

Photoredox-Catalyzed Acylation/Cyclization of 2-Isocyanobiaryls with Oxime Esters for the Synthesis of 6-Acyl Phenanthridines

Abstract
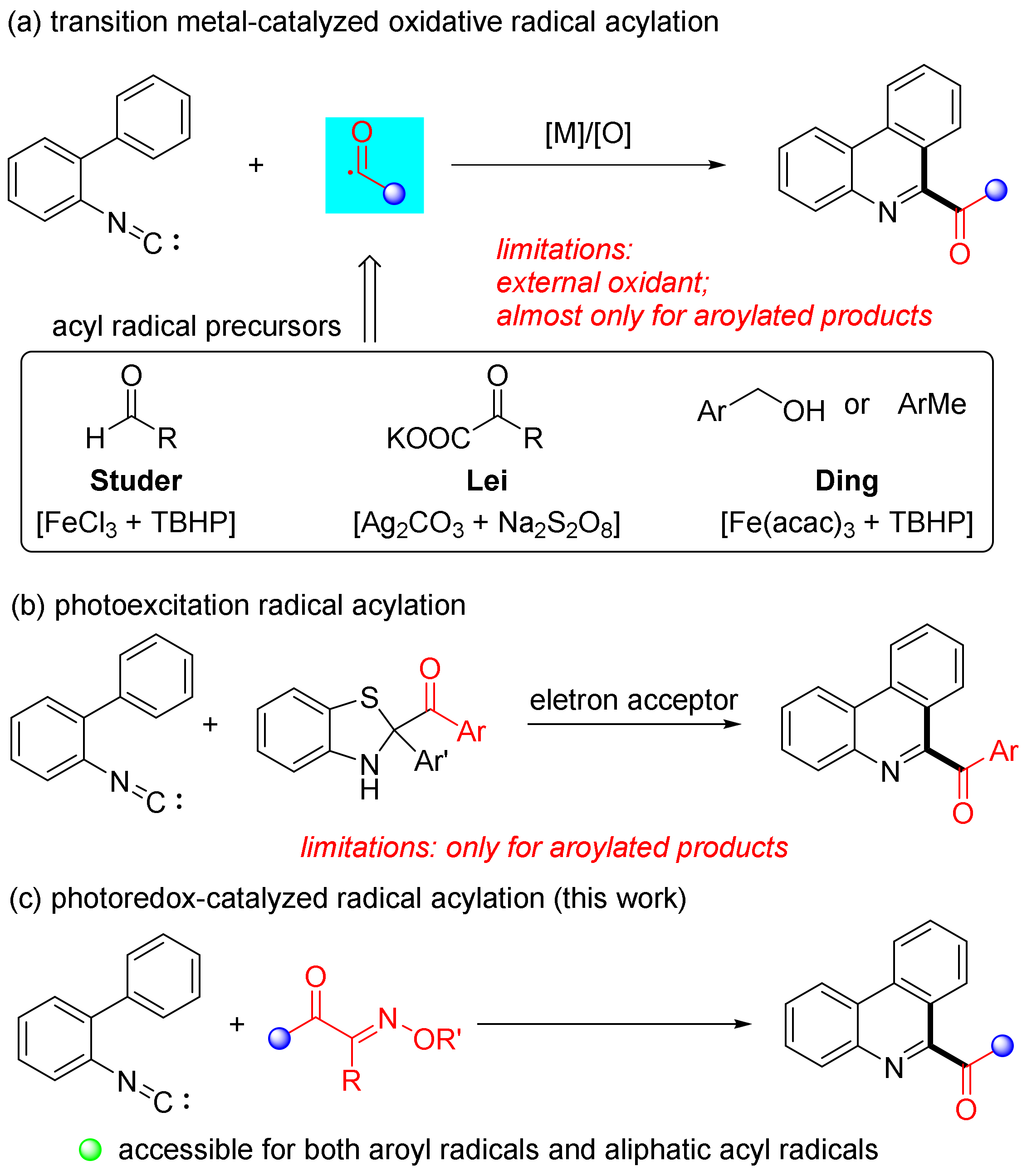
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of 3
3.3. Characterization Data for 6-Acyl Phenanthridines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Abdel-Halim, O.B.; Morikawa, T.; Ando, S.; Matsuda, H.; Yoshikawa, M. New Crinine-Type Alkaloids with Inhibitory Effect on Induction of Inducible Nitric Oxide Synthase from Crinum yemense. J. Nat. Prod. 2004, 67, 1119–1124. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Long, H.; Cui, X.; Zhou, J.; Xu, M.; Yuan, G. Exploring the Formation and Recognition of an Important G-Quadruplex in a HIF1α Promoter and Its Transcriptional Inhibition by a Benzo[c]phenanthridine Derivative. J. Am. Chem. Soc. 2014, 136, 2583–2591. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Zhou, J.; Qing, Z.; Kang, W.; Liu, S.; Liu, W.; Xie, H.; Zeng, J. Synthesis of 5-methyl phenanthridium derivatives: A new class of human DOPA decarboxylase inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 2712–2716. [Google Scholar] [CrossRef] [PubMed]
- Pictet, A.; Hubert, A. Ueber eine neue Synthese der Phenanthridinbasen. Ber. Dtsch. Chem. Ges. 1896, 29, 1182–1189. [Google Scholar] [CrossRef] [Green Version]
- Morgan, G.T.; Walls, L.P. CCCXXXV.—Researches in the phenanthridine series. Part I. A new synthesis of phenanthridine homologues and derivatives. J. Chem. Soc. 1931, 2447–2456. [Google Scholar] [CrossRef]
- Lorsbach, B.A.; Kurth, M.J. Carbon−Carbon Bond Forming Solid-Phase Reactions. Chem. Rev. 1999, 99, 1549–1582. [Google Scholar] [CrossRef] [PubMed]
- Mehta, B.K.; Yanagisawa, K.; Shiro, M.; Kotsuki, H. Unprecedented Hydrothermal Reaction of O-Phenylaniline and Related Derivatives with Cyclic Ketones. A Novel Approach to the Construction of Phenanthridine and Quinoline Ring Systems. Org. Lett. 2003, 5, 1605–1608. [Google Scholar] [CrossRef]
- Shou, W.-G.; Yang, Y.-Y.; Wang, Y.-G. Cascade Approach to Substituted 6-Aryl-phenanthridines from Aromatic Aldehydes, Anilines, and Benzenediazonium-2-carboxylate. J. Org. Chem. 2006, 71, 9241–9243. [Google Scholar] [CrossRef] [PubMed]
- Sripada, L.; Teske, J.A.; Deiters, A. Phenanthridine synthesis via [2 + 2 + 2] cyclotrimerization reactions. Org. Biomol. Chem. 2008, 6, 263–265. [Google Scholar] [CrossRef]
- Yanada, R.; Hashimoto, K.; Tokizane, R.; Miwa, Y.; Minami, H.; Yanada, K.; Ishikura, M.; Takemoto, Y. Indium(III)-Catalyzed Tandem Reaction with Alkynylbenzaldehydes and Alkynylanilines to Heteroaromatic Compounds. J. Org. Chem. 2008, 73, 5135–5138. [Google Scholar] [CrossRef] [PubMed]
- Gerfaud, T.; Neuville, L.; Zhu, J. Palladium-Catalyzed Annulation of Acyloximes with Arynes (or Alkynes): Synthesis of Phenanthridines and Isoquinolines. Angew. Chem. Int. Ed. 2009, 48, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Chen, T.; Chen, C.; Li, B. Palladium-Catalyzed Intramolecular C–H Activation/C–C Bond Formation: A Straightforward Synthesis of Phenanthridines. J. Org. Chem. 2011, 76, 9507–9513. [Google Scholar] [CrossRef] [PubMed]
