Newton, M.A.; Sushkevich, V.L.; Zimmermann, P.P.; Pinar, A.B.; van Bokhoven, J.A. The Influence of Zeolite Morphology on the Conversion of Methane to Methanol on Copper-Exchanged Omega Zeolite (MAZ). Catal. Sci. Technol. 2019, 9, 2806–2811. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Lee, M.-S.; Tao, L.; Ikuno, T.; Khare, R.; Jentys, A.; Huthwelker, T.; Borca, C.N.; Kalinko, A.; Gutiérrez, O.Y.; et al. Activity of Cu–Al–Oxo Extra-Framework Clusters for Selective Methane Oxidation on Cu-Exchanged Zeolites. JACS Au 2021, 1, 1412–1421. [Google Scholar] [CrossRef]
- Shan, J.; Huang, W.; Nguyen, L.; Yu, Y.; Zhang, S.; Li, Y.; Frenkel, A.I.; Tao, F. Conversion of Methane to Methanol with a Bent Mono(μ-Oxo)Dinickel Anchored on the Internal Surfaces of Micropores. Langmuir 2014, 30, 8558–8569. [Google Scholar] [CrossRef] [PubMed]
- Mlekodaj, K.; Lemishka, M.; Sklenak, S.; Dedecek, J.; Tabor, E. Dioxygen Splitting at Room Temperature over Distant Binuclear Transition Metal Centers in Zeolites for Direct Oxid. of Methane to Methanol. Chem. Commun. 2021, 57, 3472–3475. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Yuan, M.; Li, X.; Bian, S.; Mi, L.; Gao, Z.; Shi, Q.; Huang, W.; Zuo, Z. The Influence of UiO-bpy Skeleton for the Direct Methane-to-Methanol Conversion on Cu@UiO-bpy: Importance of the Encapsulation Effect. ChemCatChem 2021, 13, 4897–4902. [Google Scholar] [CrossRef]
- Jovanovic, Z.R.; Lange, J.-P.; Ravi, M.; Knorpp, A.J.; Sushkevich, V.L.; Newton, M.A.; Palagin, D.; van Bokhoven, J.A. Oxidation of Methane to Methanol over Cu-Exchanged Zeolites: Scientia Gratia Scientiae or Paradigm Shift in Natural Gas Valorization? J. Catal. 2020, 385, 238–245. [Google Scholar] [CrossRef]











| Catalysts | Pretreatment (gas/P/T/t) | Reaction (gas/P/T/t) | Methanol Extraction (gas/P/T/t) | Methanol Yield | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| Name | Active Metal (Me) Loading (Wt%) | Me/Al | Si/Al | mmol/gcat | mmol/molCu | ||||
| Cu-ZSM-5 (MFI) | 4.0 | 0.58 | 12 | O2/1 bar/500 °C | CH4/1 bar/200 °C/0.25 h | Liquid water/1 bar/RT/1 h | 8.2 | 13.1 | [192] |
| Cu-mordenite (MOR) | 4.3 | 0.4 | 11 | O2/1 bar/400 °C/4 h | CH4/1 bar/200 °C/0.33 h | Steam in He/1 bar/RT/2 h | 13 | 19.3 | [204] |
| Cu-mordenite (MOR) | / | 0.4 | 6.5 | He/1 bar/400 °C/1 h | CH4/7 bar/200 °C/0.5 h | 2.4% steam in He/1 bar/200 °C/2–4 h | / | 204 | [205] |
| Cu-mordenite (MOR) | / | 0.6 | 46 | O2/1 bar/400 °C/1 h | CH4/7 bar/200 °C/0.5 h | 2.4% steam in He/1 bar/200 °C/2–4 h | / | 316 | [206] |
| Cu-mordenite (MOR) | 2.0 | 0.4 | 20 | 10% N2O/1 bar/600 °C/2 h | CH4/1 bar/150 °C/1 h | 7% steam in N2/1 bar/135 °C | 97 | 310 | [197] |
| Cu-mordenite (MOR) | 2.0 | 0.4 | 20 | O2/1 bar/450 °C/2 h | CH4/1 bar/150 °C/1 h | 7% steam in N2/1 bar/135 °C | 67 | 214 | [197] |
| Cu-mordenite (MOR) | 4.1 | 0.38 | 8.5 | O2/1 bar/450 °C/4 h | 5% CH4/1 bar/200 °C/0.5 h | Liquid water/1 bar/25 °C/2 h | 19 | 29.7 | [134] |
