Pyridine-Chelated Imidazo[1,5-a]Pyridine N-Heterocyclic Carbene Nickel(II) Complexes for Acrylate Synthesis from Ethylene and CO2

 , , and
, , and
Abstract
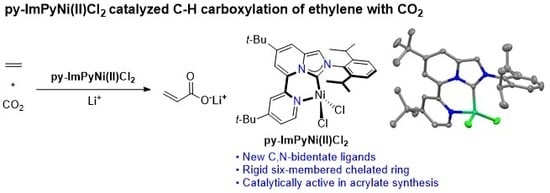
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Ligand Precursors
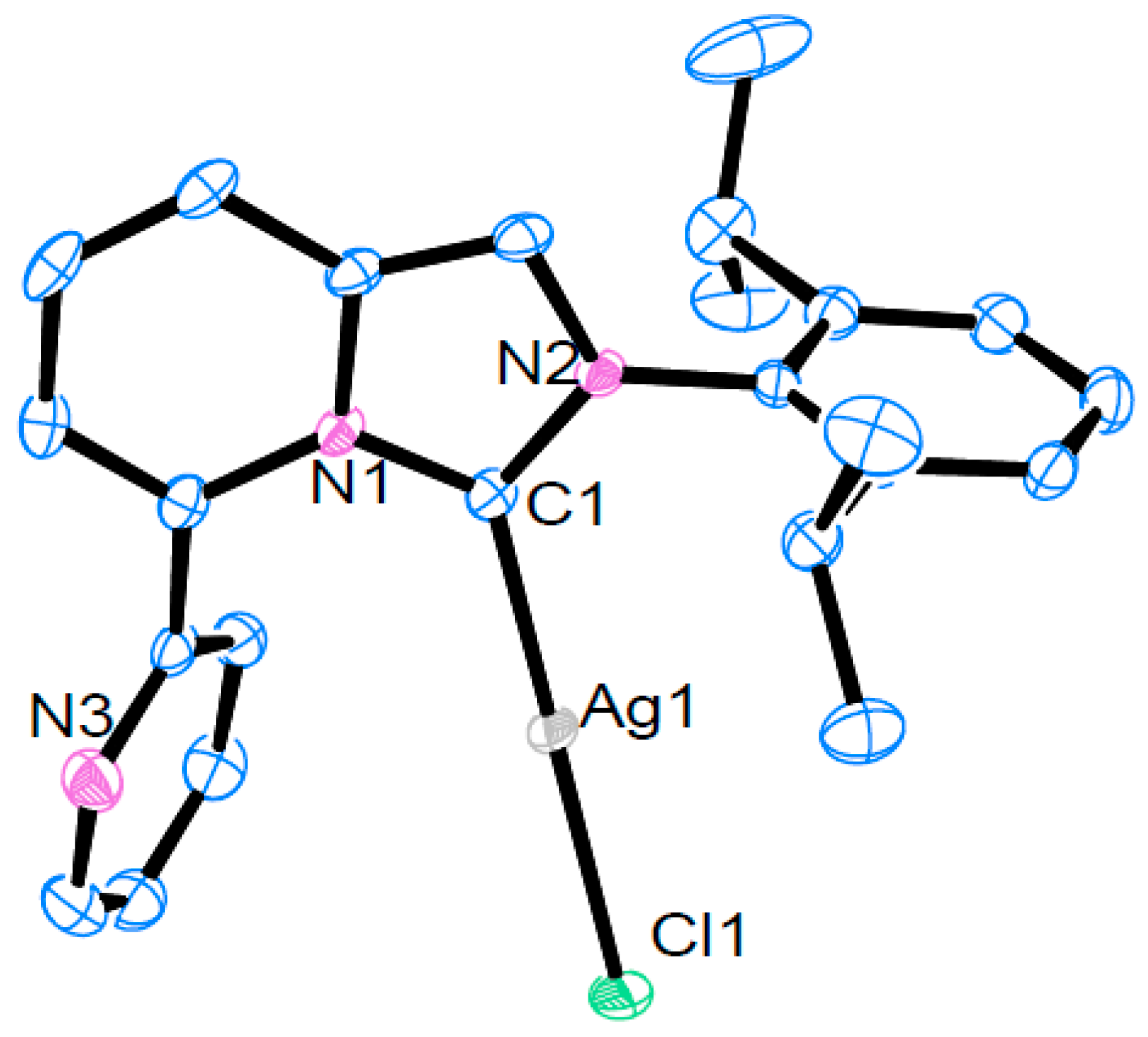
2.2. Synthesis and Structural Analysis of Ag(I) Complexes
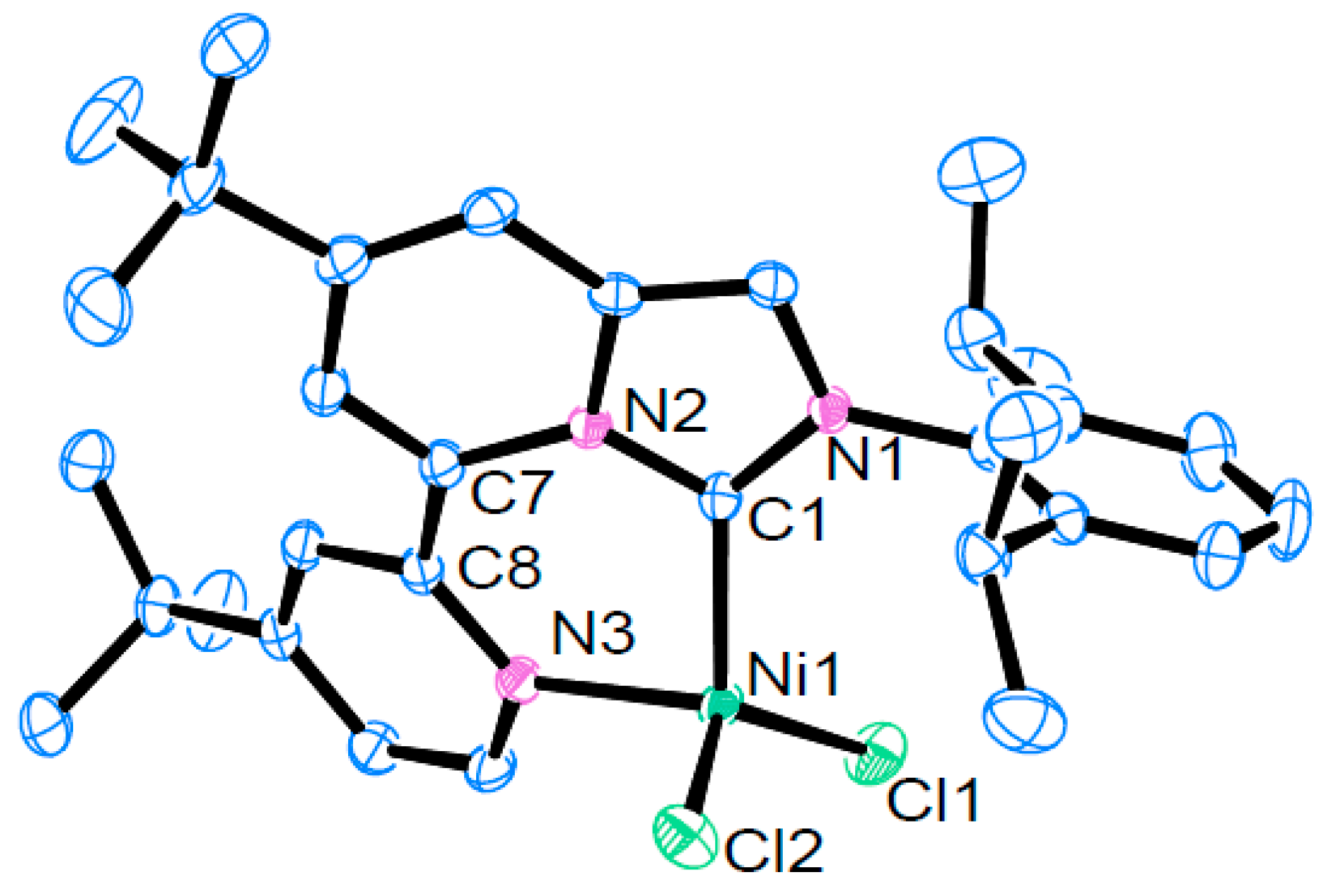
2.3. Synthesis and Structural Analysis of Nickel(II) Complexes
2.4. Nickel Mediated Acrylate Synthesis from Ethylene Using CO2
2.5. Optimization of Reaction Conditions Using the Combination 8b and PCy3
3. Materials and Methods
3.1. General Remarks
3.2. Materials
3.3. Synthesis of Ligand Precursors. General Procedure
3.4. Synthesis of Ag(I) Complexes. General Procedure
3.5. Synthesis of Ni(II) Complexes. General Procedure
3.6. General Procedure for the Synthesis of Lithium Acrylate Using Ethylene and CO2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W.A.; Kühn, F.E. Transformation of carbon dioxide with homogeneous transition-metal catalysts: A molecular solution to a global challenge? Angew. Chem. Int. Ed. 2011, 50, 8510–8537. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Diao, Z.-F.; Guo, C.-X.; He, L.-N. Carboxylation of olefins/alkynes with CO2 to industrially relevant acrylic acid derivatives. J. CO2 Util. 2013, 1, 60–68. [Google Scholar] [CrossRef]
- Kraus, S.; Rieger, B. Ni-Catalyzed Synthesis of Acrylic Acid Derivatives from CO2 and Ethylene. In Carbon Dioxide and Organometallics; Springer: Cham, Switzerland, 2015; pp. 199–223. [Google Scholar]
- Limbach, M. Acrylates from alkenes and CO2, the stuff that dreams are made of. In Advances in Organometallic Chemistry; Elsevier: Amsterdam, The Netherlands, 2015; Volume 63, pp. 175–202. [Google Scholar]
- Hollering, M.; Dutta, B.; Kühn, F.E. Transition metal mediated coupling of carbon dioxide and ethene to acrylic acid/acrylates. Coord. Chem. Rev. 2016, 309, 51–67. [Google Scholar] [CrossRef]
- Alper, E.; Orhan, O.Y. CO2 utilization: Developments in conversion processes. Petroleum 2017, 3, 109–126. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Sun, Y. Synthesis of acrylic acid derivatives from CO2 and ethylene. Chem 2017, 3, 211–228. [Google Scholar] [CrossRef]
- Wu, X.-F.; Zheng, F. Synthesis of Carboxylic Acids and Esters from CO2. Top. Curr. Chem. 2017, 375, 4. [Google Scholar] [CrossRef]
- Schmitz, M.; Solmi, M.V.; Leitner, W. Catalytic Processes Combining CO2 and Alkenes into Value-Added Chemicals. In Organometallics for Green Catalysis; Springer: Berlin, Germany, 2018; pp. 17–38. [Google Scholar]
- Dabral, S.; Schaub, T. The use of carbon dioxide (CO2) as a building block in organic synthesis from an industrial perspective. Adv. Synth. Catal. 2019, 361, 223–246. [Google Scholar] [CrossRef] [Green Version]
- Solmi, M.V.; Schmitz, M.; Leitner, W. CO2 as a Building Block for the Catalytic Synthesis of Carboxylic Acids. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 2019; Volume 178, pp. 105–124. [Google Scholar]
- Weissermel, K.; Arpe, H. Propene Conversion Products. Ind. Org. Chem. 2008, 4, 267–312. [Google Scholar]
- Hoberg, H.; Schaefer, D. Nickel (0)-induzierte C C-verknüpfung zwischen kohlendioxid und ethylen sowie mono-oder di-substituierten alkenen. J. Organomet. Chem. 1983, 251, c51–c53. [Google Scholar] [CrossRef]
