
From Cell-Free Protein Synthesis to Whole-Cell Biotransformation: Screening and Identification of Novel α-Ketoglutarate-Dependent Dioxygenases for Preparative-Scale Synthesis of Hydroxy-l-Lysine

 , and
, and
Abstract
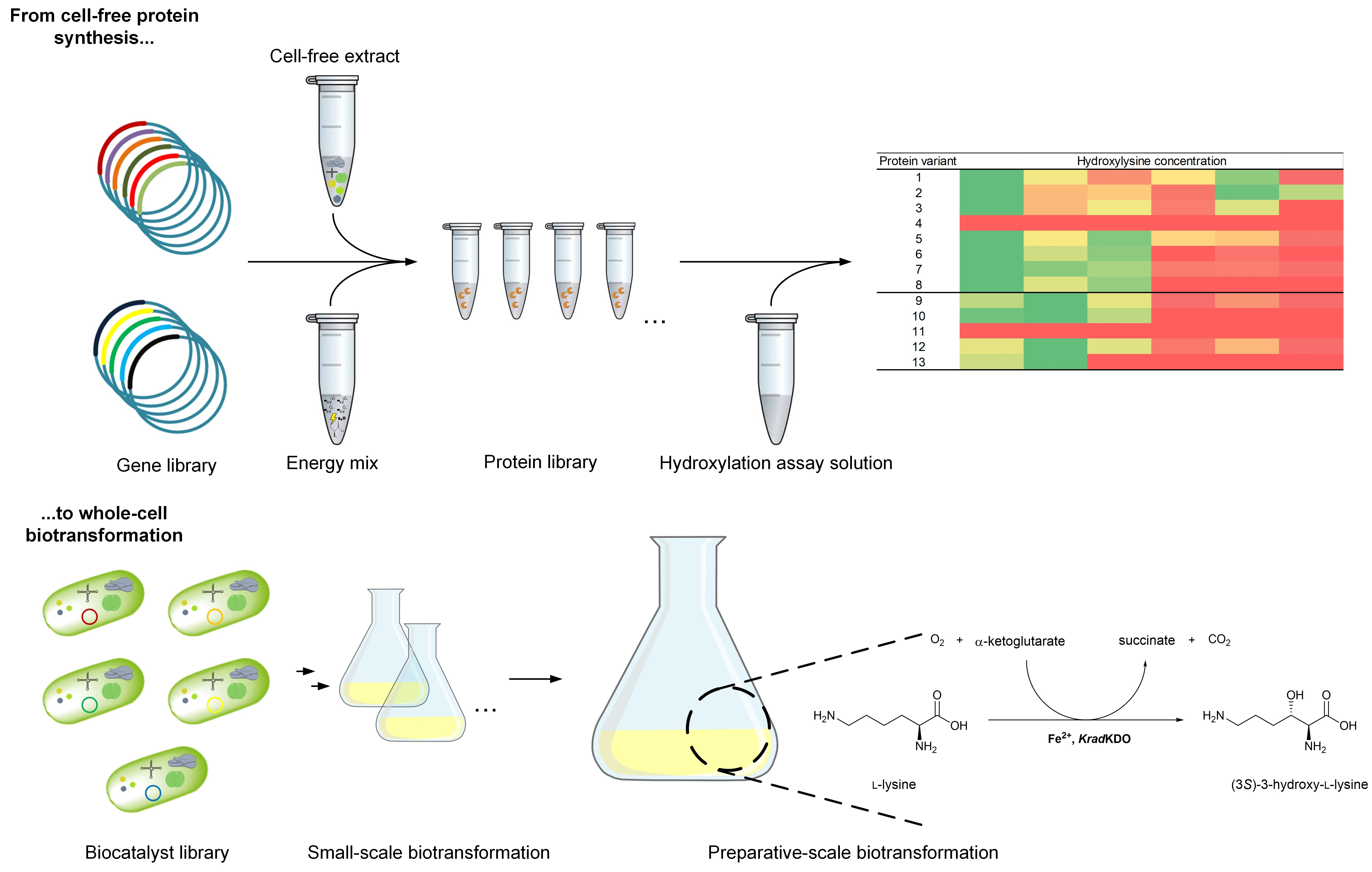
: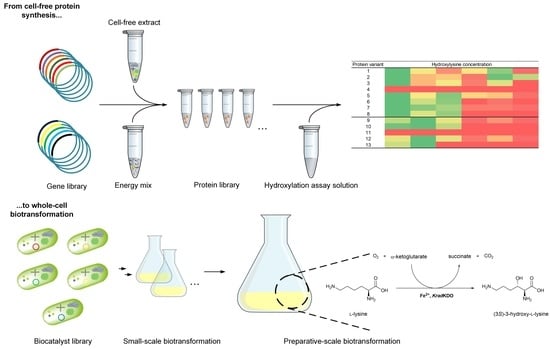
1. Introduction
2. Results
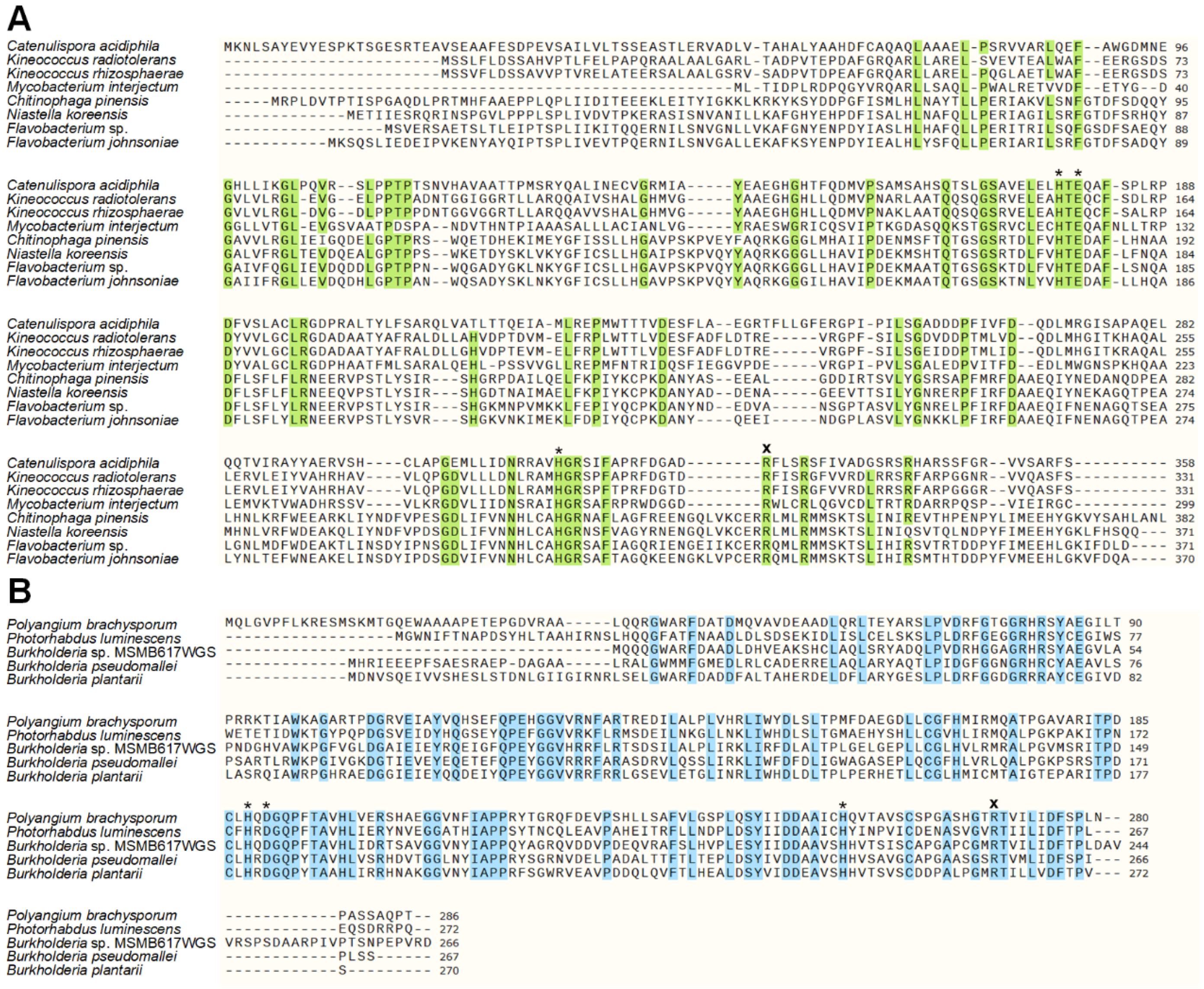
2.1. Sequence Similarity Search for Novel KDOs in Bacteria
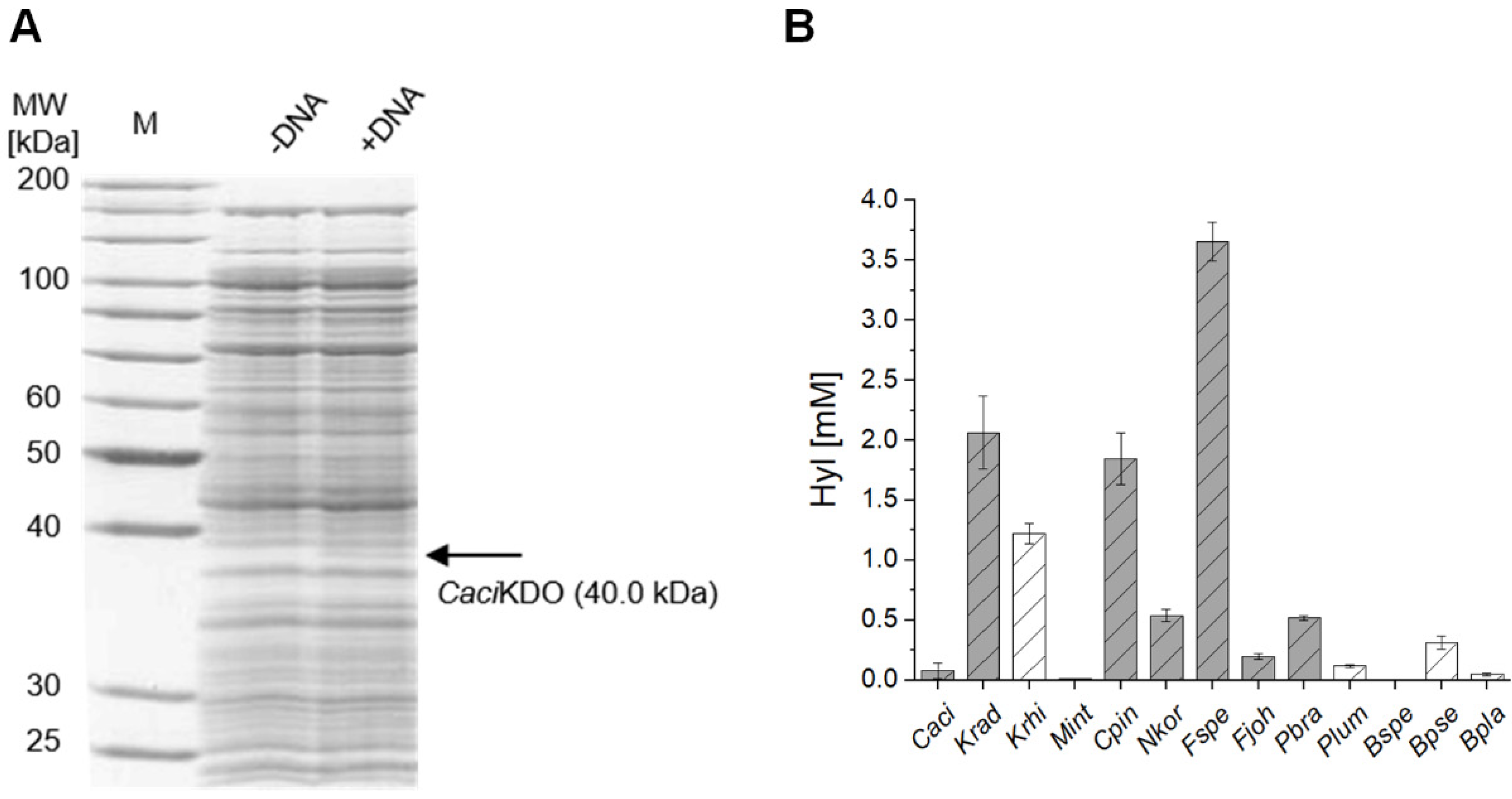
2.2. Cell-Free Protein Synthesis Identifies Novel KDOs
2.3. Chaperone-Assisted Expression Can Improve the Productivity of Cell-Free Synthesized KDOs
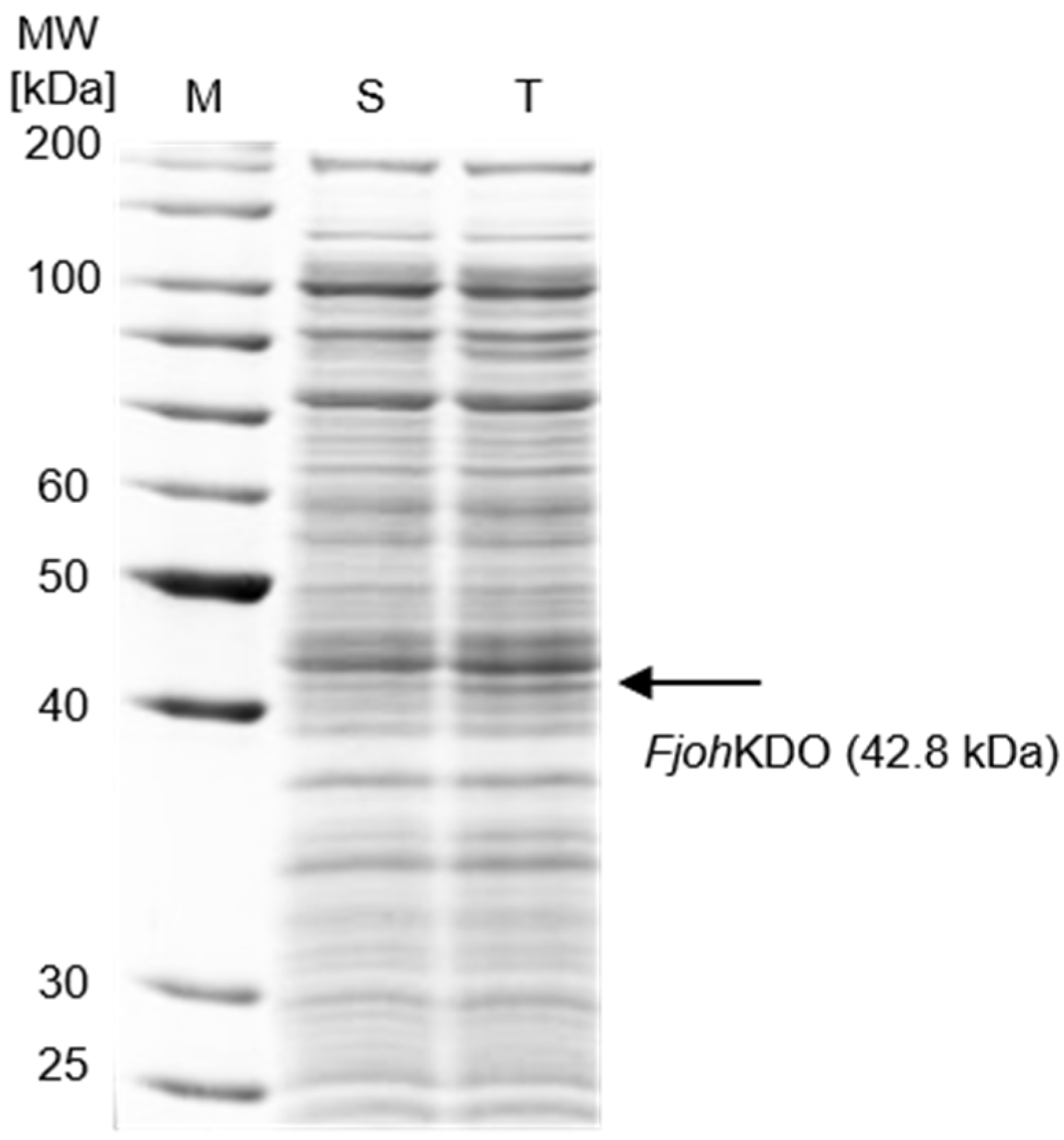
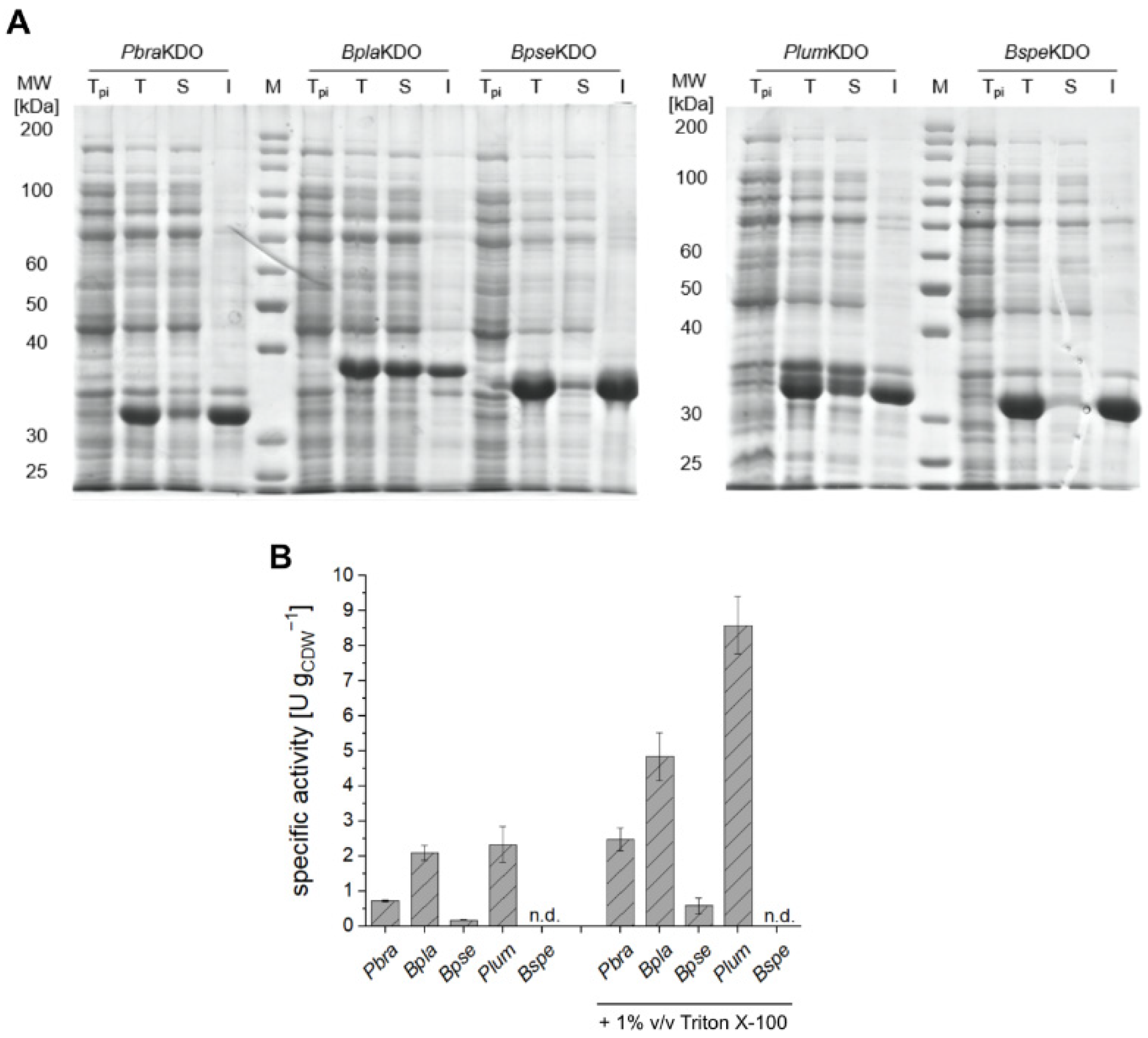
2.4. Heterologous Expression of Novel KDOs
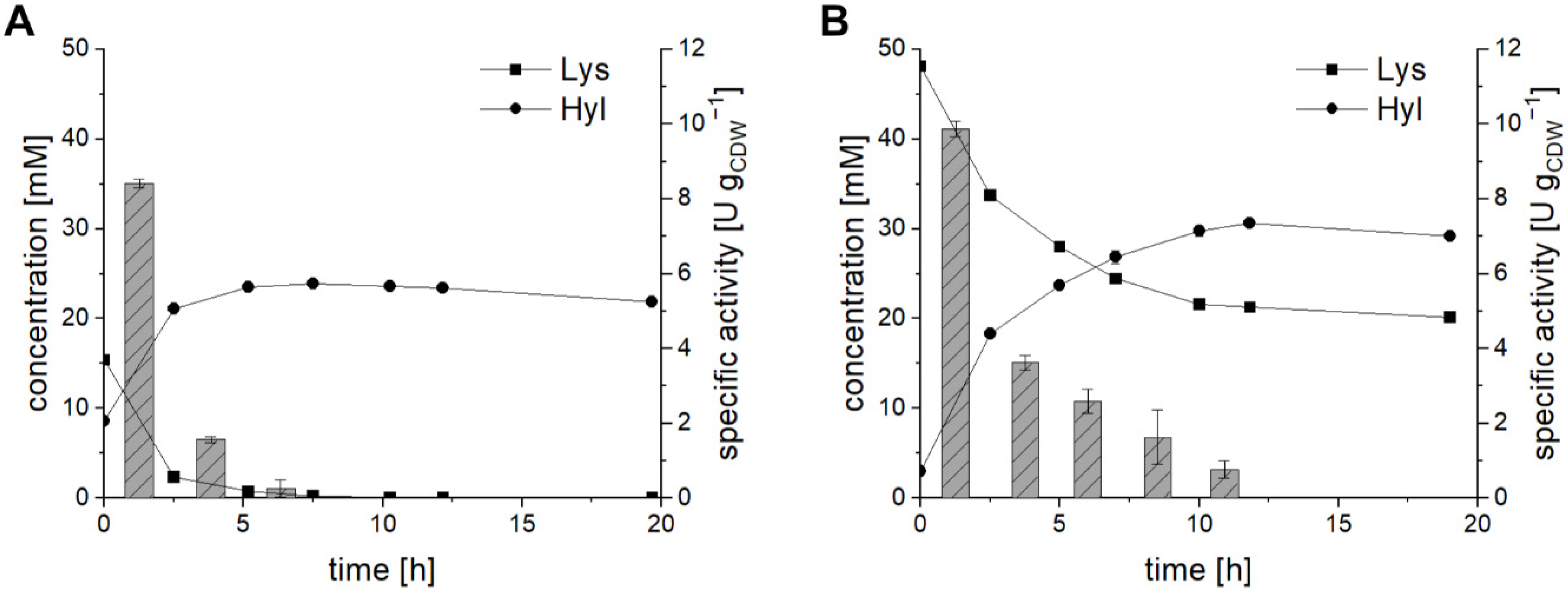
2.5. Preparative-Scale Production of Hydroxy-l-Lysine
3. Materials and Methods
3.1. Chemicals/Strains and Plasmids
3.2. Cloning

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Information | ||
|---|---|---|---|
| Escherichia coli DH5α | F-, Φ80dlacZΔM15, Δ(lacZYA-argF)U169, deoR, recA1, endA1, hsdR17(rK-mK+), phoA, supE44, λ-, thi 1, gyrA96, relA1 | ||
| Escherichia coli BL21 (DE3) | F-, ompT, hsdSB(rB-mB-), gal (c1875, ind1, Sam7, nin5, lacUV5-T7 gene1), dcm (DE3) | ||
| Kineococcusradiotolerans | wild-type, DSM No. 14245 | ||
| Kineococcusrhizospharae | wild-type, DSM No. 19711 | ||
| Plasmid | Protein | Description | Reference |
| pET-22b(+)-CaciKDO | CaciKDO | KDO, l-Lysine 3-hydroxylase | [3] |
| pET-22b(+)-CpinKDO | CpinKDO | KDO, l-Lysine 4-hydroxylase | [3] |
| pET-22b(+)-FjohKDO | FjohKDO | KDO, l-Lysine 4-hydroxylase | [3] |
| pET-22b(+)-NkorKDO | NkorKDO | KDO, l-Lysine 4-hydroxylase | [3] |
| pET-22b(+)-FspeKDO | FspeKDO | KDO, l-Lysine 4-hydroxylase | [3] |
