

Pt2 Dimer Anchored Vertically in Defective BN Monolayer as an Efficient Catalyst for N2 Reduction: A DFT Study

Abstract
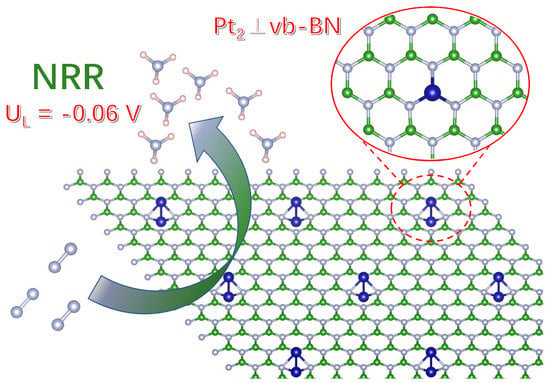
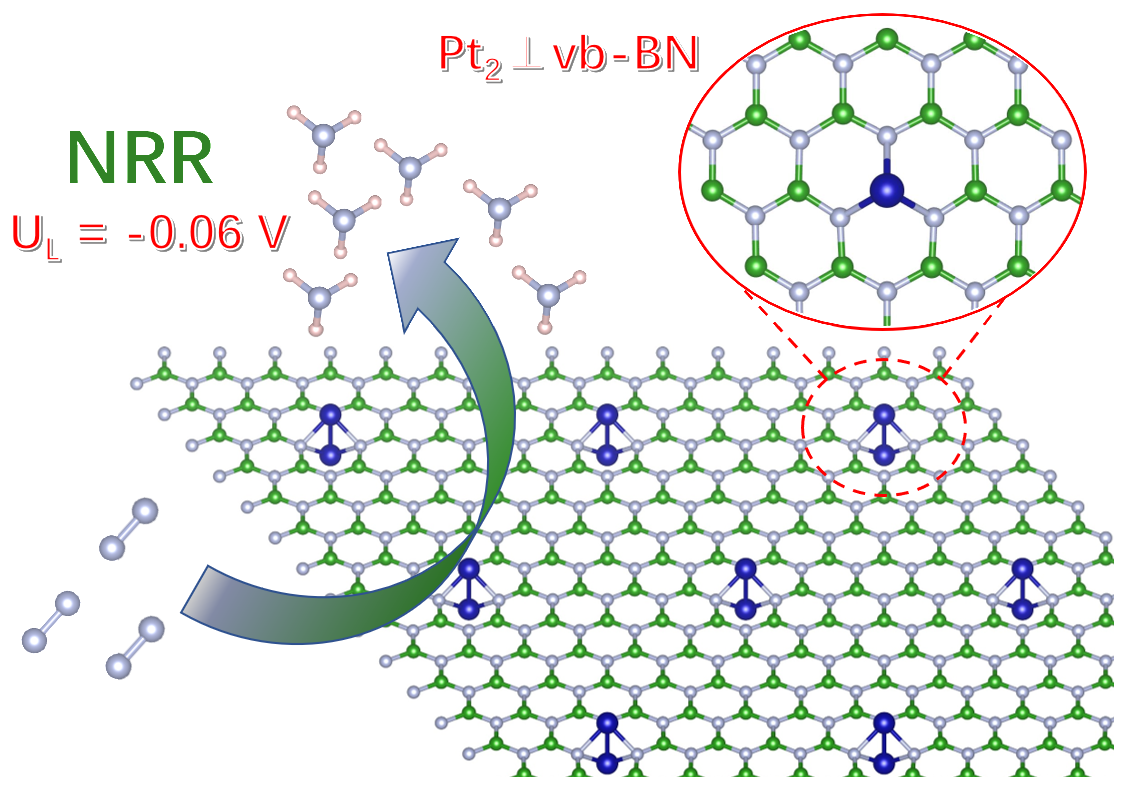
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
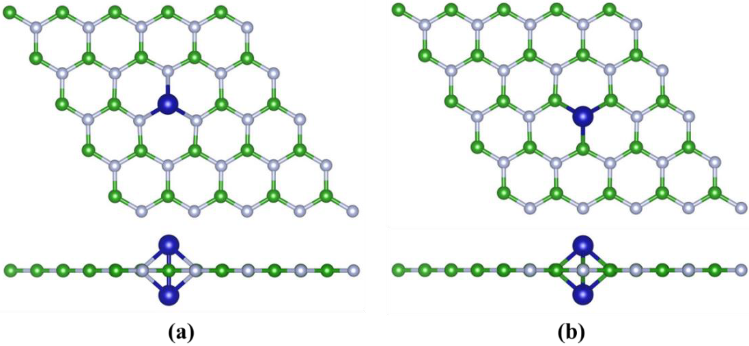
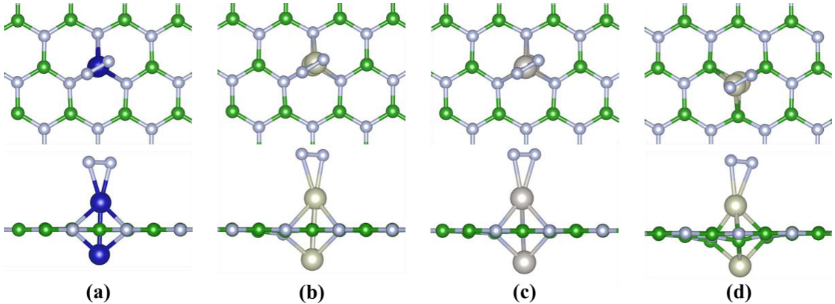
2.1. The Stability, Magnetic and Electronic Properties of DACs
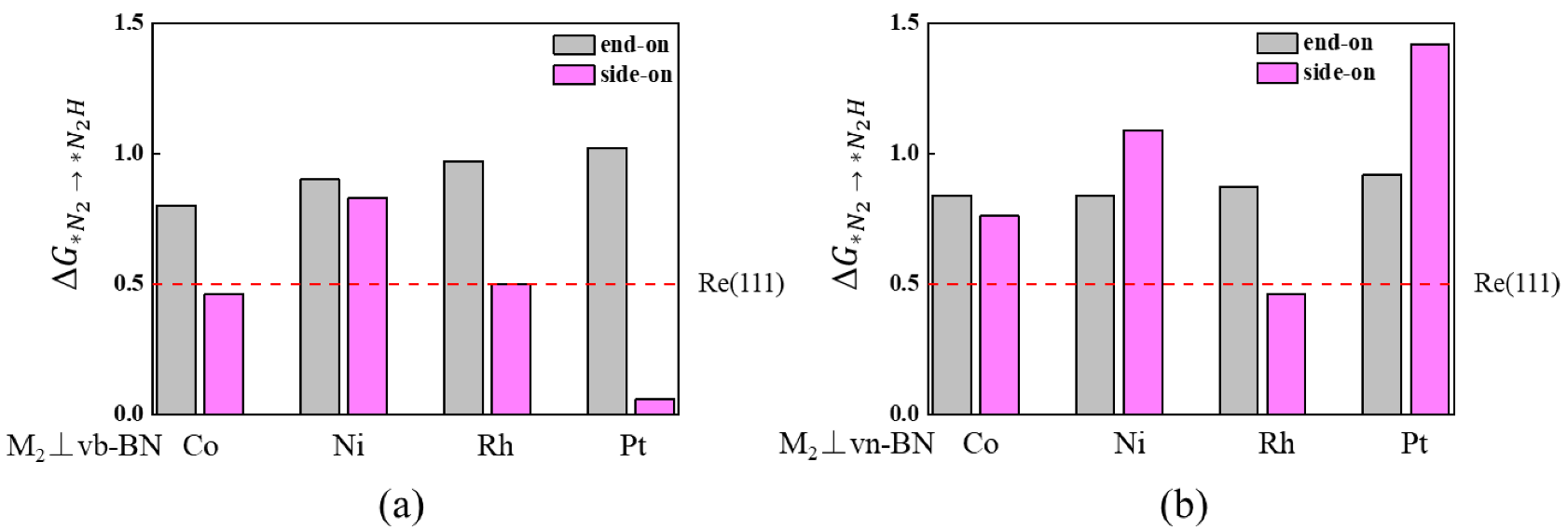
2.2. Screening of the DACs
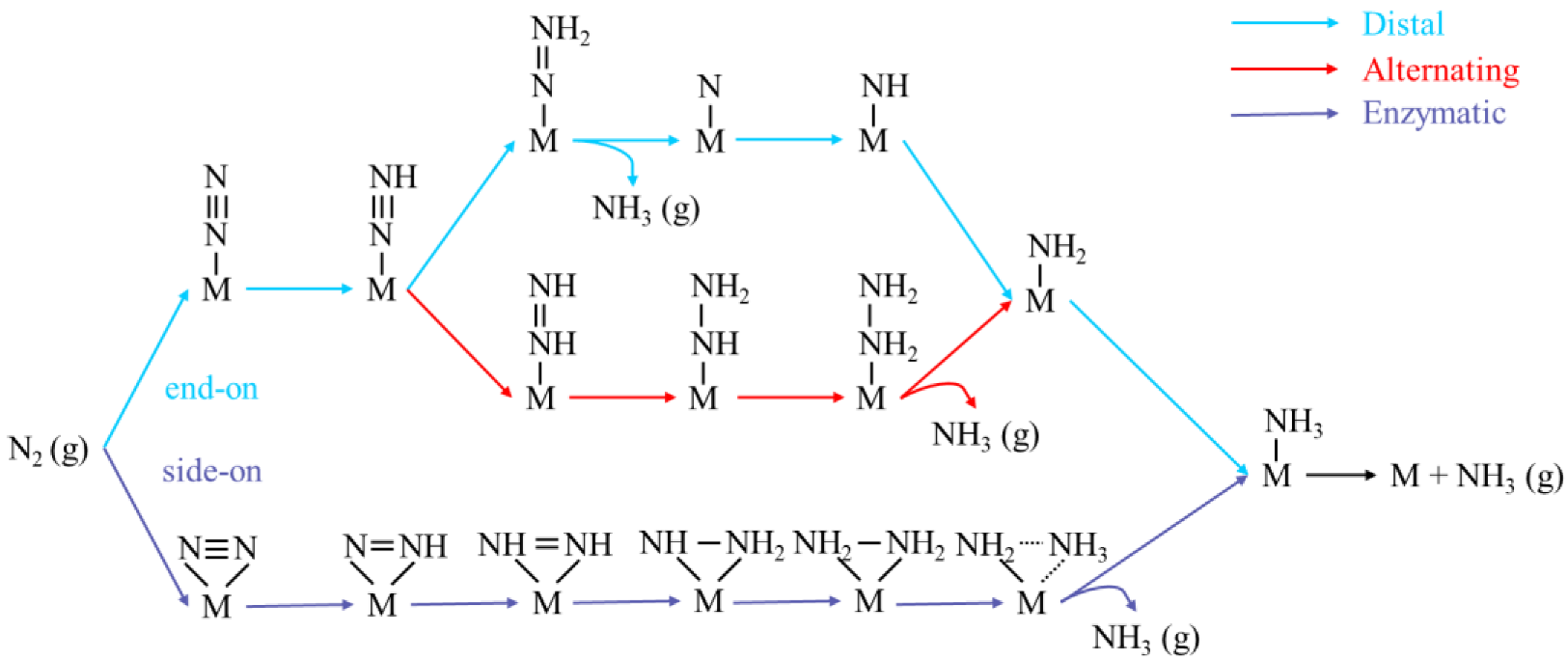
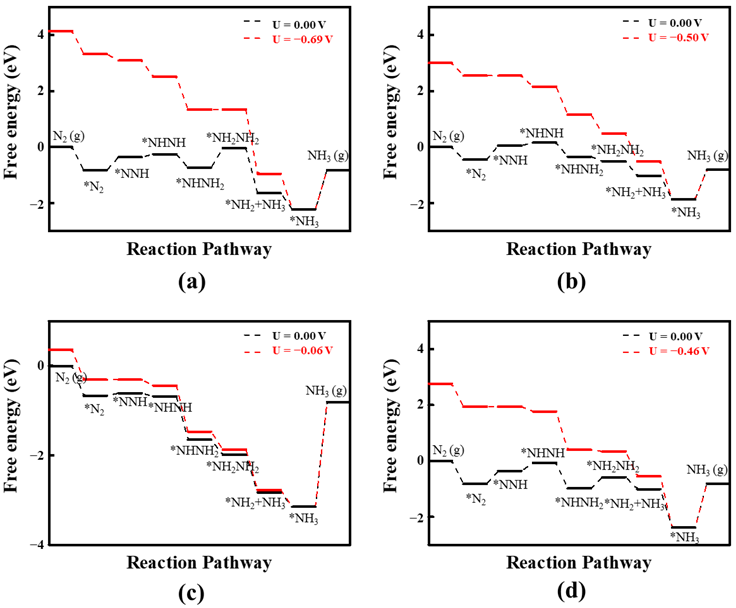
2.3. The Reaction Pathway of NRR on Four Promising DACs
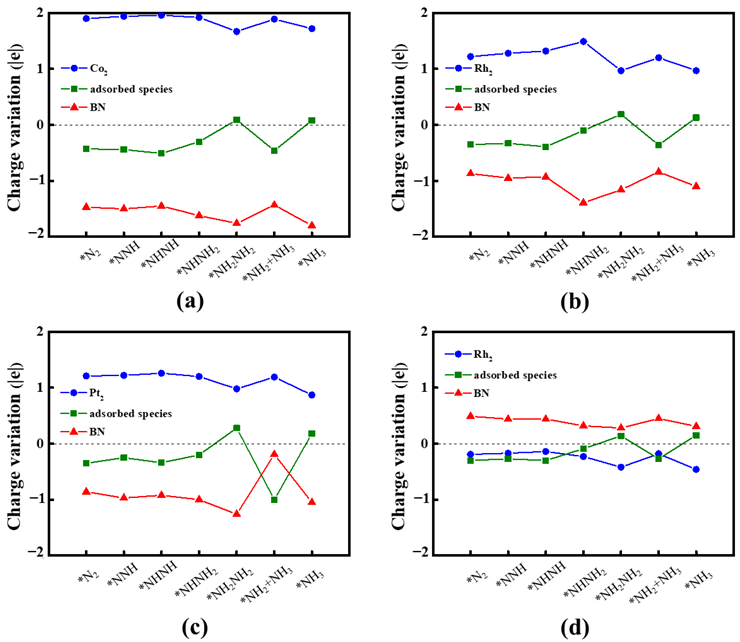
2.4. Origin of NRR Activity
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, H.; Xu, G.; Zhang, L.; Wang, W.; Miao, W.; Chen, K.; Cheng, L.; Li, Y.; Han, S. Ultrafine cobalt nanoparticles supported on carbon nanospheres for hydrolysis of sodium borohydride. Renew. Energy 2020, 162, 345–354. [Google Scholar] [CrossRef]
- Rapson, T.D.; Gregg, C.M.; Allen, R.S.; Ju, H.; Doherty, C.M.; Mulet, X.; Giddey, S.; Wood, C.C. Insights into nitrogenase bioelectrocatalysis for green ammonia production. ChemSusChem 2020, 13, 4856–4865. [Google Scholar] [CrossRef] [PubMed]
- Karl, D.; Letelier, R.; Tupas, L.; Dore, J.; Christian, J.; Hebel, D. The role of nitrogen fixation in biogeochemical cycling in the subtropical north pacific ocean. Nature 1997, 388, 533. [Google Scholar] [CrossRef]
- Hoffman, B.M.; Lukoyanov, D.; Yang, Z.-Y.; Dean, D.R.; Seefeldt, L.C. Mechanism of nitrogen fixation by nitrogenase: The next stage. Chem. Rev. 2014, 114, 4041–4062. [Google Scholar] [CrossRef]
