
Intra-Molecular Electrical Field Regulated Nonlinear Catalyst Charge Transfer in the Organic Conjugated Molecular System

 and
and
Abstract
:1. Introduction
2. Results and Discussion
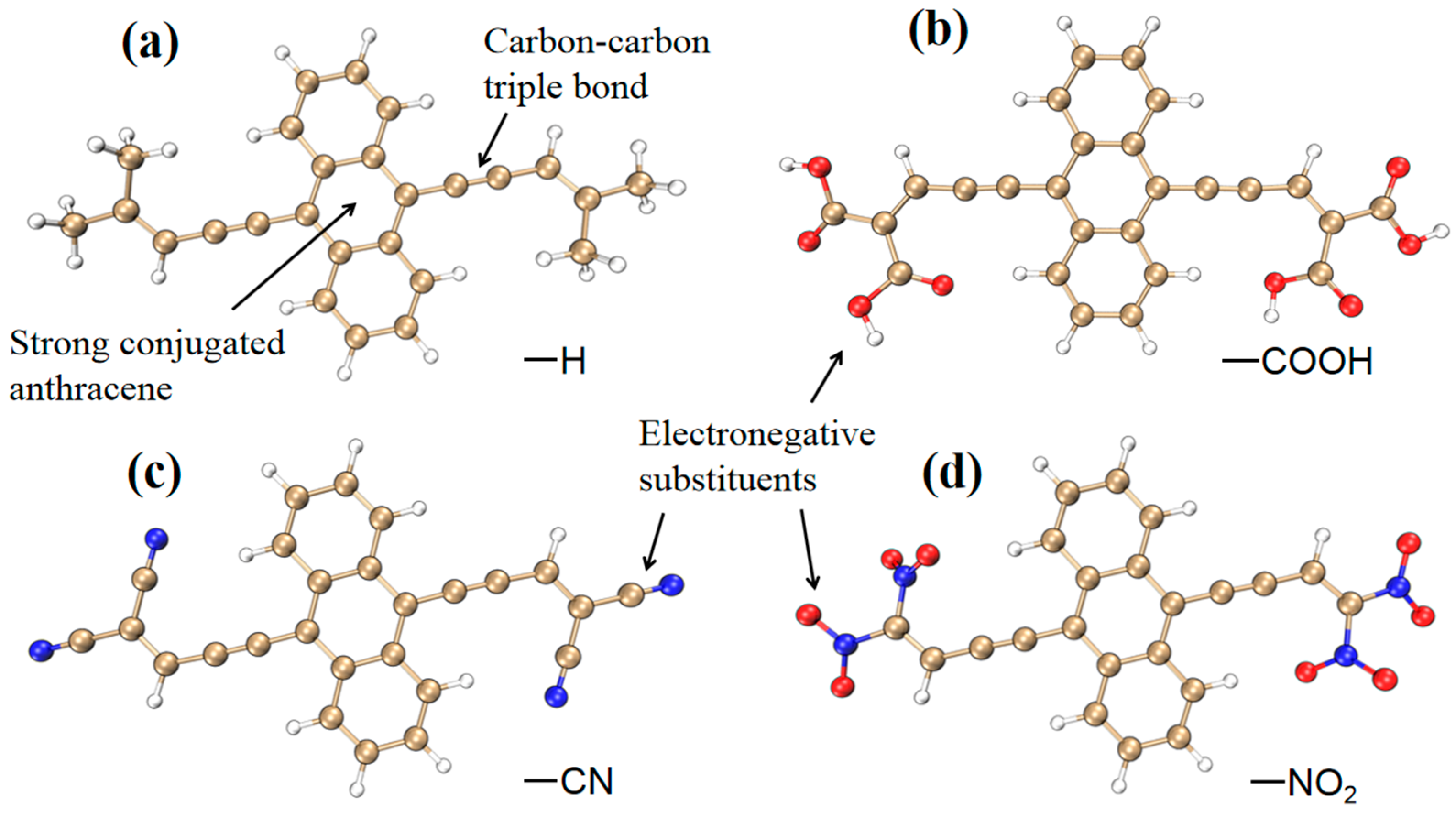
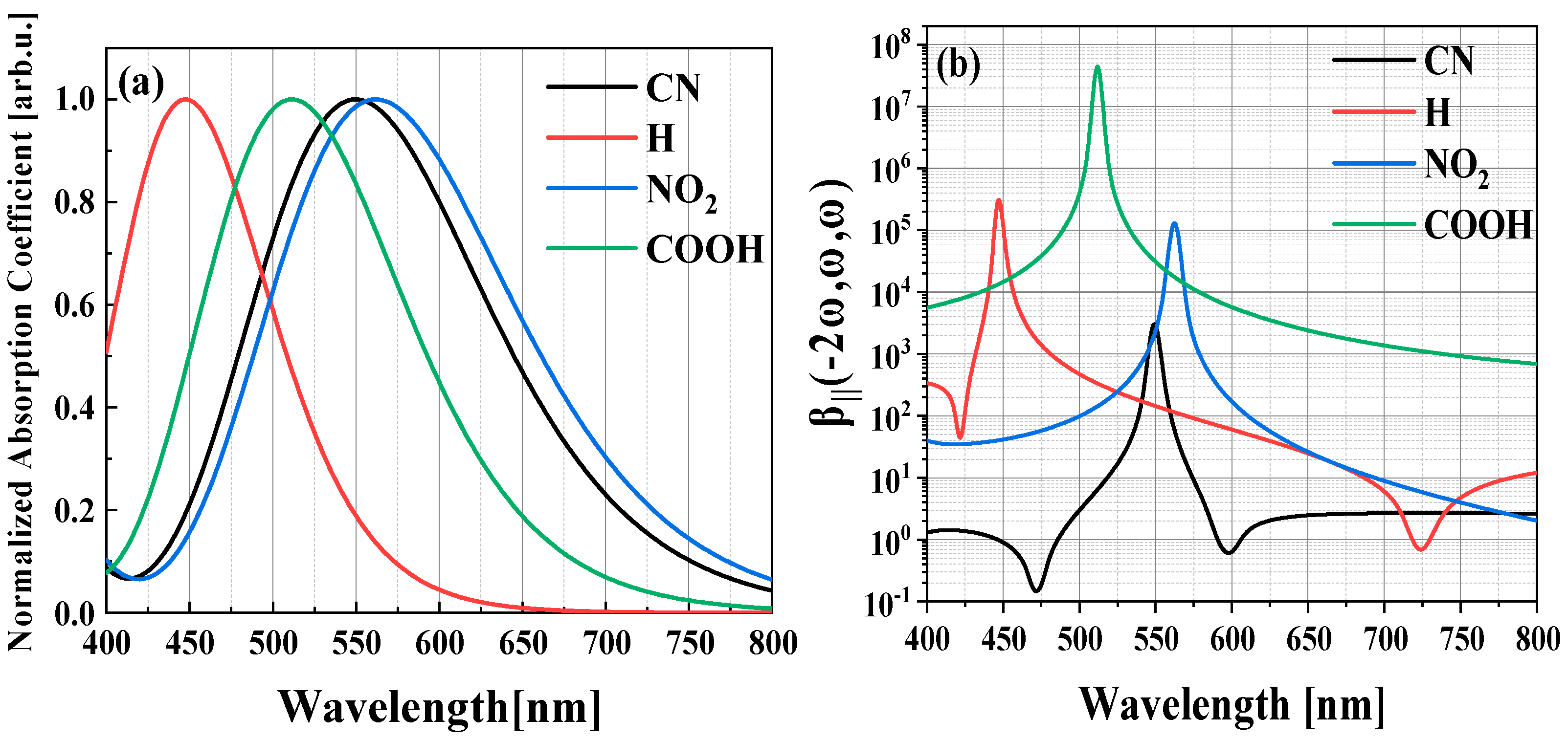
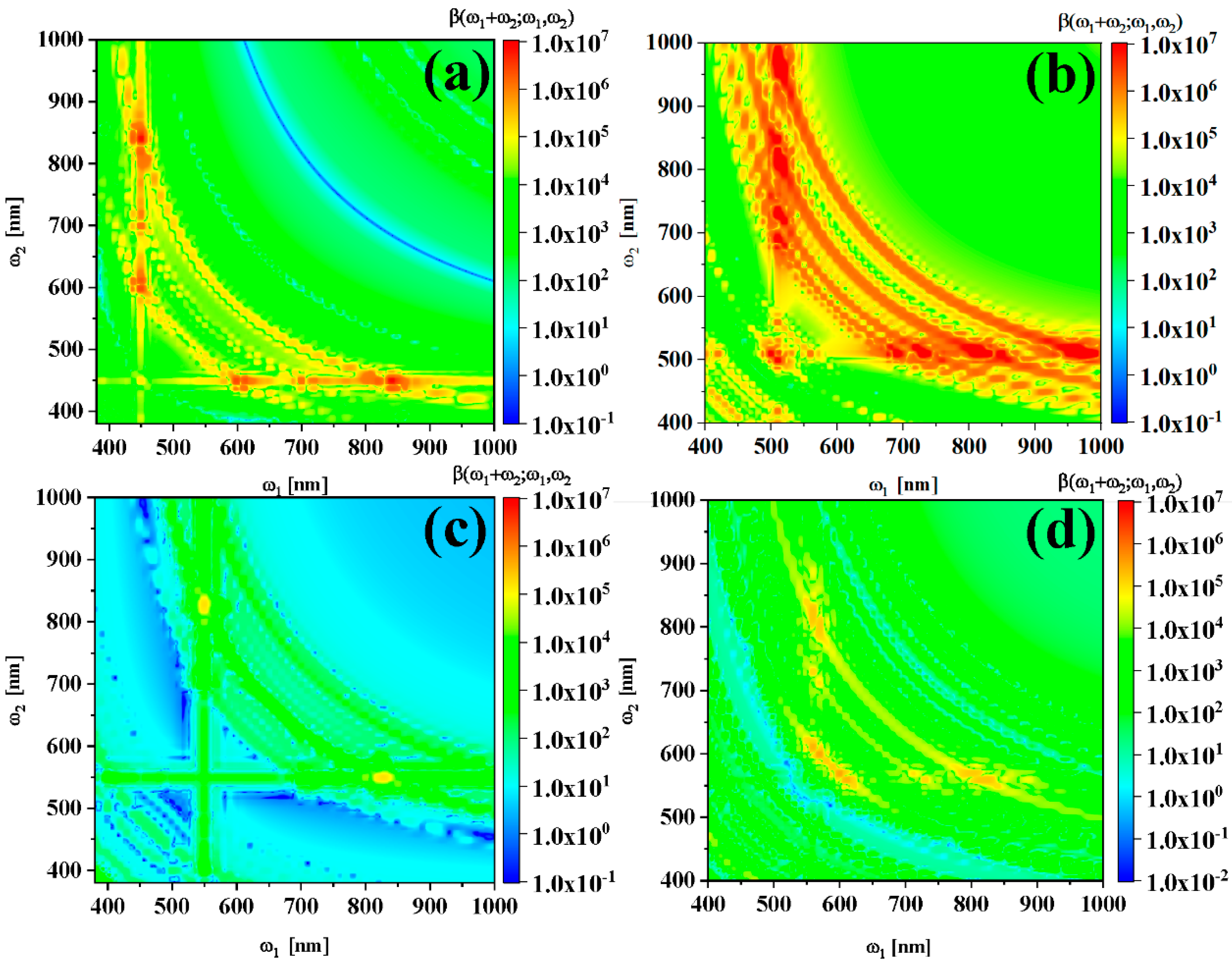
2.1. Molecular Structure, Linear and Nonlinear Optical Properties
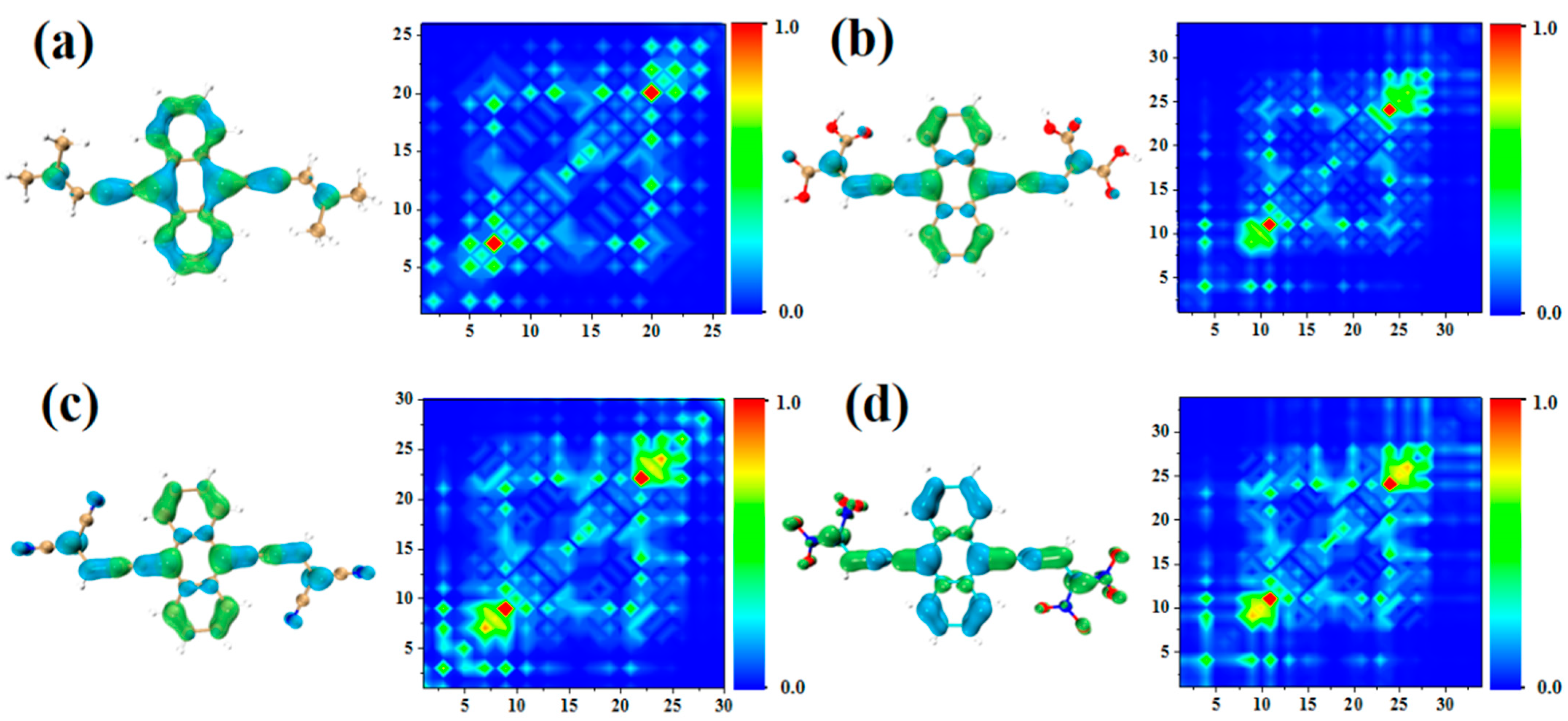
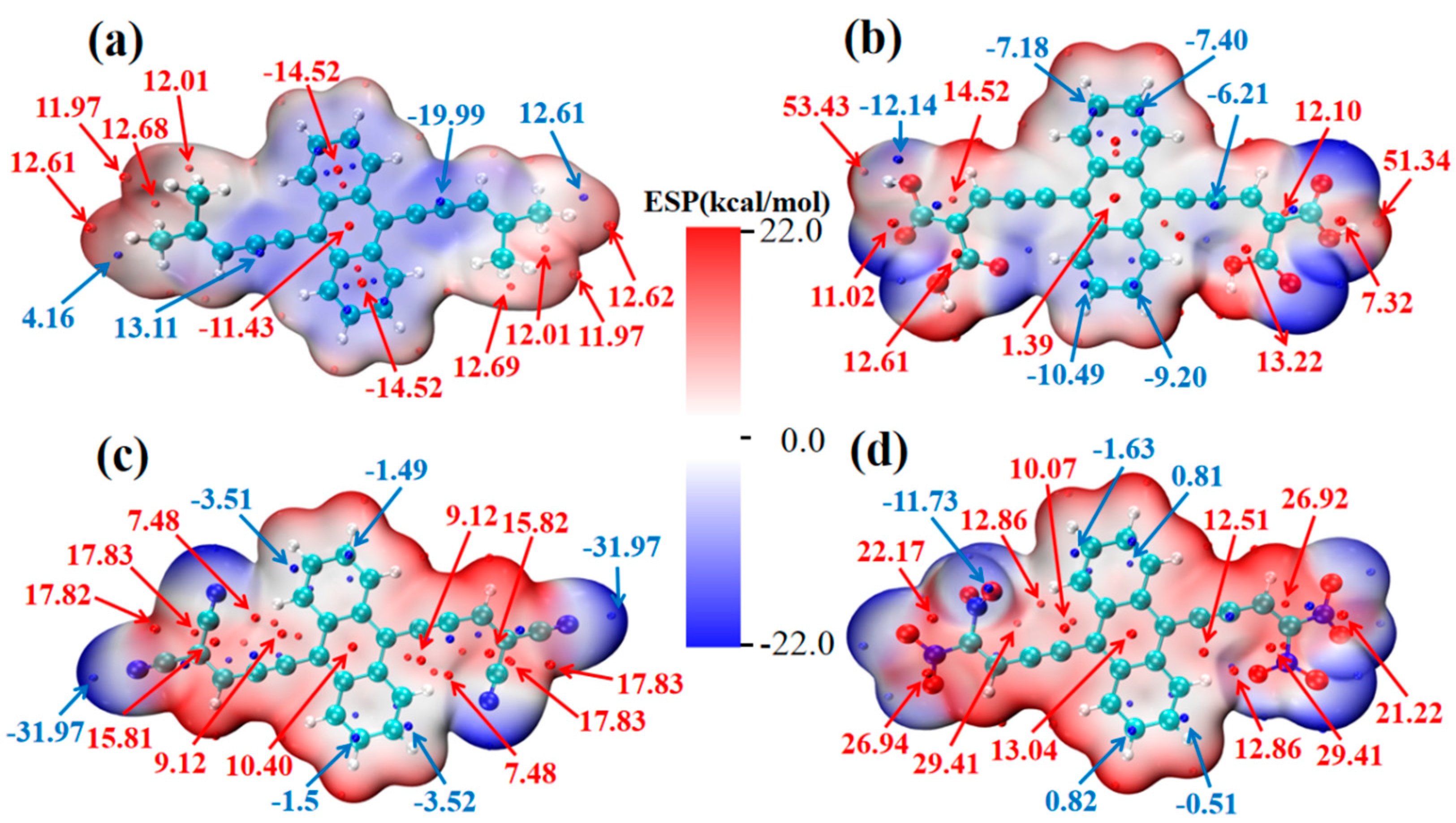
2.2. Physical Mechanism and Transition Characteristic
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dou, C.; Liu, J.; Wang, L. Conjugated polymers containing B←N unit as electron acceptors for all-polymer solar cells. Sci. China Chem. 2017, 60, 450–459. [Google Scholar] [CrossRef]
- Miao, W.; Wang, L.; Mu, X.; Wang, J. The magical photoelectric and optoelectronic properties of grapheme nanoribbons and their applications. J. Mater. Chem. C 2021, 9, 13600. [Google Scholar] [CrossRef]
- Kim, D.E.; Baeg, K.J. Reduction Treatment of Molecular-Doped Polymer Semiconductors for High-Performance N-Channel Organic Field-Effect Transistors. J. Korean Phys. Soc. 2019, 75, 821–826. [Google Scholar] [CrossRef]