- Grigg, R.D.; Van Hoveln, R.; Schomaker, J.M. Copper-Catalyzed Recycling of Halogen Activating Groups via 1, 3-Halogen Migration. J. Am. Chem. Soc. 2012, 134, 16131–16134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, X.; Yao, W.; Zhu, Q.; Xu, Y. Synthesis of 6-Substituted Phenanthridine Derivatives by Palladium-Catalysed Domino Suzuki–Miyaura/Aza-Michael Reactions. Eur. J. Org. Chem. 2014, 2014, 7443–7450. [Google Scholar] [CrossRef]
- Fan, J.; Li, L.; Zhang, J.; Xie, M. Expeditious synthesis of phenanthridines through a Pd/MnO2-mediated C–H arylation/oxidative annulation cascade from aldehydes, aryl iodides and amino acids. Chem. Commun. 2020, 56, 2775–2778. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Hou, J.; Tian, J.; Yu, W.; Chang, J. Synthesis of phenanthridines by I2-mediated sp3 C–H amination. Org. Biomol. Chem. 2020, 18, 3312–3323. [Google Scholar] [CrossRef]
- Li, G.; Kong, X.; Liang, Q.; Lin, L.; Yu, K.; Xu, B.; Chen, Q. Metal-Free Electrochemical Coupling of Vinyl Azides: Synthesis of Phenanthridines and β-Ketosulfones. Eur. J. Org. Chem. 2020, 2020, 6135–6145. [Google Scholar] [CrossRef]
- Lang, S. Unravelling the labyrinth of palladium-catalysed reactions involving isocyanides. Chem. Soc. Rev. 2013, 42, 4867–4880. [Google Scholar] [CrossRef]
- Qiu, G.; Ding, Q.; Wu, J. Recent advances in isocyanide insertion chemistry. Chem. Soc. Rev. 2013, 42, 5257–5269. [Google Scholar] [CrossRef]
- Vlaar, T.; Ruijter, E.; Maes, B.U.W.; Orru, R.V.A. Palladium-Catalyzed Migratory Insertion of Isocyanides: An Emerging Platform in Cross-Coupling Chemistry. Angew. Chem. Int. Ed. 2013, 52, 7084–7097. [Google Scholar] [CrossRef] [PubMed]
- Sumi, S.; Matsumoto, K.; Tokuyama, H.; Fukuyama, T. Enantioselective Total Synthesis of Aspidophytine. Org. Lett. 2003, 5, 1891–1893. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, T.; Tsuboi, Y.; Iwata, K.; Tsuchii, K.; Nomoto, A.; Sonoda, M.; Ogawa, A. Photoinduced thiotelluration of isocyanides by using a (PhS)2–(PhTe)2 mixed system, and its application to bisthiolation via radical cyclization. Tetrahedron Lett. 2007, 48, 5953–5957. [Google Scholar] [CrossRef]
- Mitamura, T.; Iwata, K.; Ogawa, A. (PhTe)2-Mediated Intramolecular Radical Cyclization of o-Ethynylaryl Isocyanides Leading to Bistellurated Quinolines upon Visible-Light Irradiation. Org. Lett. 2009, 11, 3422–3424. [Google Scholar] [CrossRef]
- Sha, W.; Yu, J.-T.; Jiang, Y.; Yang, H.; Cheng, J. The benzoyl peroxide-promoted functionalization of simple alkanes with 2-aryl phenyl isonitrile. Chem. Commun. 2014, 50, 9179–9181. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sha, W.; Dai, Q.; Feng, X.; Wu, W.; Peng, H.; Chen, B.; Cheng, J. The Benzoyl Peroxide Promoted Dual C–C Bond Formation via Dual C–H Bond Cleavage: α-Phenanthridinylation of Ether by Isocyanide. Org. Lett. 2014, 16, 2088–2091. [Google Scholar] [CrossRef]