- Guo, X.; Gu, J.; Lin, S.; Zhang, S.; Chen, Z.; Huang, S. Tackling the activity and selectivity challenges of electrocatalysts toward the nitrogen reduction reaction via atomically dispersed biatom catalysts. J. Am. Chem. Soc. 2020, 142, 5709–5721. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, A.; Zhang, Z.; Zhou, Z. Double-atom catalysts: Transition metal dimeranchored C2N monolayers as N2 fixation electrocatalysts. J. Mater. Chem. A 2018, 6, 18599–18604. [Google Scholar] [CrossRef]
- Erisman, J.W.; Sutton, M.A.; Galloway, J.; Klimont, Z.; Winiwarter, W. How a century of ammonia synthesis changed the world. Nat. Geosci. 2008, 1, 636. [Google Scholar] [CrossRef]
- Ferrara, M.; Melchionna, M.; Fornasiero, P.; Bevilacqua, M. The role of structured carbon in downsized transition metal-based electrocatalysts toward a green nitrogen fixation. Catalysts 2021, 11, 1529. [Google Scholar] [CrossRef]
- Ham, C.J.M.; Koper, M.T.M.; Hetterscheid, D.G.H. Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 2014, 43, 5183–5191. [Google Scholar]
- Tanabe, Y.; Nishibayashi, Y. Developing more sustainable processes for ammonia synthesis. Coord. Chem. Rev. 2013, 257, 2551–2564. [Google Scholar] [CrossRef]
- Wang, S.; Ichihara, F.; Pang, H.; Chen, H.; Ye, J. Nitrogen fixation reaction derived from nanostructured catalytic materials. Adv. Funct. Mater. 2018, 28, 1803309. [Google Scholar] [CrossRef]
- Kyriakou, V.; Garagounis, I.; Vasileiou, E.; Vourros, A.; Stoukides, M. Progress in the electrochemical synthesis of ammonia. Catal. Today 2017, 286, 2–13. [Google Scholar] [CrossRef]
- Liu, X.; Jiao, Y.; Zheng, Y.; Qiao, S.-Z. Isolated boron sites for electroreduction of dinitrogen to ammonia. ACS Catal. 2020, 10, 1847–1854. [Google Scholar] [CrossRef]