- Wendlandt, A.E.; Stahl, S.S. Quinone-catalyzed selective oxidation of organic molecules. Angew. Chem. Int. Ed. 2015, 54, 14638–14658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrest, S.R. The path to ubiquitous and low-cost organic electronic appliances on plastic. Nature 2004, 428, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Lv, X.; Zhang, P.; Chuong, T.T.; Wu, B.; Feng, X.; Shan, C.; Liu, J.; Tang, Y. Plant Sunscreen and Co (II)/(III) Porphyrins for UV-Resistant and Thermally Stable Perovskite Solar Cells: From Natural to Artificial. Adv. Mater. 2018, 30, 1800568. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Chen, R.; Nan, Z.A.; Lv, X.; Meng, R.; Cao, J.; Tang, Y. Perfection of Perovskite Grain Boundary Passivation by Eu-Porphyrin Complex for Overall-Stable Perovskite Solar Cells. Adv. Sci. 2019, 6, 1802040. [Google Scholar] [CrossRef]
- Li, X.; Li, C.; Wu, Y.; Cao, J.; Tang, Y. A reaction-and-assembly approach using monoamine zinc porphyrin for highly stable large-area perovskite solar cells. Sci. China Chem. 2020, 63, 777–784. [Google Scholar] [CrossRef]