| Cu-mordenite (MOR) | 2.3 | 0.18 | 7 | O2/1 bar/500 °C/8 h | CH4/1 bar/200 °C/6 h | 10% steam/1 bar/200 °C/1 h | 169 | 470 | [207] |
| Cu-SSZ-13 (CHA) | 3.9 | 0.5 | 5 | O2/1 bar/500 °C/2 h | CH4/1 bar/200 °C/1 h | 10% steam/1 bar/200 °C/2 h | 125 | 200 | [208] |
| Cu-ZSM-5 (MFI) | 3.3 | 0.5 | 17 | O2/1 bar/450 °C/1 h | CH4/1 bar/200 °C/8 h | Steam/1 bar/135 °C/2 h | 89 | 172 | [209] |
| Cu-mordenite (MOR) | 2.2 | 0.28 | 10 | O2/1 bar/450 °C/2 h | CH4/35 bar/200 °C/20 h | Steam/1 bar/200 °C/2 h | / | 390 | [210] |
| Cu-SSZ-13 (CHA) | 2.2 | 0.25 | 11 | He/1 bar/400 °C/0.5 h | CH4/1 bar/200 °C/5 h | 3.2% steam in He/1 bar/200 °C | 60 | 174 | [211] |
| Cu-SSZ-13 (CHA) | 2.0 | 0.22 | 10 | He/1 bar/400 °C/0.5 h | CH4/1 bar/200 °C/15 h | 3.2% steam in He/1 bar/200 °C | 25 | 76.3 | [212] |
| Cu-SUZ-4 (SZR) | 4.3 | 0.43 | 8.2 | O2/1 bar/450 °C/4 h | CH4/1 bar/200 °C/0.5 h | Liquid water/1 bar/RT/2 h | 14.4 | 11.5 | [213] |
| Cu-omega (MAZ) | 5.9 | 0.29 | 3.2 | O2/1 bar/450 °C/4 h | CH4/1 bar/200 °C/0.5 h | Liquid water/1 bar/RT/2 h | 86.1 | 92.6 | [213] |
| Cu-UZM-22 (MEI) | 4.3 | 0.32 | 4.8 | O2/1 bar/550 °C/4 h | CH4/1 bar/200 °C/0.5 h | Liquid water/1 bar/RT/2 h | 16.1 | 23.8 | [213] |
| Cu-omega (MAZ) | 4.64 | / | 4.0 | O2/1 bar/450 °C/4 h | CH4/6 bar/200 °C/0.5 h | Liquid water/1 bar/RT/2 h | 144.8 | 210 | [214] |
| Cu-mordenite (MOR) | 1.8 | / | 9.3 | O2/1 bar/500 °C/1 h | CH4/1 bar/200 °C/4 h | 50% Steam/1 bar/135 °C | 160 | 565 | [215] |
| Cu-ZSM-5 (MFI) | 2.5 | 0.37 | 11.5 | O2/1 bar/550 °C/0.5 h | CH4/1 bar/210 °C/0.5 h | H2O + O2 + CH4/1 bar/210 °C/0.5 h | 82 | 210 | [169] |
| Cu-SSZ-13 (CHA) | 3.2 | 0.4 | 12 | O2/1 bar/450 °C/2 h | CH4/1 bar/200 °C/1 h | Steam/1 bar/200°C/1 h | 45 | 90 | [198] |
| Cu-SSZ-13 (CHA) | 3.2 | 0.4 | 12 | N2O/1 bar/450 °C/2 h | CH4/1 bar/200 °C/1 h | Steam/1 bar/200°C/1 h | 35 | 70 | [198] |
| Ni-ZSM-5 (MFI) | 5.0 | 0.1 | 15 | O2/1 bar/550 °C/3 h | CH4/1 bar/175 °C/0.75 h | Liquid water/1 bar/RT/24 h | 5.8 | 6.9 | [216] |
| Ni-ferrierite (FER) | 1.0 | 0.1 | 8.6 | Ar/1 bar/450 °C/3 h—O2/1 bar/RT/1 h | CH4/1 bar/RT | / | 116 | 680 | [217] |
| Cu@UiO-bpy | 19.2 | / | / | O2/1 bar/200 °C/3 h | CH4/1 bar/200 °C/3 h | Steam with He/1 bar | 24.3 | 8.1 | [218] |
| Fe-SSZ-13 (CHA) | 2.7 | 0.43 | 13 | He/1 bar/900 °C/5 h—N2O/1 bar/180 °C/0.42 h | CH4/1 bar/RT/0.17 h | Steam with He/1bar/RT/25 h | 134 | 270 | [179] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Z.C.; Park, E.D. Gas-Phase Selective Oxidation of Methane into Methane Oxygenates. Catalysts 2022, 12, 314. https://doi.org/10.3390/catal12030314
Xu ZC, Park ED. Gas-Phase Selective Oxidation of Methane into Methane Oxygenates. Catalysts. 2022; 12(3):314. https://doi.org/10.3390/catal12030314
Chicago/Turabian StyleXu, Zhen Chao, and Eun Duck Park. 2022. "Gas-Phase Selective Oxidation of Methane into Methane Oxygenates" Catalysts 12, no. 3: 314. https://doi.org/10.3390/catal12030314