- Li, L.; Tang, C.; Xia, B.; Jin, H.; Zheng, Y.; Qiao, S.-Z. Two-dimensional mosaic bismuth nanosheets for highly selective ambient electrocatalytic nitrogen reduction. ACS Catal. 2019, 9, 2902–2908. [Google Scholar] [CrossRef]
- Yan, Z.; Ji, M.; Xia, J.; Zhu, H. Recent advanced materials for electrochemical and photoelectrochemical synthesis of ammonia from dinitrogen: One step closer to a sustainable energy future. Adv. Energy Mater. 2020, 10, 1902020. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Jeong, H.; Shin, S.; Lee, H. Heterogeneous atomic catalysts overcoming the limitations of single-atom catalysts. ACS Nano 2020, 14, 14355–14374. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Li, F.; Zhao, J.; Chen, Z. Revisiting catalytic performance of supported metal dimers for oxygen reduction reaction via magnetic coupling from first principles. Adv. Powder Mater. 2022, 1, 100031. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, Y.; Zhang, Y.-X.; Niu, Z. Dual-atom catalysts: Controllable synthesis and electrocatalytic applications. Sci. China Chem. 2021, 64, 1908–1922. [Google Scholar] [CrossRef]
- Lü, F.; Zhao, S.; Guo, R.; He, J.; Peng, X.; Bao, H.; Fu, J.; Han, L.; Qi, G.; Luo, J.; et al. Nitrogen-coordinated single Fe sites for efficient electrocatalytic N2 fixation in neutral media. Nano Energy 2019, 61, 420–427. [Google Scholar] [CrossRef]
- Yin, H.; Li, S.-L.; Gan, L.-Y.; Wang, P. Pt-embedded in monolayer g-C3N4 as a promising single-atom electrocatalyst for ammonia synthesis. J. Mater. Chem. A 2019, 7, 11908–11914. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, Z. Single Mo atom supported on defective boron nitride monolayer as an efficient electrocatalyst for nitrogen fixation: A computational study. J. Am. Chem. Soc. 2017, 139, 12480–12487. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Meng, H.; Li, F.; Zhao, J. Fe3 cluster anchored on the C2N monolayer for efficient electrochemical nitrogen fixation. Catalysts 2020, 10, 974. [Google Scholar] [CrossRef]
