
Enzymatic Hydrogen Electrosynthesis at Enhanced Current Density Using a Redox Polymer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Optimization of Electrochemical Buffer Conditions
2.2. Hydrogenase Activity Using Methyl Viologen Assays
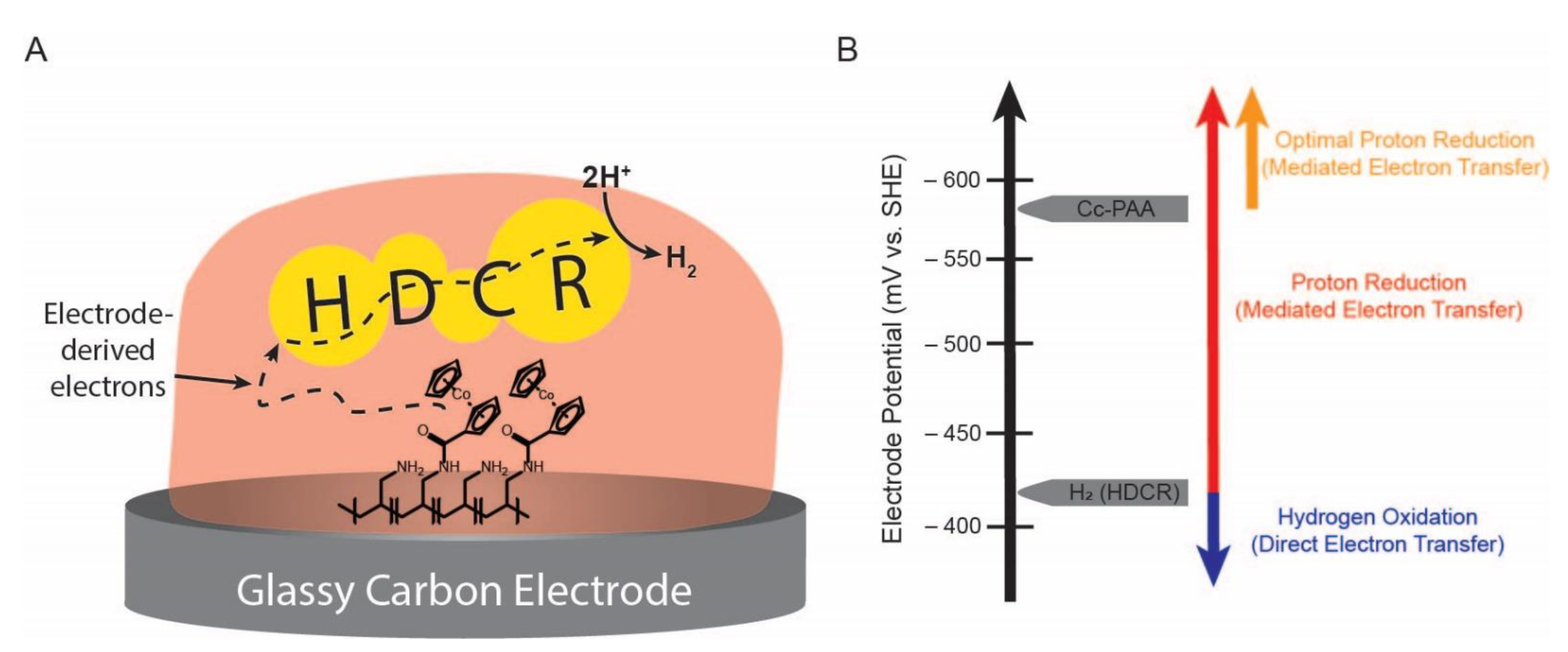
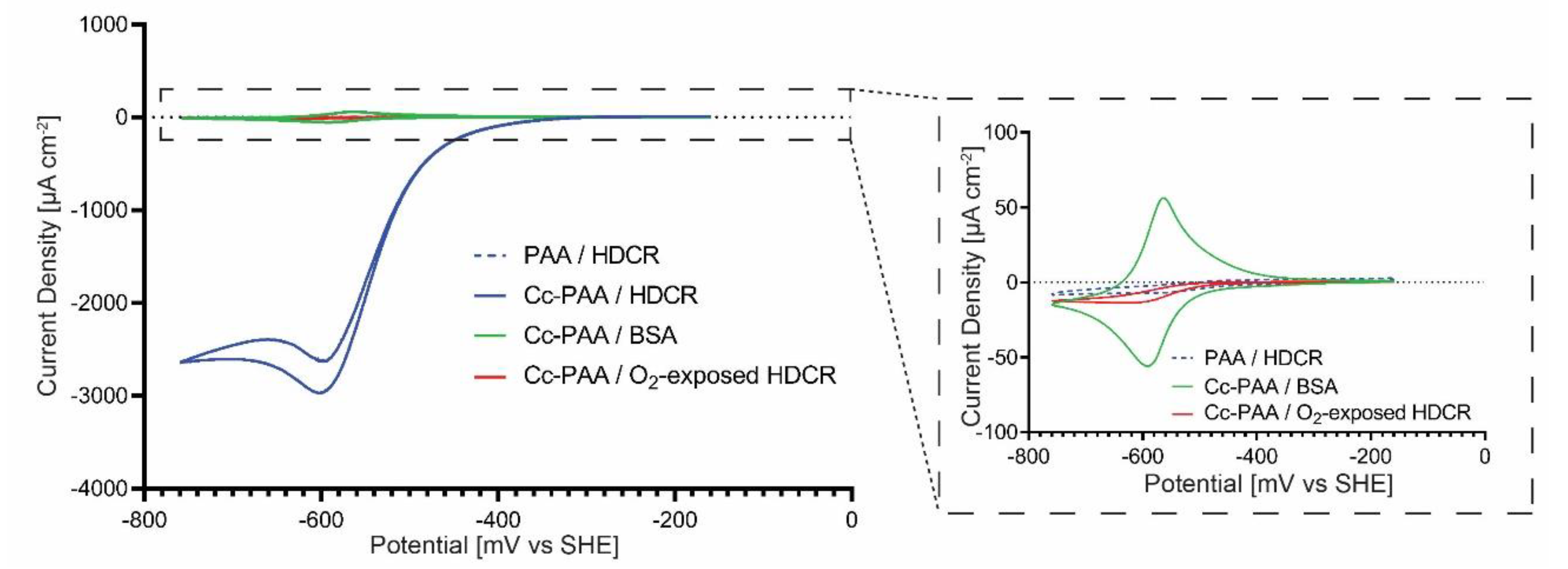
2.3. Cobaltocene-Mediated Hydrogen Evolution in Cyclic Voltammetry Experiments