| pET-24a(+)-KradKDO | KradKDO | KDO, l-Lysine 3-hydroxylase | This study |
| pET-24a(+)-KrhiKDO | KrhiKDO | Putative KDO | This study |
| pET-24a(+)-MintKDO | MintKDO | Putative KDO | This study |
| pET-24a(+)-PbraKDO | PbraKDO | KDO, l-Lysine 4-hydroxylase | This study |
| pET-24a(+)-BplaKDO | BplaKDO | Putative KDO | This study |
| pET-24a(+)-BpseKDO | BpseKDO | Putative KDO | This study |
| pET-24a(+)-PlumKDO | PlumKDO | Putative KDO | This study |
| pET-24a(+)-BspeKDO | BspeKDO | Putative KDO | This study |
| pAR1219 | T7RNAP | T7 RNA-polymerase | [33] |
| pG-KJE8 | DnaK, DnaJ, GrpE, GroES, GroEL | Molecular chaperones | [24] |
| pKJE7 | DnaK, DnaJ, GrpE | Molecular chaperones | [24] |
| pGro7 | GroES, GroEL | Molecular chaperones | [24] |
| pG-Tf2 | GroES, GroEL, tig | Molecular chaperones | [34] |
| pTf16 | tig | Molecular chaperones | [34] |
3.3. E. coli Extract Preparation
3.4. Cell-Free Protein Synthesis (CFPS)
3.5. In Vitro Biotransformations
3.6. Resting-Cell Biotransformations
3.7. Preparative-Scale Biotransformation
3.8. Analytics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peters, C.; Buller, R. Industrial Application of 2-Oxoglutarate-Dependent Oxygenases. Catalysts 2019, 9, 221. [Google Scholar] [CrossRef] [Green Version]
- Herr, C.Q.; Hausinger, R.P. Amazing Diversity in Biochemical Roles of Fe(II)/2-Oxoglutarate Oxygenases. Trends Biochem. Sci. 2018, 43, 517–532. [Google Scholar] [CrossRef]
- Baud, D.; Saaidi, P.-L.; Monfleur, A.; Harari, M.; Cuccaro, J.; Fossey, A.; Besnard, M.; Debard, A.; Mariage, A.; Pellouin, V.; et al. Synthesis of Mono- and Dihydroxylated Amino Acids with New α-Ketoglutarate-Dependent Dioxygenases: Biocatalytic Oxidation of C-H Bonds. ChemCatChem 2014, 6, 3012–3017. [Google Scholar] [CrossRef] [Green Version]