- Hoberg, H.; Peres, Y.; Milchereit, A. C–C-Verknüpfung von alkenen mit CO2 an nickel (O); n-pentesäuren aus ethen. J. Organomet. Chem. 1986, 307, C41–C43. [Google Scholar] [CrossRef]
- Hoberg, H.; Jenni, K.; Angermund, K.; Krüger, C. CC-Linkages of Ethene with CO2 on an Iron (0) Complex—Synthesis and Crystal Structure Analysis of [(PEt3)2Fe(C2H4)2]. Angew. Chem. Int. Ed. Engl. 1987, 26, 153–155. [Google Scholar] [CrossRef]
- Hoberg, H.; Peres, Y.; Krüger, C.; Tsay, Y.H. A 1-Oxa-2-nickela-5-cyclopentanone from Ethene and Carbon Dioxide: Preparation, Structure, and Reactivity. Angew. Chem. Int. Ed. Engl. 1987, 26, 771–773. [Google Scholar] [CrossRef]
- Hoberg, H.; Ballesteros, A.; Sigan, A.; Jégat, C.; Bärhausen, D.; Milchereit, A. Ligandgesteuerte Ringkontraktion von Nickela-fünf-in Vierringkomplexe—Neuartige startsysteme für die präparative chemie. J. Organomet. Chem. 1991, 407, C23–C29. [Google Scholar] [CrossRef]
- Alvarez, R.; Carmona, E.; Cole-Hamilton, D.J.; Galindo, A.; Gutierrez-Puebla, E.; Monge, A.; Poveda, M.L.; Ruiz, C. Formation of acrylic acid derivatives from the reaction of carbon dioxide with ethylene complexes of molybdenum and tungsten. J. Am. Chem. Soc. 1985, 107, 5529–5531. [Google Scholar] [CrossRef]
- Alvarez, R.; Carmona, E.; Galindo, A.; Gutierrez, E.; Marin, J.M.; Monge, A.; Poveda, M.L.; Ruiz, C.; Savariault, J.M. Formation of carboxylate complexes from the reactions of carbon dioxide with ethylene complexes of molybdenum and tungsten. X-ray and neutron diffraction studies. Organometallics 1989, 8, 2430–2439. [Google Scholar] [CrossRef]
- Galindo, A.; Pastor, A.; Perez, P.J.; Carmona, E. Bis(ethylene) complexes of molybdenum and tungsten and their reactivity toward carbon dioxide. New examples of acrylate formation by coupling of ethylene and carbon dioxide. Organometallics 1993, 12, 4443–4451. [Google Scholar] [CrossRef]
- Fischer, R.; Langer, J.; Malassa, A.; Walther, D.; Görls, H.; Vaughan, G. A key step in the formation of acrylic acid from CO2 and ethylene: The transformation of a nickelalactone into a nickel-acrylate complex. Chem. Commun. 2006, 23, 2510–2512. [Google Scholar] [CrossRef]
- Bruckmeier, C.; Lehenmeier, M.W.; Reichardt, R.; Vagin, S.; Rieger, B. Formation of methyl acrylate from CO2 and ethylene via methylation of nickelalactones. Organometallics 2010, 29, 2199–2202. [Google Scholar] [CrossRef]
- Lee, S.T.; Cokoja, M.; Drees, M.; Li, Y.; Mink, J.; Herrmann, W.A.; Kühn, F.E. Transformation of nickelalactones to methyl acrylate: On the way to a catalytic conversion of carbon dioxide. ChemSusChem 2011, 4, 1275–1279. [Google Scholar] [CrossRef]
- Jin, D.; Schmeier, T.J.; Williard, P.G.; Hazari, N.; Bernskoetter, W.H. Lewis acid induced β-elimination from a nickelalactone: Efforts toward acrylate production from CO2 and ethylene. Organometallics 2013, 32, 2152–2159. [Google Scholar] [CrossRef]
- Lee, S.T.; Ghani, A.A.; D’Elia, V.; Cokoja, M.; Herrmann, W.A.; Basset, J.-M.; Kühn, F.E. Liberation of methyl acrylate from metallalactone complexes via M–O ring opening (M = Ni, Pd) with methylation agents. New J. Chem. 2013, 37, 3512–3517. [Google Scholar] [CrossRef]