- Arachchige, L.J.; Xu, Y.; Dai, Z.; Zhang, X.; Wang, F.; Sun, C. Theoretical investigation of single and double transition metals anchored on graphene monolayer for nitrogen reduction reaction. J. Phys. Chem. C 2020, 124, 15295–15301. [Google Scholar] [CrossRef]
- Ma, L.; Zeng, X.C. Catalytic directional cutting of hexagonal boron nitride: The roles of interface and etching agents. Nano Lett. 2017, 17, 3208–3214. [Google Scholar] [CrossRef]
- Xu, D.; Liu, Y.; Zhao, J.; Cai, Q.; Wang, X. Theoretical study of the deposition of Pt clusters on defective hexagonal boron nitride (h-BN) sheets: Morphologies, electronic structures, and interactions with O. J. Phys. Chem. C 2014, 118, 8868–8876. [Google Scholar] [CrossRef]
- Lin, Y.; Bunker, C.E.; Fernando, K.A.S.; Connell, J.W. Aqueously dispersed silver nanoparticle-decorated boron nitride nanosheets for reusable, Thermal oxidation-resistant surface enhanced raman spectroscopy (SERS) devices. ACS Appl. Mater. Interfaces 2012, 4, 1110–1117. [Google Scholar] [CrossRef]
- Cao, L.; Dai, P.; Tang, J.; Li, D.; Chen, R.; Liu, D.; Gu, X.; Li, L.; Bando, Y.; Ok, Y.S.; et al. Spherical superstructure of boron nitride nanosheets derived from boron-containing metal-organic frameworks. J. Am. Chem. Soc. 2020, 142, 8755–8762. [Google Scholar] [CrossRef]