- Hara, R.; Yamagata, K.; Miyake, R.; Kawabata, H.; Uehara, H.; Kino, K. Discovery of Lysine Hydroxylases in the Clavaminic Acid Synthase-Like Superfamily for Efficient Hydroxylysine Bioproduction. Appl. Environ. Microbiol. 2017, 83, e00693-17. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Zhu, M.; Song, Z.; Li, C.; Wang, Y.; Zhu, Z.; Sun, D.; Lu, F.; Qin, H.-M. Reshaping the Binding Pocket of Lysine Hydroxylase for Enhanced Activity. ACS Catal. 2020, 10, 13946–13956. [Google Scholar] [CrossRef]
- Marin, J.; Didierjean, C.; Aubry, A.; Casimir, J.R.; Briand, J.P.; Guichard, G. Synthesis of Enantiopure 4-Hydroxypipecolate and 4-Hydroxylysine Derivatives from a Common 4,6-Dioxopiperidinecarboxylate Precursor. J. Org. Chem. 2004, 69, 130–141. [Google Scholar] [CrossRef]
- Zhang, X.; King-Smith, E.; Renata, H. Total Synthesis of Tambromycin by Combining Chemocatalytic and Biocatalytic C−H Functionalization. Angew. Chem. Int. Ed. 2018, 57, 5037–5041. [Google Scholar] [CrossRef]
- Amatuni, A.; Shuster, A.; Adibekian, A.; Renata, H. Concise Chemoenzymatic Total Synthesis and Identification of Cellular Targets of Cepafungin I. Cell Chem. Biol. 2020, 27, 1318–1326.e18. [Google Scholar] [CrossRef] [PubMed]
- Amatuni, A.; Renata, H. Identification of a Lysine 4-Hydroxylase from the Glidobactin Biosynthesis and Evaluation of Its Biocatalytic Potential. Org. Biomol. Chem. 2019, 17, 1736–1739. [Google Scholar] [CrossRef] [PubMed]
- Baud, D.; Peruch, O.; Saaidi, P.-L.; Fossey, A.; Mariage, A.; Petit, J.-L.; Salanoubat, M.; Vergne-Vaxelaire, C.; de Berardinis, V.; Zaparucha, A. Biocatalytic Approaches towards the Synthesis of Chiral Amino Alcohols from Lysine: Cascade Reactions Combining Alpha-Keto Acid Oxygenase Hydroxylation with Pyridoxal Phosphate- Dependent Decarboxylation. Adv. Synth. Catal. 2017, 359, 1563–1569. [Google Scholar] [CrossRef]