- Plessow, P.N.; Weigel, L.; Lindner, R.; Schäfer, A.; Rominger, F.; Limbach, M.; Hofmann, P. Mechanistic details of the nickel-mediated formation of acrylates from CO2, ethylene and methyl iodide. Organometallics 2013, 32, 3327–3338. [Google Scholar] [CrossRef]
- Jin, D.; Williard, P.G.; Hazari, N.; Bernskoetter, W.H. Effect of Sodium Cation on Metallacycle β-Hydride Elimination in CO2–Ethylene Coupling to Acrylates. Chem. A Eur. J. 2014, 20, 3205–3211. [Google Scholar] [CrossRef]
- Pápai, I.; Schubert, G.; Mayer, I.; Besenyei, G.; Aresta, M. Mechanistic details of nickel (0)-assisted oxidative coupling of CO2 with C2H4. Organometallics 2004, 23, 5252–5259. [Google Scholar] [CrossRef]
- Graham, D.C.; Mitchell, C.; Bruce, M.I.; Metha, G.F.; Bowie, J.H.; Buntine, M.A. Production of Acrylic Acid through Nickel-Mediated Coupling of Ethylene and Carbon Dioxide–A DFT Study. Organometallics 2007, 26, 6784–6792. [Google Scholar] [CrossRef]
- Plessow, P.N.; Schäfer, A.; Limbach, M.; Hofmann, P. Acrylate formation from CO2 and ethylene mediated by nickel complexes: A theoretical study. Organometallics 2014, 33, 3657–3668. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Z.; Cheng, R.; Liu, B. Mechanistic Aspects of Acrylic Acid Formation from CO2–Ethylene Coupling over Palladium-and Nickel-based Catalysts. ChemCatChem 2018, 10, 1420–1430. [Google Scholar] [CrossRef]
- Hendriksen, C.; Pidko, E.A.; Yang, G.; Schäffner, B.; Vogt, D. Catalytic formation of acrylate from carbon dioxide and ethene. Chem. A Eur. J. 2014, 20, 12037–12040. [Google Scholar] [CrossRef]
- Lejkowski, M.L.; Lindner, R.; Kageyama, T.; Bódizs, G.É.; Plessow, P.N.; Müller, I.B.; Schäfer, A.; Rominger, F.; Hofmann, P.; Futter, C.; et al. The first catalytic synthesis of an acrylate from CO2 and an alkene—A rational approach. Chem. A Eur. J. 2012, 18, 14017–14025. [Google Scholar] [CrossRef]
- Huguet, N.; Jevtovikj, I.; Gordillo, A.; Lejkowski, M.L.; Lindner, R.; Bru, M.; Khalimon, A.Y.; Rominger, F.; Schunk, S.A.; Hofmann, P.; et al. Nickel-Catalyzed Direct Carboxylation of Olefins with CO2: One-Pot Synthesis of α, β-Unsaturated Carboxylic Acid Salts. Chem. A Eur. J. 2014, 20, 16858–16862. [Google Scholar] [CrossRef]
- Manzini, S.; Huguet, N.; Trapp, O.; Schaub, T. Palladium-and Nickel-Catalyzed Synthesis of Sodium Acrylate from Ethylene, CO2, and Phenolate Bases: Optimization of the Catalytic System for a Potential Process. Eur. J. Org. Chem. 2015, 2015, 7122–7130. [Google Scholar] [CrossRef]
- Knopf, I.; Tofan, D.; Beetstra, D.; Al-Nezari, A.; Al-Bahily, K.; Cummins, C.C. A family of cis-macrocyclic diphosphines: Modular, stereoselective synthesis and application in catalytic CO2/ethylene coupling. Chem. Sci. 2017, 8, 1463–1468. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, M.N.; Shimmei, K.; Uttley, K.B.; Bernskoetter, W.H. Synthesis and Reactivity of 1, 2-Bis(di-iso-propylphosphino)benzene Nickel Complexes: A Study of Catalytic CO2–Ethylene Coupling. Organometallics 2018, 37, 3573–3580. [Google Scholar] [CrossRef]
- Vavasori, A.; Calgaro, L.; Pietrobon, L.; Ronchin, L. The coupling of carbon dioxide with ethene to produce acrylic acid sodium salt in one pot by using Ni (II) and Pd (II)-phosphine complexes as precatalysts. Pure Appl. Chem. 2018, 90, 315–326. [Google Scholar] [CrossRef]