- Xiong, J.; Di, J.; Zhu, W.; Li, H. Hexagonal boron nitride adsorbent: Synthesis, performance tailoring and applications. J. Energy Chem. 2020, 40, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.; He, R.; Shen, W.; Li, M. Theoretical analysis of oxygen reduction reaction activity on single metal (Ni, Pd, Pt, Cu, Ag, Au) atom supported on defective two-dimensional boron nitride materials. Phys. Chem. Chem. Phys. 2019, 21, 18589–18594. [Google Scholar] [CrossRef]
- Deng, C.; He, R.; Shen, W.; Li, M.; Zhang, T. A single-atom catalyst of cobalt supported on a defective two-dimensional boron nitride material as a promising electrocatalyst for the oxygen reduction reaction: A DFT study. Phys. Chem. Chem. Phys. 2019, 21, 6900–6907. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Meng, Y.; Koso, A.; Yao, Y.; Tang, H.; Xia, S. The mechanism of nitrogen reduction reaction on defective boron nitride (BN) monolayer doped with monatomic Co, Ni, and Mo—A first principles study. Colloid. Surfqce. A 2022, 647, 129072. [Google Scholar] [CrossRef]
- Huang, B.; Wu, Y.; Luo, Y.; Zhou, N. Double atom-anchored defective boron nitride catalyst for efficient electroreduction of CO2 to CH4: A first principles study. Chem. Phys. Lett. 2020, 756, 137852. [Google Scholar] [CrossRef]
- Cui, Q.; Qin, G.; Wang, W.; Sun, L.; Du, A.; Sun, Q. Mo-doped boron nitride monolayer as a promising single-atom electrocatalyst for CO2 conversion. Beilstein J. Nanotechnol. 2019, 10, 540–548. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Xin, Y.; Yuan, J.; Wang, L.; Zhang, W. Direct conversion of methane to methanol on boron nitride-supported copper single atoms. Nanoscale 2022, 14, 5447–5453. [Google Scholar] [CrossRef]
- Yu, L.; Li, F. Metal dimers embedded vertically in defectgraphene as gas sensors: A first-principles study. Phys. Chem. Chem. Phys. 2022, 24, 9842–9847. [Google Scholar] [CrossRef]