- Fossey-Jouenne, A.; Vergne-Vaxelaire, C.; Zaparucha, A. Enzymatic Cascade Reactions for the Synthesis of Chiral Amino Alcohols from L-Lysine. J. Vis. Exp. 2018, 132, 56926. [Google Scholar] [CrossRef] [PubMed]
- Rolf, J.; Rosenthal, K.; Lütz, S. Application of Cell-Free Protein Synthesis for Faster Biocatalyst Development. Catalysts 2019, 9, 190. [Google Scholar] [CrossRef] [Green Version]
- Quertinmont, L.T.; Orru, R.; Lutz, S. RApid Parallel Protein EvaluatoR (RAPPER), from Gene to Enzyme Function in One Day. Chem. Commun. 2015, 51, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Silverman, A.D.; Karim, A.S.; Jewett, M.C. Cell-Free Gene Expression: An Expanded Repertoire of Applications. Nat. Rev. Genet. 2020, 21, 151–170. [Google Scholar] [CrossRef]
- Rolf, J.; Siedentop, R.; Lütz, S.; Rosenthal, K. Screening and Identification of Novel CGAS Homologues Using a Combination of in Vitro and In Vivo Protein Synthesis. Int. J. Mol. Sci. 2019, 21, 105. [Google Scholar] [CrossRef] [Green Version]
- Stech, M.; Kubick, S. Cell-Free Synthesis Meets Antibody Production: A Review. Antibodies 2015, 4, 12–33. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.-H.; Kim, D.-M.; Kim, H.-J.; Jun, S.-Y.; Lee, K.-Y.; Kim, H.-J. Cell-Free Production of Aggregation-Prone Proteins in Soluble and Active Forms. Biotechnol. Prog. 2005, 21, 1412–1419. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-J.; Lee, K.-H.; Lim, H.J.; Kim, D.-M. Tandem Cell-Free Protein Synthesis as a Tool for Rapid Screening of Optimal Molecular Chaperones. Biotechnol. J. 2019, 14, 1800523. [Google Scholar] [CrossRef] [PubMed]