- Stieber, S.C.E.; Huguet, N.; Kageyama, T.; Jevtovikj, I.; Ariyananda, P.; Gordillo, A.; Schunk, S.A.; Rominger, F.; Hofmann, P.; Limbach, M. Acrylate formation from CO2 and ethylene: Catalysis with palladium and mechanistic insight. Chem. Commun. 2015, 51, 10907–10909. [Google Scholar] [CrossRef] [PubMed]
- Manzini, S.; Cadu, A.; Schmidt, A.C.; Huguet, N.; Trapp, O.; Paciello, R.; Schaub, T. Enhanced activity and recyclability of palladium complexes in the catalytic synthesis of sodium acrylate from carbon dioxide and ethylene. ChemCatChem 2017, 9, 2269–2274. [Google Scholar] [CrossRef]
- Manzini, S.; Huguet, N.; Trapp, O.; Paciello, R.A.; Schaub, T. Synthesis of acrylates from olefins and CO2 using sodium alkoxides as bases. Catal. Today 2017, 281, 379–386. [Google Scholar] [CrossRef]
- Knopf, I.; Courtemanche, M.-A.; Cummins, C.C. Cobalt Complexes Supported by cis-Macrocyclic Diphosphines: Synthesis, Reactivity, and Activity toward Coupling Carbon Dioxide and Ethylene. Organometallics 2017, 36, 4834–4843. [Google Scholar] [CrossRef]
- Ito, T.; Takahashi, K.; Iwasawa, N. Reactivity of a Ruthenium (0) Complex Bearing a Tetradentate Phosphine Ligand: Applications to Catalytic Acrylate Salt Synthesis from Ethylene and CO2. Organometallics 2018, 38, 205–209. [Google Scholar] [CrossRef]
- Rummelt, S.M.; Zhong, H.; Korobkov, I.; Chirik, P.J. Iron-mediated coupling of carbon dioxide and ethylene: Macrocyclic metallalactones enable access to various carboxylates. J. Am. Chem. Soc. 2018, 140, 11589–11593. [Google Scholar] [CrossRef]
- Buchwald, S.L.; Milstein, D. Ligand Design in Metal Chemistry: Reactivity and Catalysis; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Joannou, M.V.; Bezdek, M.J.; Albahily, K.; Korobkov, I.; Chirik, P.J. Synthesis and Reactivity of Reduced α-Diimine Nickel Complexes Relevant to Acrylic Acid Synthesis. Organometallics 2018, 37, 3389–3393. [Google Scholar] [CrossRef]
- Takahashi, K.; Cho, K.; Iwai, A.; Ito, T.; Iwasawa, N. Development of N-Phosphinomethyl-Substituted NHC-Nickel (0) Complexes as Robust Catalysts for Acrylate Salt Synthesis from Ethylene and CO2. Chem. A Eur. J. 2019, 25, 13504–13508. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Hirsch-Weil, D.; Abboud, K.A.; Hong, S. Development of Biisoquinoline-Based Chiral Diaminocarbene Ligands: Enantioselective SN2′ Allylic Alkylation Catalyzed by Copper−Carbene Complexes. J. Org. Chem. 2008, 73, 1983–1986. [Google Scholar] [CrossRef]
- Snead, D.R.; Seo, H.; Hong, S. Recent developments of chiral diaminocarbene-metal complexes for asymmetric catalysis. Curr. Org. Chem. 2008, 12, 1370–1387. [Google Scholar] [CrossRef]
- Hirsch-Weil, D.; Abboud, K.A.; Hong, S. Isoquinoline-based chiral monodentate N-heterocyclic carbenes. Chem. Commun. 2010, 46, 7525–7527. [Google Scholar] [CrossRef]
- Rodig, M.J.; Seo, H.; Hirsch-Weil, D.; Abboud, K.A.; Hong, S. Isoquinoline-based diimine ligands for Cu (II)–Catalyzed enantioselective nitroaldol (Henry) reactions. Tetrahedron Asymmetry 2011, 22, 1097–1102. [Google Scholar] [CrossRef]
- Jaiswal, A.S.; Hirsch-Weil, D.; Proulx, E.R.; Hong, S.; Narayan, S. Anti-tumor activity of novel biisoquinoline derivatives against breast cancers. Bioorg. Med. Chem. Lett. 2014, 24, 4850–4853. [Google Scholar] [CrossRef]