- Rohe, S.; McCallum, T.; Morris, A.O.; Barriault, L. Transformations of Isonitriles with Bromoalkanes Using Photoredox Gold Catalysis. J. Org. Chem. 2018, 83, 10015–10024. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Pan, W.; Wu, S.; Bu, X.; Xin, S.; Yu, J.; Xu, H.; Yang, X. Visible-light-induced tandem radical addition-cyclization of 2-aryl phenyl isocyanides catalysed by recyclable covalent organic frameworks. Green Chem. 2019, 21, 2905–2910. [Google Scholar] [CrossRef]
- Zhuang, Y.-J.; Qu, J.-P.; Kang, Y.-B. Deprotonated Salicylaldehyde as Visible Light Photocatalyst. J. Org. Chem. 2020, 85, 4386–4397. [Google Scholar] [CrossRef]
- Zhang, Y.; Jin, Y.; Wang, L.; Zhang, Q.; Meng, C.; Duan, C. Selective C(sp3)–H activation of simple alkanes: Visible light-induced metal-free synthesis of phenanthridines with H2O2 as a sustainable oxidant. Green Chem. 2021, 23, 6926–6930. [Google Scholar] [CrossRef]
- Ye, H.-B.; Zhou, X.-Y.; Li, L.; He, X.-K.; Xuan, J. Photochemical Synthesis of Succinic Ester-Containing Phenanthridines from Diazo Compounds as 1,4-Dicarbonyl Precursors. Org. Lett. 2022, 24, 6018–6023. [Google Scholar] [CrossRef]
- Gu, L.; Jin, C.; Liu, J.; Ding, H.; Fan, B. Transition-metal-free, visible-light induced cyclization of arylsulfonyl chlorides with 2-isocyanobiphenyls to produce phenanthridines. Chem. Commun. 2014, 50, 4643–4645. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Huang, J.; He, Y.; Zhao, J.; Lei, J.; Zhu, Q. Arylative Cyclization of 2-Isocyanobiphenyls with Anilines: One-Pot Synthesis of 6-Arylphenanthridines via Competitive Reaction Pathways. Org. Lett. 2014, 16, 2546–2549. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Li, L.; Lin, G.; Wang, Q.; Zhang, P.; Mao, Z.-W.; Zhou, L. Synthesis of 6-substituted phenanthridines by metal-free, visible-light induced aerobic oxidative cyclization of 2-isocyanobiphenyls with hydrazines. Green Chem. 2014, 16, 2418–2421. [Google Scholar] [CrossRef]
- Pan, C.; Zhang, H.; Han, J.; Cheng, Y.; Zhu, C. Metal-free radical oxidative decarboxylation/cyclization of acyl peroxides and 2-isocyanobiphenyls. Chem. Commun. 2015, 51, 3786–3788. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yu, S. Visible-Light-Mediated Fluoroalkylation of Isocyanides with Ethyl Bromofluoroacetates: Unified Synthesis of Mono- and Difluoromethylated Phenanthridine Derivatives. Org. Lett. 2014, 16, 2938–2941. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, X.; Dolbier, W.R. Photoredox-Catalyzed Tandem Insertion/Cyclization Reactions of Difluoromethyl and 1,1-Difluoroalkyl Radicals with Biphenyl Isocyanides. Org. Lett. 2015, 17, 4401–4403. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Shen, W.-G.; Ao, G.-Z.; Liu, F. Transition-metal-free radical fluoroalkylation of isocyanides for the synthesis of tri-/di-/monofluoromethylated phenanthridines. Org. Chem. Front. 