- Bastard, K.; Isabet, T.; Stura, E.A.; Legrand, P.; Zaparucha, A. Structural Studies Based on Two Lysine Dioxygenases with Distinct Regioselectivity Brings Insights Into Enzyme Specificity within the Clavaminate Synthase-Like Family. Sci. Rep. 2018, 8, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Yu, Y.; Liang, J.; Zhang, Y.; Bian, X.; Zhi, X.; Ding, X. Reclassification of ’Polyangium Brachysporum’ DSM 7029 as Schlegelella Brevitalea Sp. Nov. Int. J. Syst. Evol. Microbiol. 2019, 69, 2877–2883. [Google Scholar] [CrossRef] [PubMed]
- Koehntop, K.D.; Emerson, J.P.; Que, L. The 2-His-1-Carboxylate Facial Triad: A Versatile Platform for Dioxygen Activation by Mononuclear Non-Heme Iron(II) Enzymes. J. Biol. Inorg. Chem. 2005, 10, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Schinn, S.M.; Broadbent, A.; Bradley, W.T.; Bundy, B.C. Protein Synthesis Directly from PCR: Progress and Applications of Cell-Free Protein Synthesis with Linear DNA. N. Biotechnol. 2016, 33, 480–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishihara, K.; Kanemori, M.; Kitagawa, M.; Yanagi, H.; Yura, T. Chaperone Coexpression Plasmids: Differential and Synergistic Roles of DnaK-DnaJ-GrpE and GroEL-GroES in Assisting Folding of an Allergen of Japanese Cedar Pollen, Cryj2, in Escherichia Coli. Appl. Environ. Microbiol. 1998, 64, 1694–1699. [Google Scholar] [CrossRef] [Green Version]
- Falcioni, F.; Blank, L.M.; Frick, O.; Karau, A.; Bühler, B.; Schmida, A. Proline Availability Regulates Proline-4-Hydroxylase Synthesis and Substrate Uptake in Proline-Hydroxylating Recombinant Escherichia Coli. Appl. Environ. Microbiol. 2013, 79, 3091–3100. [Google Scholar] [CrossRef] [Green Version]