- Yu, L.; Li, F.; Huang, J.; Sumpter, B.G.; Mustain, W.E.; Chen, Z. Double-atom catalysts featuring inverse sandwich structure for CO2 reduction reaction: A synergetic first-principles and machine learning investigation. Adv. Funct. Mater. 2022. under review. [Google Scholar]
- Si, M.S.; Xue, D.S. Magnetic properties of vacancies in a graphitic boron nitride sheet by first-principles pseudopotential calculations. Phys. Rev. B 2007, 75, 193409. [Google Scholar] [CrossRef]
- Zeng, H.; Liu, X.; Chen, F.; Chen, Z.; Fan, X.; Lau, W. Single atoms on a nitrogen-doped boron phosphide monolayer: A new promising bifunctional electrocatalyst for ORR and OER. ACS Appl. Mater. Interfaces 2020, 12, 52549–52559. [Google Scholar] [CrossRef]
- Ali, S.; Haneef, M.; Akbar, J.; Ullah, I.; Ullah, S.; Samad, A. Single Au atom supported defect mediated boron nitride monolayer as an efficient catalyst for acetylene hydrochlorination: A first principles study. Mol. Catal. 2021, 511, 111753. [Google Scholar] [CrossRef]
- Chatt, J.; Dilworth, J.R.; Richards, R.L. Recent advances in the chemistry of nitrogen fixation. Chem. Rev. 1978, 78, 589–625. [Google Scholar] [CrossRef]
- Seefeldt, L.C.; Hoffman, B.M.; Dean, D.R. Mechanism of Mo-dependent nitrogenase. Annu. Rev. Biochem. 2009, 78, 701–722. [Google Scholar] [CrossRef] [PubMed]
- Hinnemann, B.; Nørskov, J.K. Catalysis by enzymes: The biological ammonia synthesis. Top. Catal. 2006, 37, 55–70. [Google Scholar] [CrossRef]
- Zheng, G.; Li, L.; Hao, S.; Zhang, X.; Tian, Z.; Chen, L. Double atom catalysts: Heteronuclear transition metal dimer anchored on nitrogen-doped graphene as superior electrocatalyst for nitrogen reduction reaction. Adv. Theory Simul. 2020, 3, 2000190. [Google Scholar] [CrossRef]