- Sharma, K.; Sharma, V.; Sharma, S.S. Dye-sensitized solar cells: Fundamentals and current status. Nanoscale Res. Lett. 2018, 13, 1–46. [Google Scholar] [CrossRef]
- Huang, Z.S.; Cai, C.; Zang, X.F.; Iqbal, Z.; Zeng, H.; Kuang, D.B.; Wang, L.; Meier, H.; Cao, D. Effect of the linkage location in double branched organic dyes on the photovoltaic performance of DSSCs. J. Mater. Chem. A 2015, 3, 1333–1344. [Google Scholar] [CrossRef]
- Hara, K.; Sato, T.; Katoh, R.; Furube, A.; Yoshihara, T.; Murai, M.; Kurashige, M.; Ito, S.; Shinpo, A.; Suga, S. Novel conjugated organic dyes for efficient dye-sensitized solar cells. Adv. Funct. Mater. 2005, 15, 246–252. [Google Scholar] [CrossRef]
- Ko, S.; Choi, H.; Kang, M.S.; Hwang, H.; Ji, H.; Kim, J.; Ko, J.; Kang, Y. Silole-spaced triarylamine derivatives as highly efficient organic sensitizers in dye-sensitized solar cells (DSSCs). J. Mater. Chem. 2010, 20, 2391–2399. [Google Scholar] [CrossRef]
- Lu, Q.C.; Wang, Q.G.; Song, P.; Ma, F.C.; Yang, Y.; Li, Y. PCDTBT8-doped PffBT4T-2OD-based ternary solar cells with enhanced open-circuit voltage, fill factor and charge separation efficiency. Solar RRL 2021, 10, 1002. [Google Scholar] [CrossRef]