- Hara, R.; Nishikawa, T.; Okuhara, T.; Koketsu, K.; Kino, K. Ectoine Hydroxylase Displays Selective Trans-3-Hydroxylation Activity towards l-Proline. Appl. Microbiol. Biotechnol. 2019, 103, 5689–5698. [Google Scholar] [CrossRef]
- Zhang, C.; Ma, J.; Li, Z.; Liang, Y.; Xu, Q.; Xie, X.; Chen, N. A Strategy for L-Isoleucine Dioxygenase Screening and 4-Hydroxyisoleucine Production by Resting Cells. Bioengineered 2017, 9, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Xu, J.; Jiang, T.; Ge, Y.; Liu, P.; Zhang, M.; Su, Z.; Gao, C.; Ma, C.; Xu, P. Overexpression of Transport Proteins Improves the Production of 5-Aminovalerate from l-Lysine in Escherichia Coli. Sci. Rep. 2016, 6, 30884. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Huang, Y.; Mi, L.; Chen, W.; Wang, D.; Wang, Q. An Economically and Environmentally Acceptable Synthesis of Chiral Drug Intermediate L-Pipecolic Acid from Biomass-Derived Lysine via Artificially Engineered Microbes. J. Ind. Microbiol. Biotechnol. 2018, 45, 405–415. [Google Scholar] [CrossRef]
- Mantri, M.; Zhang, Z.; McDonough, M.A.; Schofield, C.J. Autocatalysed Oxidative Modifications to 2-Oxoglutarate Dependent Oxygenases. FEBS J. 2012, 279, 1563–1575. [Google Scholar] [CrossRef]
- Kadisch, M.; Willrodt, C.; Hillen, M.; Bühler, B.; Schmid, A. Maximizing the Stability of Metabolic Engineering-Derived Whole-Cell Biocatalysts. Biotechnol. J. 2017, 12, 1600170. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic Assembly of DNA Molecules up to Several Hundred Kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Davanloo, P.; Rosenberg, A.H.; Dunn, J.J.; Studier, F.W. Cloning and Expression of the Gene for Bacteriophage T7 RNA Polymerase. Proc. Natl. Acad. Sci. USA 1984, 81, 2035–2039. [Google Scholar] [CrossRef] [Green Version]