2017, 4, 2049–2053. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Li, G.-X.; He, G.; Chen, G. Halogen-Bond-Promoted Photoactivation of Perfluoroalkyl Iodides: A Photochemical Protocol for Perfluoroalkylation Reactions. Org. Lett. 2017, 19, 1442–1445. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, C.; Zhang, J.; Shi, Y.; Zhang, S.; Geng, Y.; Tung, C.-H.; Wang, W. Cobalt-catalyzed radical cyclization of isocyanides forming phenanthridine derivatives. Org. Chem. Front. 2018, 5, 2997–3002. [Google Scholar] [CrossRef]
- Cheng, Y.; Jiang, H.; Zhang, Y.; Yu, S. Isocyanide Insertion: De Novo Synthesis of Trifluoromethylated Phenanthridine Derivatives. Org. Lett. 2013, 15, 5520–5523. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Dong, X.; Xiao, T.; Zhou, L. PhI(OAc)2-Mediated Synthesis of 6-(Trifluoromethyl) phenanthridines by Oxidative Cyclization of 2-Isocyanobiphenyls with CF3SiMe3 under Metal-Free Conditions. Org. Lett. 2013, 15, 4846–4849. [Google Scholar] [CrossRef] [PubMed]
- Lübbesmeyer, M.; Leifert, D.; Schäfer, H.; Studer, A. Electrochemical initiation of electron-catalyzed phenanthridine synthesis by trifluoromethylation of isonitriles. Chem. Commun. 2018, 54, 2240–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Song, X.; Guo, J.; Chen, S.; Gao, J.; Jiang, J.; Gao, F.; Li, Y.; Wang, M. Synthesis of Chloro(phenyl)trifluoromethyliodane and Catalyst-Free Electrophilic Trifluoromethylations. Org. Lett. 2018, 20, 3933–3937. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.-J.; Zhu, T.-H.; Gu, Z.-Y.; Hao, W.-J.; Wang, S.-Y.; Ji, S.-J. Silver-catalyzed 2-isocyanobiaryls insertion/cyclization with phosphine oxides: Synthesis of 6-phosphorylated phenanthridines. Tetrahedron 2014, 70, 6985–6990. [Google Scholar] [CrossRef]
- Noël-Duchesneau, L.; Lagadic, E.; Morlet-Savary, F.; Lohier, J.-F.; Chataigner, I.; Breugst, M.; Lalevée, J.; Gaumont, A.-C.; Lakhdar, S. Metal-Free Synthesis of 6-Phosphorylated Phenanthridines: Synthetic and Mechanistic Insights. Org. Lett. 2016, 18, 5900–5903. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.-L.; Li, X.-Y.; Zhu, S.-S.; Li, S.-J.; Song, Y.; Qu, L.-B.; Yu, B. 4CzIPN-tBu-Catalyzed Proton-Coupled Electron Transfer for Photosynthesis of Phosphorylated N-Heteroaromatics. J. Am. Chem. Soc. 2021, 143, 964–972. [Google Scholar] [CrossRef]
- Tu, H.-Y.; Liu, Y.-R.; Chu, J.-J.; Hu, B.-L.; Zhang, X.-G. FeCl3-Promoted Carboxamidation and Cyclization of Aryl Isonitriles with Formamides toward Phenanthridine-6-carboxamides. J. Org. Chem. 2014, 79, 9907–9912. [Google Scholar] [CrossRef]
- Yang, Z.; Song, X.; Wei, Z.; Cao, J.; Liang, D.; Duan, H.; Lin, Y. Metal-free oxidative cascade cyclization of isocyanides with thiols: A new pathway for constructing 6-aryl(alkyl)thiophenanthridines. Tetrahedron Lett. 2016, 57, 2410–2413. [Google Scholar] [CrossRef]