- Park, D.-A.; Ryu, J.Y.; Lee, J.; Hong, S. Bifunctional N-heterocyclic carbene ligands for Cu-catalyzed direct C–H carboxylation with CO2. RSC Adv. 2017, 7, 52496–52502. [Google Scholar] [CrossRef] [Green Version]
- Byun, S.; Seo, H.; Choi, J.-H.; Ryu, J.Y.; Lee, J.; Chung, W.-J.; Hong, S. Fluoro-imidazopyridinylidene Ruthenium Catalysts for Cross Metathesis with Ethylene. Organometallics 2019, 38, 4121–4132. [Google Scholar] [CrossRef]
- Park, D.-A.; Byun, S.; Ryu, J.Y.; Lee, J.; Lee, J.; Hong, S. Abnormal N-Heterocyclic Carbene–Palladium Complexes for the Copolymerization of Ethylene and Polar Monomers. ACS Catal. 2020, 10, 5443–5453. [Google Scholar] [CrossRef]
- Diez-Gonzalez, S.; Marion, N.; Nolan, S.P. N-heterocyclic carbenes in late transition metal catalysis. Chem. Rev. 2009, 109, 3612–3676. [Google Scholar] [CrossRef]
- Dröge, T.; Glorius, F. The Measure of All Rings—N-Heterocyclic Carbenes. Angew. Chem. Int. Ed. 2010, 49, 6940–6952. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.J.; Nolan, S.P. Quantifying and understanding the electronic properties of N-heterocyclic carbenes. Chem. Soc. Rev. 2013, 42, 6723–6753. [Google Scholar] [CrossRef] [PubMed]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Prakasham, A.; Ghosh, P. Nickel N-heterocyclic carbene complexes and their utility in homogeneous catalysis. Inorg. Chim. Acta 2015, 431, 61–100. [Google Scholar] [CrossRef]
- Danopoulos, A.A.; Simler, T.; Braunstein, P. N-heterocyclic carbene complexes of copper, nickel, and cobalt. Chem. Rev. 2019, 119, 3730–3961. [Google Scholar] [CrossRef]
- Nolan, S.P. The development and catalytic uses of N-heterocyclic carbene gold complexes. Acc. Chem. Res. 2011, 44, 91–100. [Google Scholar] [CrossRef]
- Froese, R.D.; Lombardi, C.; Pompeo, M.; Rucker, R.P.; Organ, M.G. Designing Pd–N-heterocyclic carbene complexes for high reactivity and selectivity for cross-coupling applications. Acc. Chem. Res. 2017, 50, 2244–2253. [Google Scholar] [CrossRef]
- Ogba, O.; Warner, N.; O’Leary, D.; Grubbs, R. Recent advances in ruthenium-based olefin metathesis. Chem. Soc. Rev. 2018, 47, 4510–4544. [Google Scholar] [CrossRef] [Green Version]
- Peris, E.; Crabtree, R.H. Recent homogeneous catalytic applications of chelate and pincer N-heterocyclic carbenes. Coord. Chem. Rev. 2004, 248, 2239–2246. [Google Scholar] [CrossRef]
- Normand, A.T.; Cavell, K.J. Donor-Functionalised N-Heterocyclic Carbene Complexes of Group 9 and 10 Metals in Catalysis: Trends and Directions. Eur. J. Inorg. Chem. 2008, 2008, 2781–2800. [Google Scholar] [CrossRef]
- Poyatos, M.; Mata, J.A.; Peris, E. Complexes with poly (N-heterocyclic carbene) ligands: Structural features and catalytic applications. Chem. Rev. 2009, 109, 3677–3707. [Google Scholar] [CrossRef]
- Biffis, A.; Baron, M.; Tubaro, C. Poly-NHC Complexes of transition metals: Recent applications and new trends. In Advances in Organometallic Chemistry; Elsevier: Amsterdam, The Netherlands, 2015; Volume 63, pp. 203–288. [Google Scholar]