2.4. Stability of Cc-PAA at Elevated Temperatures
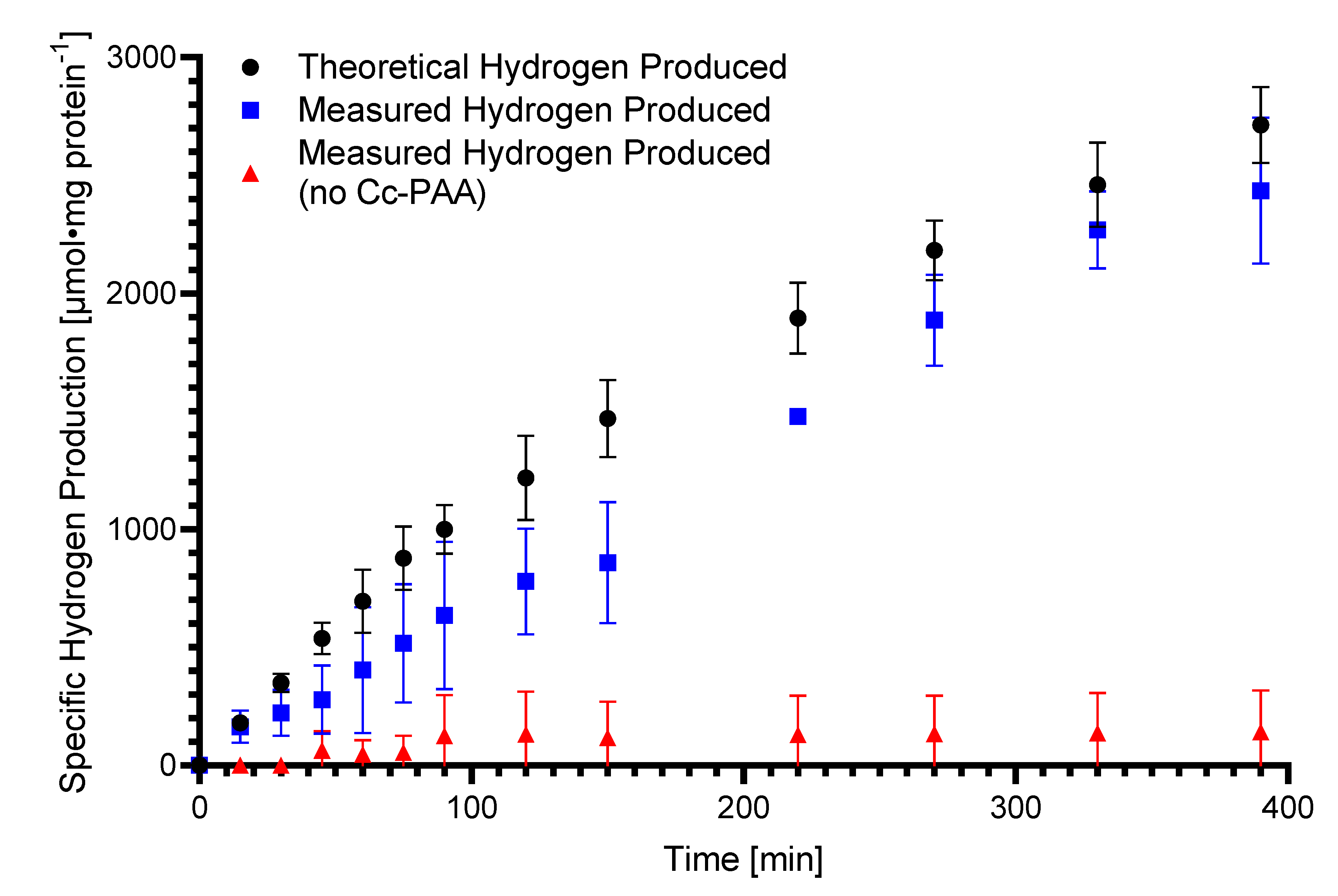
2.5. Electrochemical Hydrogen Evolution Rates by HDCR and Faradaic Efficiency
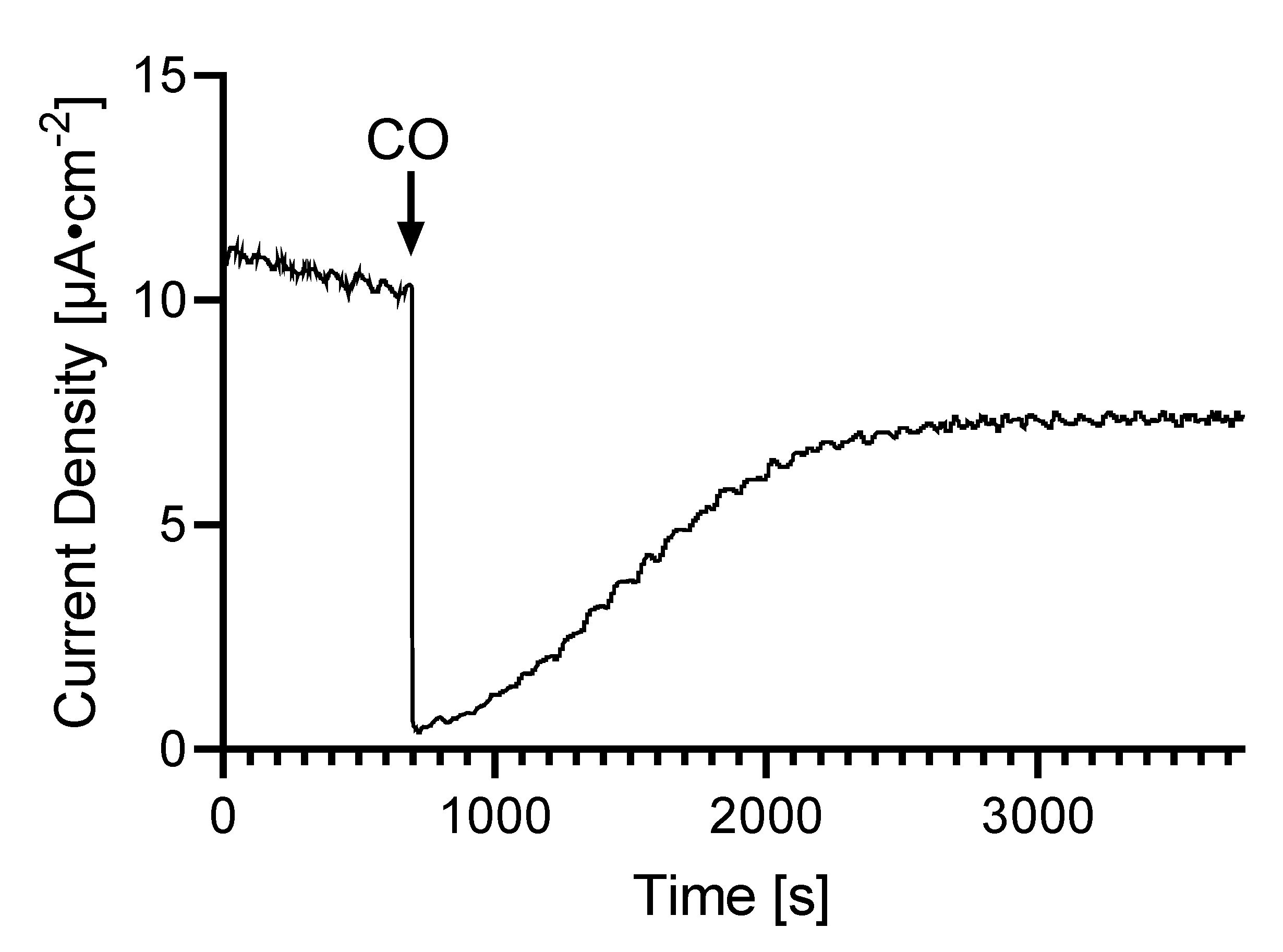
2.6. Reversible HDCR Inactivation by Carbon Monoxide
3. Materials and Methods
3.1. Materials and Chemicals
3.2. HDCR Expression and Purification
3.3. Synthesis of Cobaltocene-Functionalized Poly(allylamine) (Cc-PAA)
3.4. Preparation of HDCR-Embedded Electrodes
3.5. Electrochemical Methods
3.6. Synthesis of Ti(III) Citrate
3.7. Methyl Viologen Assay for Quantification of Chemical Hydrogen Evolution Rates
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HDCR | Hydrogen-Dependent CO2 Reductase; |
| Cc-PAA | cobaltocene-functionalized poly(allylamine); |
| RDE | rotating disk electrode; |
| SHE | standard hydrogen electrode; |
| CV | cyclic voltammetry. |
References
- Lindstrom, P.; Fritsch, D.; Fickling, M.; Chase, N.; Martin, L.; Anti, L.; Hansom, S.; Gross, P.; Palguta, J.; Dyl, K.; et al. Annual Energy Outlook 2020; U.S. Energy Information Administration: Washington, DC, USA, 2020.