- Zheng, G.; Li, L.; Tian, Z.; Zhang, X.; Chen, L. Heterogeneous single-cluster catalysts (Mn3, Fe3, Co3, and Mo3) supported on nitrogen-doped graphene for robust electrochemical nitrogen reduction. J. Energy Chem. 2021, 54, 612–619. [Google Scholar] [CrossRef]
- Montoya, J.H.; Tsai, C.; Vojvodic, A.; Nørskov, J.K. The challenge of electrochemical ammonia synthesis: A new perspective on the role of nitrogen scaling relations. ChemSusChem 2015, 8, 2180–2186. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Back, S.; Kim, N.-Y.; Lim, J.; Kim, Y.-H.; Jung, Y. Suppression of hydrogen evolution reaction in electrochemical N2 reduction using single-atom catalysts: A computational guideline. ACS Catal. 2018, 8, 7517–7525. [Google Scholar] [CrossRef]
- Ling, C.; Shi, L.; Ouyang, Y.; Zeng, X.C.; Wang, J. Nanosheet supported single-metal atom bifunctional catalyst for overall water splitting. Nano Lett. 2017, 17, 5133–5139. [Google Scholar] [CrossRef]
- Li, F.; Liu, X.; Chen, Z. 1+1ʹ > 2: Heteronuclear bi-atom catalyst outperforms its homonuclear counterparts for CO oxidation. Small Methods 2019, 3, 1800480. [Google Scholar] [CrossRef]
- Kresse, G.G.; Furthmüller, J.J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.G.; Furthmüller, J.J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Bučko, T.; Hafner, J.; Lebègue., S.; Ángyán, J.G. Improved description of the structure of molecular and layered crystals: Ab initio DFT calculations with van der Waals corrections. J. Phys. Chem. A 2010, 114, 11814–11824. [Google Scholar] [CrossRef] [PubMed]
- Mathew, K.; Sundararaman, R.; Letchworth-Weaver, K.; Arias, T.A.; Hennig, R.G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkelman, G.; Arnaldsson, A.; Jόnsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M.E. Nose–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635. [Google Scholar] [CrossRef]