- Hameury, S.; de Frémont, P.; Braunstein, P. Metal complexes with oxygen-functionalized NHC ligands: Synthesis and applications. Chem. Soc. Rev. 2017, 46, 632–733. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, M.G.; Ho, C.C. Recent advances in bidentate bis(N-heterocyclic carbene) transition metal complexes and their applications in metal-mediated reactions. Coord. Chem. Rev. 2018, 375, 373–388. [Google Scholar] [CrossRef]
- Alcarazo, M.; Roseblade, S.J.; Cowley, A.R.; Fernández, R.; Brown, J.M.; Lassaletta, J.M. Imidazo[1,5-a]pyridine: A versatile architecture for stable N-heterocyclic carbenes. J. Am. Chem. Soc. 2005, 127, 3290–3291. [Google Scholar] [CrossRef] [PubMed]
- Burstein, C.; Lehmann, C.W.; Glorius, F. Imidazo[1,5-a]pyridine-3-ylidenes—Pyridine derived N-heterocyclic carbene ligands. Tetrahedron 2005, 61, 6207–6217. [Google Scholar] [CrossRef]
- Check, C.T.; Jang, K.P.; Schwamb, C.B.; Wong, A.S.; Wang, M.H.; Scheidt, K.A. Ferrocene-Based Planar Chiral Imidazopyridinium Salts for Catalysis. Angew. Chem. Int. Ed. 2015, 54, 4264–4268. [Google Scholar] [CrossRef]
- Espina, M.; Rivilla, I.; Conde, A.; Díaz-Requejo, M.M.; Pérez, P.J.; Álvarez, E.; Fernández, R.; Lassaletta, J.M. Chiral, Sterically Demanding N-Heterocyclic Carbenes Fused into a Heterobiaryl Skeleton: Design, Synthesis, and Structural Analysis. Organometallics 2015, 34, 1328–1338. [Google Scholar] [CrossRef]
- Grande-Carmona, F.; Iglesias-Sigüenza, J.; Álvarez, E.; Díez, E.; Fernández, R.; Lassaletta, J.M. Synthesis and characterization of axially chiral imidazoisoquinolin-2-ylidene silver and gold complexes. Organometallics 2015, 34, 5073–5080. [Google Scholar] [CrossRef]
- Iglesias-Sigüenza, J.; Izquierdo, C.; Díez, E.; Fernández, R.; Lassaletta, J.M. Chirality and catalysis with aromatic N-fused heterobicyclic carbenes. Dalton Trans. 2016, 45, 10113–10117. [Google Scholar] [CrossRef] [Green Version]
- Schwamb, C.B.; Fitzpatrick, K.P.; Brueckner, A.C.; Richardson, H.C.; Cheong, P.H.-Y.; Scheidt, K.A. Enantioselective synthesis of α-amidoboronates catalyzed by planar-Chiral NHC-Cu (I) complexes. J. Am. Chem. Soc. 2018, 140, 10644–10648. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chen, K.; Chen, W.; Liu, M.; Wu, H. Well-Designed N-Heterocyclic Carbene Ligands for Palladium-Catalyzed Denitrative C–N Coupling of Nitroarenes with Amines. ACS Catal. 2019, 9, 8110–8115. [Google Scholar] [CrossRef]
- Tang, Y.; Benaissa, I.; Huynh, M.; Vendier, L.; Lugan, N.; Bastin, S.; Belmont, P.; César, V.; Michelet, V. An original L-shape, tunable N-Heterocyclic Carbene platform for efficient gold (I) catalysis. Angew. Chem. Int. Ed. 2019, 58, 7977–7981. [Google Scholar] [CrossRef]
- Zhang, J.-Q.; Liu, Y.; Wang, X.-W.; Zhang, L. Synthesis of Chiral Bifunctional NHC Ligands and Survey of Their Utilities in Asymmetric Gold Catalysis. Organometallics 2019, 38, 3931–3938. [Google Scholar] [CrossRef]
- Grohmann, C.; Hashimoto, T.; Fröhlich, R.; Ohki, Y.; Tatsumi, K.; Glorius, F. An iron (II) complex of a diamine-bridged bis-N-heterocyclic carbene. Organometallics 2012, 31, 8047–8050. [Google Scholar] [CrossRef]
- Kriechbaum, M.; List, M.; JF Berger, R.; Patzschke, M.; Monkowius, U. Silver and Gold Complexes with a New 1, 10-Phenanthroline Analogue N-Heterocyclic Carbene: A Combined Structural, Theoretical, and Photophysical Study. Chem. A Eur. J. 2012, 18, 5506–5509. [Google Scholar] [CrossRef]