- Dresselhaus, M.S.; Thomas, I.L. Alternative energy technologies. Nature 2001, 414, 332–337. [Google Scholar] [CrossRef]
- Ruth, J.C.; Spormann, A.M. Enzyme Electrochemistry for Industrial Energy Applications—A Perspective on Future Areas of Focus. ACS Catal. 2021, 11, 5951–5967. [Google Scholar] [CrossRef]
- Abe, J.O.; Popoola, A.P.I.; Ajenifuja, E.; Popoola, O.M. Hydrogen energy, economy and storage: Review and recommendation. Int. J. Hydrogen Energy 2019, 44, 15072–15086. [Google Scholar] [CrossRef]
- Gotovsky, M.A.; Gotovsky, A.M.; Mikhailov, V.E.; Lychakov, V.D.; Sukhorukov, Y.G.; Sukhorukova, E.A. Formate: The Third Way in Green Energy. Int. J. Chem. Eng. Appl. 2019, 10, 189–194. [Google Scholar] [CrossRef]
- Blanco, H.; Cazzola, P.; Dulac, J.; Fukui, H.; Kim, T.-Y.; Kurban, Z.; Levi, P.; Malischek, R.; McGlade, C.; Petrosyan, K.; et al. The Future of Hydrogen; International Energy Agency: Paris, France, 2019. [Google Scholar]
- Cadoux, C.M.; Milton, R.D. Recent enzymatic electrochemistry for reductive reactions. ChemElectroChem 2020, 7, 1974–1986. [Google Scholar] [CrossRef]
- Ceccaldi, P.; Schuchmann, K.; Müller, V.; Elliott, S.J. The Hydrogen Dependent CO2 reductase: The first completely co tolerant fefe-hydrogenase. Energy Environ. Sci. 2017, 10, 503–508. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, F.M.; Ciurus, S.; Jain, S.; Baum, C.; Wiechmann, A.; Basen, M.; Müller, V. Revealing formate production from carbon monoxide in wild type and mutants of Rnf- and Ech-containing acetogens, Acetobacterium woodii and Thermoanaerobacter kivui. Microb. Biotechnol. 2020, 13, 2044–2056. [Google Scholar] [CrossRef]
- Schwarz, F.M.; Schuchmann, K.; Müller, V. Hydrogenation of CO2 at ambient pressure catalyzed by a highly active thermostable biocatalyst. Biotechnol. Biofuels 2018, 11, 237. [Google Scholar] [CrossRef]
- Müller, V. New Horizons in Acetogenic Conversion of One-Carbon Substrates and Biological Hydrogen Storage. Trends Biotechnol. 2019, 37, 1344–1354. [Google Scholar] [CrossRef]
- Lubitz, W.; Ogata, H.; Rudiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef]
- Lienemann, M.; Deutzmann, J.S.; Milton, R.D.; Sahin, M.; Spormann, A.M. Mediator-free enzymatic electrosynthesis of formate by the Methanococcus maripaludis heterodisulfide reductase supercomplex. Bioresour. Technol. 2018, 254, 278–283. [Google Scholar] [CrossRef]
- Basso, A.; Serban, S. Industrial applications of immobilized enzymes—A review. Mol. Catal. 2019, 479, 110607. [Google Scholar] [CrossRef]
- Srikanth, S.; Alvarez-Gallego, Y.; Vanbroekhoven, K.; Pant, D. Enzymatic Electrosynthesis of Formic Acid through Carbon Dioxide Reduction in a Bioelectrochemical System: Effect of Immobilization and Carbonic Anhydrase Addition. ChemPhysChem 2017, 18, 3174–3181. [Google Scholar] [CrossRef]
- Sakai, K.; Kitazumi, Y.; Shirai, O.; Takagi, K.; Kano, K. Efficient bioelectrocatalytic CO2 reduction on gas-diffusion-type biocathode with tungsten-containing formate dehydrogenase. Electrochem. Commun. 2016, 73, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-Y.; Park, H.S.; Fontecilla-Camps, J.C.; Reisner, E. Photoelectrochemical H2 Evolution with a Hydrogenase Immobilized on a TiO2-Protected Silicon Electrode. Angew. Chem. Int. Ed. 2016, 55, 5971–5974. [Google Scholar] [CrossRef] [Green Version]