- Singh, M.; Yadav, A.K.; Yadav, L.D.S.; Singh, R.K.P. Direct synthesis of 6-sulfonylated phenanthridines via silver-catalyzed radical sulfonylation-cyclization of 2-isocyanobiphenyls. Tetrahedron Lett. 2018, 59, 3198–3201. [Google Scholar] [CrossRef]
- Leifert, D.; Daniliuc, C.G.; Studer, A. 6-Aroylated Phenanthridines via Base Promoted Homolytic Aromatic Substitution (BHAS). Org. Lett. 2013, 15, 6286–6289. [Google Scholar] [CrossRef]
- Liu, J.; Fan, C.; Yin, H.; Qin, C.; Zhang, G.; Zhang, X.; Yi, H.; Lei, A. Synthesis of 6-acyl phenanthridines by oxidative radical decarboxylation–cyclization of α-oxocarboxylates and isocyanides. Chem. Commun. 2014, 50, 2145–2147. [Google Scholar] [CrossRef] [Green Version]
- Nie, Z.; Ding, Q.; Peng, Y. Synthesis of 6-aroyl phenanthridines by Fe-catalyzed oxidative radical cyclization of 2-isocyanobiphenyls with benzylic alcohols. Tetrahedron 2016, 72, 8350–8357. [Google Scholar] [CrossRef]
- He, X.-K.; Lu, J.; Ye, H.-B.; Li, L.; Xuan, J. Direct Photoexcitation of Benzothiazolines: Acyl Radical Generation and Application to Access Heterocycles. Molecules 2021, 26, 6843. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Kleinmans, R.; Daniliuc, C.G.; Glorius, F. Synthesis of Polysubstituted 2-Oxabicyclo [2.1.1] hexanes via Visible-Light-Induced Energy Transfer. J. Am. Chem. Soc. 2022, 144, 20207–20213. [Google Scholar] [CrossRef]
- Lacker, C.R.; DeLano, T.J.; Chen, E.P.; Kong, J.; Belyk, K.M.; Piou, T.; Reisman, S.E. Enantioselective Synthesis of N-Benzylic Heterocycles by Ni/Photoredox Dual Catalysis. J. Am. Chem. Soc. 2022, 144, 20190–20195. [Google Scholar] [CrossRef] [PubMed]
- He, B.-Q.; Yu, X.-Y.; Wang, P.-Z.; Chen, J.-R.; Xiao, W.-J. A photoredox catalyzed iminyl radical-triggered C–C bond cleavage/addition/Kornblum oxidation cascade of oxime esters and styrenes: Synthesis of ketonitriles. Chem. Commun. 2018, 54, 12262–12265. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-Y.; Chen, J.-R.; Wang, P.-Z.; Yang, M.-N.; Liang, D.; Xiao, W.-J. A Visible-Light-Driven Iminyl Radical-Mediated C−C Single Bond Cleavage/Radical Addition Cascade of Oxime Esters. Angew. Chem. Int. Ed. 2018, 57, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; He, B.-Q.; Wang, P.-Z.; Yu, X.-Y.; Zhao, Q.-Q.; Chen, J.-R.; Xiao, W.-J. Photoinduced, Copper-Catalyzed Radical Cross-Coupling of Cycloketone Oxime Esters, Alkenes, and Terminal Alkynes. Org. Lett. 2019, 21, 4359–4364. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, P.-Z.; Lu, B.; Liang, D.; Yu, X.-Y.; Xiao, W.-J.; Chen, J.-R. Enantioselective Radical Ring-Opening Cyanation of Oxime Esters by Dual Photoredox and Copper Catalysis. Org. Lett. 2019, 21, 9763–9768. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Cheng, Y.; Chen, L.-Y.; Chen, J.-R.; Xiao, W.-J. Photoinduced Copper-Catalyzed Radical Aminocarbonylation of Cycloketone Oxime Esters. ACS Catal. 2019, 9, 8159–8164. [Google Scholar] [CrossRef]