- Kriechbaum, M.; Winterleitner, G.; Gerisch, A.; List, M.; Monkowius, U. Synthesis, Characterization and Luminescence of Gold Complexes Bearing an NHC Ligand Based on the Imidazo[1,5-a]quinolinol Scaffold. Eur. J. Inorg. Chem. 2013, 2013, 5567–5575. [Google Scholar] [CrossRef]
- Nakano, R.; Nozaki, K. Copolymerization of propylene and polar monomers using Pd/IzQO catalysts. J. Am. Chem. Soc. 2015, 137, 10934–10937. [Google Scholar] [CrossRef]
- Tao, W.J.; Nakano, R.; Ito, S.; Nozaki, K. Copolymerization of ethylene and polar monomers by using Ni/IzQO catalysts. Angew. Chem. Int. Ed. 2016, 55, 2835–2839. [Google Scholar] [CrossRef]
- Tao, W.; Akita, S.; Nakano, R.; Ito, S.; Hoshimoto, Y.; Ogoshi, S.; Nozaki, K. Copolymerisation of ethylene with polar monomers by using palladium catalysts bearing an N-heterocyclic carbene–phosphine oxide bidentate ligand. Chem. Commun. 2017, 53, 2630–2633. [Google Scholar] [CrossRef]
- Azouzi, K.; Duhayon, C.; Benaissa, I.; Lugan, N.l.; Canac, Y.; Bastin, S.P.; César, V. Bidentate iminophosphorane-NHC ligand derived from the imidazo[1,5-a]pyridin-3-ylidene scaffold. Organometallics 2018, 37, 4726–4735. [Google Scholar] [CrossRef]
- Yasuda, H.; Nakano, R.; Ito, S.; Nozaki, K. Palladium/IzQO-catalyzed coordination–insertion copolymerization of ethylene and 1, 1-disubstituted ethylenes bearing a polar functional group. J. Am. Chem. Soc. 2018, 140, 1876–1883. [Google Scholar] [CrossRef]
- Dong, J.; Li, M.; Wang, B. Synthesis, Structures, and Norbornene Polymerization Behavior of Imidazo[1,5-a]pyridine-sulfonate-Ligated Palladacycles. Organometallics 2019, 38, 3786–3795. [Google Scholar] [CrossRef]
- Tao, W.; Wang, X.; Ito, S.; Nozaki, K. Palladium complexes bearing an N-heterocyclic carbene–sulfonamide ligand for cooligomerization of ethylene and polar monomers. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 474–477. [Google Scholar] [CrossRef]
- Greenburg, Z.R.; Jin, D.; Williard, P.G.; Bernskoetter, W.H. Nickel promoted functionalization of CO2 to anhydrides and ketoacids. Dalton Trans. 2014, 43, 15990–15996. [Google Scholar] [CrossRef]
- Gaydou, M.; Moragas, T.; Juliá-Hernández, F.; Martin, R. Site-selective catalytic carboxylation of unsaturated hydrocarbons with CO2 and water. J. Am. Chem. Soc. 2017, 139, 12161–12164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aresta, M.; Pastore, C.; Giannoccaro, P.; Kovács, G.; Dibenedetto, A.; Pápai, I. Evidence for Spontaneous Release of Acrylates from a Transition-Metal Complex Upon Coupling Ethene or Propene with a Carboxylic Moiety or CO2. Chem. A Eur. J. 2007, 13, 9028–9034. [Google Scholar] [CrossRef] [PubMed]
- Loch, J.A.; Albrecht, M.; Peris, E.; Mata, J.; Faller, J.W.; Crabtree, R.H. Palladium complexes with tridentate pincer bis-carbene ligands as efficient catalysts for C–C coupling. Organometallics 2002, 21, 700–706. [Google Scholar] [CrossRef]
- Langer, J.; Fischer, R.; Görls, H.; Walther, D. A new set of nickelacyclic carboxylates (“nickelalactones”) containing pyridine as supporting ligand: Synthesis, structures and application in C–C–and C–S–linkage reactions. J. Organomet. Chem. 2004, 689, 2952–2962. [Google Scholar] [CrossRef]
- Ebisu, Y.; Kawamura, K.; Hayashi, M. Enantioselective copper-catalyzed 1,4-addition of dialkylzincs to enones using a novel N,N,P-Cu(II) complex. Tetrahedron Asymmetry 2012, 23, 959–964. [Google Scholar] [CrossRef]