- Impeng, S.; Junkaew, A.; Maitarad, P.; Kungwan, N.; Zhang, D.; Shi, L.; Namuangruk, S. A MnN4 moiety embedded graphene as a magnetic gas sensor for CO detection: A first principle study. Appl. Surf. Sci. 2019, 473, 820–827. [Google Scholar] [CrossRef]
- Fu, L.; Wang, R.; Zhao, C.; Huo, J.; He, C.; Kim, K.-H.; Zhang, W. Construction of Cr-embedded graphyne electrocatalyst for highly selective reduction of CO2 to CH4: A DFT study. Chem. Eng. J. 2021, 414, 128857. [Google Scholar] [CrossRef]
- Ling, C.; Zhang, Y.; Li, Q.; Bai, X.; Shi, L.; Wang, J. New mechanism for N2 reduction: The essential role of surface hydrogenation. J. Am. Chem. Soc. 2019, 141, 18264–18270. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Skulason, E.; Bligaard, T.; Gudmundsdottir, S.; Studt, F.; Rossmeisl, J.; Abild-Pedersen, F.; Vegge, T.; Jonssonac, H.; Nørskov, J.K. A theoretical evaluation of possible transition metal electro-catalysts for N2 reduction. Phys. Chem. Chem. Phys. 2012, 14, 1235–1245. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, L.; Li, F. Pt2 Dimer Anchored Vertically in Defective BN Monolayer as an Efficient Catalyst for N2 Reduction: A DFT Study. Catalysts 2022, 12, 1387. https://doi.org/10.3390/catal12111387
Yu L, Li F. Pt2 Dimer Anchored Vertically in Defective BN Monolayer as an Efficient Catalyst for N2 Reduction: A DFT Study. Catalysts. 2022; 12(11):1387. https://doi.org/10.3390/catal12111387
Chicago/Turabian StyleYu, Linke, and Fengyu Li. 2022. "Pt2 Dimer Anchored Vertically in Defective BN Monolayer as an Efficient Catalyst for N2 Reduction: A DFT Study" Catalysts 12, no. 11: 1387. https://doi.org/10.3390/catal12111387