- Cheng, Y.-Y.; Lei, T.; Su, L.; Fan, X.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Visible Light Irradiation of Acyl Oxime Esters and Styrenes Efficiently Constructs β-Carbonyl Imides by a Scission and Four-Component Reassembly Process. Org. Lett. 2019, 21, 8789–8794. [Google Scholar] [CrossRef]
- Fan, X.; Lei, T.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Photocatalytic C–C Bond Activation of Oxime Ester for Acyl Radical Generation and Application. Org. Lett. 2019, 21, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Mück-Lichtenfeld, C.; Daniliuc, C.G.; Studer, A. 6-Trifluoromethyl-Phenanthridines through Radical Trifluoromethylation of Isonitriles. Angew. Chem. Int. Ed. 2013, 52, 10792–10795. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.-Q.; Xie, J.; Chen, Z.; Xiong, B.-Q.; Liu, Y.; Tang, K.-W. Visible-Light-Induced Transition-Metal-Free Nitrogen-Centered Radical Strategy for the Synthesis of 2-Acylated 9H-Pyrrolo [1,2-a] indoles. J. Org. Chem. 2021, 86, 13720–13733. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Entry | Variation from the Standard Conditions | Yield (%) 2 |
| 1 | None | 94 |
| 2 | Without Ir(ppy)3 | 0 |
| 3 | Ru(bpy)3Cl2 instead of Ir(ppy)3 | 12 |
| 4 | Eosin Y instead of Ir(ppy)3 | 36 |
| 5 | Na2-Eosin Y instead of Ir(ppy)3 | 16 |
| 6 | Ir(ppy)3 (2 mol%) | 95 |
| 7 | Without additional light | 49 |
| 8 | 3 W blue LED light instead of 5 W blue LED light | 63 |
| 9 | 7 W blue LED light instead of 5 W blue LED light | 84 |
| 10 | 36 W compact fluorescent light instead of 5 W blue LED light | 52 |
| 11 | 2a-1 instead of 2a | 31 |
| 12 | 2a-2 instead of 2a | 46 |
| 13 | 2a-3 instead of 2a | 40 |
| 14 | 2a-4 instead of 2a | 55 |
| 15 | 2a-5 instead of 2a | 52 |
| 16 | 2a-6 instead of 2a | 13 |
| 17 | CH3CN instead of DMF | 86 |
| 18 | THF instead of DMF | 39 |
| 19 | DCE instead of DMF | 35 |
| 20 | nBuOAc instead of DMF | 21 |
| 21 | toluene instead of DMF | 48 |
| 22 | DMSO instead of DMF | 52 |
| 23 | at 80 °C | 74 |
| 24 | at 120 °C | 88 |
| 25 | for 24 h | 94 |
| 26 | Argon instead of nitrogen | 93 |
| 27 | Air instead of nitrogen | 0 |
| 28 3 | None | 70 |
 |
 |
 |
 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, B.; Ding, C.; Ou, M.; Liu, Y.; Liu, J.; Liu, Y. Photoredox-Catalyzed Acylation/Cyclization of 2-Isocyanobiaryls with Oxime Esters for the Synthesis of 6-Acyl Phenanthridines. Catalysts 2022, 12, 1446. https://doi.org/10.3390/catal12111446
Tang B, Ding C, Ou M, Liu Y, Liu J, Liu Y. Photoredox-Catalyzed Acylation/Cyclization of 2-Isocyanobiaryls with Oxime Esters for the Synthesis of 6-Acyl Phenanthridines. Catalysts. 2022; 12(11):1446. https://doi.org/10.3390/catal12111446
Chicago/Turabian StyleTang, Boxiao, Chuan Ding, Min Ou, Yu Liu, Junwei Liu, and Yilin Liu. 2022. "Photoredox-Catalyzed Acylation/Cyclization of 2-Isocyanobiaryls with Oxime Esters for the Synthesis of 6-Acyl Phenanthridines" Catalysts 12, no. 11: 1446. https://doi.org/10.3390/catal12111446