- Ruth, J.C.; Milton, R.D.; Gu, W.; Spormann, A.M. Enhanced Electrosynthetic Hydrogen Evolution by Hydrogenases Embedded in a Redox-Active Hydrogel. Chem. Eur. J. 2020, 26, 7323–7329. [Google Scholar] [CrossRef] [PubMed]
- Morra, S.; Valetti, F.; Sarasso, V.; Castrignanò, S.; Sadeghi, S.J.; Gilardi, G. Hydrogen production at high Faradaic efficiency by a bio-electrode based on TiO2 adsorption of a new [FeFe]-hydrogenase from Clostridium perfringens. Bioelectrochemistry 2015, 106, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Ruff, A. Redox polymers in bioelectrochemistry: Common playgrounds and novel concepts. Curr. Opin. Electrochem. 2017, 5, 66–73. [Google Scholar] [CrossRef]
- Hardt, S.; Stapf, S.; Filmon, D.T.; Birrell, J.A.; Rüdiger, O.; Fourmond, V.; Léger, C.; Plumeré, N. Reversible H2 oxidation and evolution by hydrogenase embedded in a redox polymer film. Nat. Catal. 2021, 4, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Shiraiwa, S.; So, K.; Sugimoto, Y.; Kitazumi, Y.; Shirai, O.; Nishikawa, K.; Higuchi, Y.; Kano, K. Reactivation of standard [NiFe]-hydrogenase and bioelectrochemical catalysis of proton reduction and hydrogen oxidation in a mediated-electron-transfer system. Bioelectrochemistry 2018, 123, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Sahin, S.; Cai, R.; Abdellaoui, S.; Hickey, D.P.; Minteer, S.D.; Milton, R.D. Creating a Low-Potential Redox Polymer for Efficient Electroenzymatic CO2 Reduction. Angew. Chem. Int. Ed. 2018, 57, 6582–6586. [Google Scholar] [CrossRef] [PubMed]
- Mayhew, S.G. The Redox Potential of Dithionite and SO−2 from Equilibrium Reactions with Flavodoxins, Methyl Viologen and Hydrogen plus Hydrogenase. Eur. J. Biochem. 1978, 85, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Katsyv, A.; Schoelmerich, M.C.; Basen, M.; Müller, V. The pyruvate:ferredoxin oxidoreductase of the thermophilic acetogen, Thermoanaerobacter kivui. FEBS Open Bio 2021, 11, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Zehnder, A.J.B.; Wuhrmann, K. Titanium(III) citrate as a nontoxic oxidation-reduction buffering system for the culture of obligate anaerobes. Science 1976, 194, 1165–1166. [Google Scholar] [CrossRef]
- Sidgwick, N.V.; Gentle, J.A.H.R. CCXX1.-The Solubilities of the Alkali Formates and Acetates in Water. J. Chem. Soc. 1922, 1837–1843. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruth, J.C.; Schwarz, F.M.; Müller, V.; Spormann, A.M. Enzymatic Hydrogen Electrosynthesis at Enhanced Current Density Using a Redox Polymer. Catalysts 2021, 11, 1197. https://doi.org/10.3390/catal11101197
Ruth JC, Schwarz FM, Müller V, Spormann AM. Enzymatic Hydrogen Electrosynthesis at Enhanced Current Density Using a Redox Polymer. Catalysts. 2021; 11(10):1197. https://doi.org/10.3390/catal11101197
Chicago/Turabian StyleRuth, John C., Fabian M. Schwarz, Volker Müller, and Alfred M. Spormann. 2021. "Enzymatic Hydrogen Electrosynthesis at Enhanced Current Density Using a Redox Polymer" Catalysts 11, no. 10: 1197. https://doi.org/10.3390/catal11101197