- Nishihara, K.; Kanemori, M.; Yanagi, H.; Yura, T. Overexpression of Trigger Factor Prevents Aggregation of Recombinant Proteins in Escherichia Coli. Appl. Environ. Microbiol. 2000, 66, 884–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Meyer, F.; Frey, R.; Ligibel, M.; Sager, E.; Schroer, K.; Snajdrova, R.; Buller, R. Modulating Chemoselectivity in a Fe(II)/α-Ketoglutarate-Dependent Dioxygenase for the Oxidative Modification of a Nonproteinogenic Amino Acid. ACS Catal. 2021, 11, 6261–6269. [Google Scholar] [CrossRef]
- Papadopoulou, A.; Meierhofer, J.; Meyer, F.; Hayashi, T.; Schneider, S.; Sager, E.; Buller, R.M.U. Re-programming and Optimization of a L-proline Cis-4-hydroxylase for the Cis-3-halogenation of Its Native Substrate. ChemCatChem 2021, 1–7. [Google Scholar] [CrossRef]
- Hüttel, W. Biocatalytic Production of Chemical Building Blocks in Technical Scale with α-Ketoglutarate-Dependent Dioxygenases. Chemie-Ingenieur-Technik 2013, 85, 809–817. [Google Scholar] [CrossRef]







| Reference | DnaK, DnaJ, GrpE, GroES, GroEL | DnaK, DnaJ, GrpE | GroES, GroEL | GroES, GroEL, tig | tig | |
|---|---|---|---|---|---|---|
| Caci | 0.08 ± 0.06 | 0.06 ± 0.00 | 0.03 ± 0.01 | 0.05 ± 0.00 | 0.07 ± 0.01 | 0.02 ± 0.00 |
| Krad | 2.07 ± 0.30 | 0.83 ± 0.54 | 0.99 ± 0.34 | 0.17 ± 0.05 | 2.02 ± 0.03 | 1.65 ± 0.09 |
| Krhi | 1.22 ± 0.08 | 0.15 ± 0.02 | 0.32 ± 0.01 | 0.06 ± 0.01 | 0.46 ± 0.01 | 0.02 ± 0.00 |
| Mint | 0.01 ± 0.00 | 0.02 ± 0.00 | 0.02 ± 0.00 | 0.01 ± 0.00 | 0.02 ± 0.00 | 0.01 ± 0.00 |
| Cpin | 1.85 ± 0.21 | 0.48 ± 0.24 | 1.55 ± 0.41 | 0.34 ± 0.01 | 0.30 ± 0.02 | 0.05 ± 0.00 |