- Qiao, W.; Yu, H.; Chen, X. Physical mechanism of special type photoinduced charge transfer in one-photon and two-photon absorption of Mobius rings. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 236, 118264. [Google Scholar] [CrossRef] [PubMed]
- Bo, W.; Zou, Y.; Wang, J. Novel electrical properties and applications in kaleidoscopic graphene nanoribbons. RSC Adv. 2021, 11, 33675–33691. [Google Scholar] [CrossRef]
- Wang, J.; Mu, X.; Wang, X.; Wang, N.; Ma, F.; Liang, W.; Sun, M. The thermal and thermoelectric properties of in-plane C-BN hybrid structures and graphene/h-BN van der Waals heterostructures. Mater. Today Phys. 2018, 5, 29–57. [Google Scholar] [CrossRef]
- Wang, J.; Xu, X.; Mu, X.; Ma, F.; Sun, M. Magnetics and spintronics on two-dimensional composite materials of graphene/hexagonal boron nitride. Mater. Today Phys. 2017, 3, 93–117. [Google Scholar] [CrossRef]
- Wang, J.; Mu, X.; Sun, M.; Mu, T. Optoelectronic properties and applications of graphene-based hybrid nanomaterials and van der Waals heterostructures. Appl. Mater. Today 2019, 16, 1–20. [Google Scholar] [CrossRef]
- Mu, X.; Wang, J.; Sun, M. Two-dimensional black phosphorus: Physical properties and applications. Mater. Today Phys. 2019, 8, 92–111. [Google Scholar] [CrossRef]
- Song, J.; Zhang, Z.; Feng, N. Electric Field Induced Twisted Bilayer Graphene Infrared Plasmon Spectrum. Nanomaterials 2021, 11, 2433. [Google Scholar] [CrossRef]
- Mu, X.; Zong, H.; Zhu, L.; Sun, M. External electric field-dependent photoinduced charge transfer in a donor-acceptor system in two-photon absorption. J. Phys. Chem. C 2020, 124, 2319–2332. [Google Scholar] [CrossRef]
- Mu, X.; Wang, X.; Quan, J.; Sun, M. Photoinduced Charge Transfer in Donor-Bridge-Acceptor in One-and Two-photon Absorption: Sequential and Superexchange Mechanisms. J. Phys. Chem. C 2020, 124, 4968–4981. [Google Scholar] [CrossRef]
- Chen, X.; Qiao, W.; Miao, W. The Dependence of implicit Solvent Model parameters and electronic Absorption Spectra and photoinduced charge transfer. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Cai, K.; Wei, W.; Li, Y.; Wang, Z.; Wang, J. Dependence of UV-Visible absorption characteristics on the migration distance and the hyperconjugation effect of a methine chain. J. Phys. Chem. C 2018, 122, 7831–7837. [Google Scholar] [CrossRef]
- Ding, F.; Kuiken, B.E.; Eichinger, B.E. An efficient method for calculating dynamical hyperpolarizabilities using real-time time-dependent density functional theory. J. Chem. Phys. 2013, 138, 064104. [Google Scholar] [CrossRef]
- Mu, X.; Wang, J.; Sun, M. Visualization of photoinduced charge transfer and electron-hole coherence in two-photon absorption. J. Phys. Chem. C 2019, 123, 14132–14143. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.; Devlin, F.; Chabalowski, C.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Petersson, a.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H. Gaussian 16, Revision A03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Furche, F.; Ahlrichs, R. Adiabatic time-dependent density functional methods for excited state properties. J. Chem. Phys. 2002, 117, 7433–7447. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substituent | -H | -COOH | -CN | -NO2 |
|---|---|---|---|---|
| Excited energy [eV] | 2.771 | 2.424 | 2.255 | 2.207 |
| Sm | 0.576 | 0.544 | 0.501 | 0.459 |
| Sr | 0.876 | 0.806 | 0.776 | 0.749 |
| D [angstrom] | 0.005 | 0.176 | 0.000 | 0.000 |
| Δσ | −0.097 | 0.678 | 0.773 | 1.107 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Chen, S.; Wang, L.; Liu, Y.; He, D.; Wang, M.; Wang, J. Intra-Molecular Electrical Field Regulated Nonlinear Catalyst Charge Transfer in the Organic Conjugated Molecular System. Catalysts 2021, 11, 1375. https://doi.org/10.3390/catal11111375
Li Q, Chen S, Wang L, Liu Y, He D, Wang M, Wang J. Intra-Molecular Electrical Field Regulated Nonlinear Catalyst Charge Transfer in the Organic Conjugated Molecular System. Catalysts. 2021; 11(11):1375. https://doi.org/10.3390/catal11111375
Chicago/Turabian StyleLi, Quanjiang, Shenghui Chen, Li Wang, Yanli Liu, Di He, Meishan Wang, and Jingang Wang. 2021. "Intra-Molecular Electrical Field Regulated Nonlinear Catalyst Charge Transfer in the Organic Conjugated Molecular System" Catalysts 11, no. 11: 1375. https://doi.org/10.3390/catal11111375