| Nkor | 0.54 ± 0.05 | 0.28 ± 0.08 | 0.44 ± 0.04 | 0.02 ± 0.00 | 0.03 ± 0.00 | 0.01 ± 0.00 |
| Fspe | 3.66 ± 0.16 | 3.13 ± 0.98 | 2.68 ± 0.20 | 0.23 ± 0.01 | 0.27 ± 0.02 | 0.09 ± 0.00 |
| Fjoh | 0.20 ± 0.02 | 0.08 ± 0.01 | 0.15 ± 0.01 | 0.01 ± 0.00 | 0.02 ± 0.00 | 0.01 ± 0.00 |
| Pbra | 0.52 ± 0.02 | 0.95 ± 0.20 | 0.31 ± 0.07 | 0.03 ± 0.02 | 0.08 ± 0.04 | 0.03 ± 0.00 |
| Plum | 0.12 ± 0.01 | 0.12 ± 0.01 | 0.08 ± 0.02 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.00 ± 0.00 |
| Bspe | 0.00 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 |
| Bpse | 0.31 ± 0.06 | 0.72 ± 0.12 | 0.33 ± 0.01 | 0.05 ± 0.01 | 0.15 ± 0.09 | 0.03 ± 0.02 |
| Bpla | 0.05 ± 0.01 | 0.11 ± 0.01 | 0.02 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rolf, J.; Nerke, P.; Britner, A.; Krick, S.; Lütz, S.; Rosenthal, K. From Cell-Free Protein Synthesis to Whole-Cell Biotransformation: Screening and Identification of Novel α-Ketoglutarate-Dependent Dioxygenases for Preparative-Scale Synthesis of Hydroxy-l-Lysine. Catalysts 2021, 11, 1038. https://doi.org/10.3390/catal11091038
Rolf J, Nerke P, Britner A, Krick S, Lütz S, Rosenthal K. From Cell-Free Protein Synthesis to Whole-Cell Biotransformation: Screening and Identification of Novel α-Ketoglutarate-Dependent Dioxygenases for Preparative-Scale Synthesis of Hydroxy-l-Lysine. Catalysts. 2021; 11(9):1038. https://doi.org/10.3390/catal11091038
Chicago/Turabian StyleRolf, Jascha, Philipp Nerke, Annette Britner, Sebastian Krick, Stephan Lütz, and Katrin Rosenthal. 2021. "From Cell-Free Protein Synthesis to Whole-Cell Biotransformation: Screening and Identification of Novel α-Ketoglutarate-Dependent Dioxygenases for Preparative-Scale Synthesis of Hydroxy-l-Lysine" Catalysts 11, no. 9: 1038. https://doi.org/10.3390/catal11091038