- Hutt, J.T.; Aron, Z.D. Efficient, single-step access to imidazo[1,5-a]pyridine N-heterocyclic carbene precursors. Org. Lett. 2011, 13, 5256–5259. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, Y.; Hur, M.Y.; Lee, E. Efficient synthesis of bulky N-Heterocyclic carbene ligands for coinage metal complexes. J. Organomet. Chem. 2016, 820, 1–7. [Google Scholar] [CrossRef]
- De Buysser, K.; Herman, G.; Bruneel, E.; Hoste, S.; Van Driessche, I. Determination of the number of unpaired electrons in metal-complexes. A comparison between the Evans’ method and susceptometer results. Chem. Phys. 2005, 315, 286–292. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.H.; Incarvito, C.D.; Crabtree, R.H. Interplay of linker, N-substituent, and counterion effects in the formation and geometrical distortion of N-heterocyclic biscarbene complexes of rhodium (I). Organometallics 2006, 25, 6099–6107. [Google Scholar] [CrossRef]
- Hartwig, J. Organotransition Metal Chemistry: From Bonding to Catalysis; Univ. Science Books: California, CA, USA, 2010. [Google Scholar]
- Ramírez-Monroy, A.; Swager, T.M. Metal chelates based on isoxazoline[60]fullerenes. Organometallics 2011, 30, 2464–2467. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Liu, Y.-H.; Peng, S.-M.; Liu, S.-T. Rhodium (I) complexes containing a bulky pyridinyl N-heterocyclic carbene ligand: Preparation and reactivity. J. Organomet. Chem. 2006, 691, 4012–4020. [Google Scholar] [CrossRef]
- Cheng, M.; Moore, D.R.; Reczek, J.J.; Chamberlain, B.M.; Lobkovsky, E.B.; Coates, G.W. Single-site β-diiminate zinc catalysts for the alternating copolymerization of CO2 and epoxides: Catalyst synthesis and unprecedented polymerization activity. J. Am. Chem. Soc. 2001, 123, 8738–8749. [Google Scholar] [CrossRef]
- Dai, S.; Sui, X.; Chen, C. Highly Robust Palladium (II) α-Diimine Catalysts for Slow-Chain-Walking Polymerization of Ethylene and Copolymerization with Methyl Acrylate. Angew. Chem. Int. Ed. 2015, 54, 9948–9953. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Ni(II) | Additives | % Acrylate a |
|---|---|---|---|
| 1 | 7 | none | 34 |
| 2 | 8a | none | 85 |
| 3 | 8b | none | 80 |
| 4 | 8c | none | 108 |
| 5 | 8d | none | 50 |
| 6 | 8e | none | 62 |
| 7 | 12 | none | n/a b |
| 8 | 7 | PCy3 | 180 |
| 9 | 8a | PCy3 | 677 |
| 10 | 8b | PCy3 | 845 |
| 11 | 8c | PCy3 | 728 |
| 12 | 8d | PCy3 | 194 |
| 13 | 8e | PCy3 | 814 |
| 14 | (DME)NiCl2 | PCy3 | n/a b |
| 15 c | (DME)NiBr2 | DPPE | 1200 |
| 16 c | (DME)NiBr2 | DCPE | 1500 |
| 17 c | (DME)NiBr2 | DPPP | 900 |
| 18 c | (DME)NiBr2 | DCPP | 1000 |
| 19 d | Ni(COD)2 | DCPE | 800 |
| 20 e | Ni(COD)2 | DCPP | 1400 |
| 21 c | (DME)NiBr2 | bpy | 0 |
| 22 c | (DME)NiBr2 | Dtbbpy | 0 |

| Entry | Base | Solvent | Red. | MX | % Acrylate a |
|---|---|---|---|---|---|
| 1 | NEt3 | PhCl | Zn | LiI | 845 |
| 2 | DIPEA | PhCl | Zn | LiI | 34 |
| 3 | py | PhCl | Zn | LiI | 80 |
| 4 | K2CO3 | PhCl | Zn | LiI | 0 |
| 5 | Cs2CO3 | PhCl | Zn | LiI | 0 |
| 6 | DABCO | PhCl | Zn | LiI | 0 |
| 7 | TMEDA | PhCl | Zn | LiI | 0 |
| 8 | 2-F-PhONa | PhCl | Zn | - | 0 |
| 9 | 2,6-Me-PhONa | PhCl | Zn | - | 0 |
| 10 | 2,6-Me-PhONa | PhCl | Zn | NaI | 0 |
| 11 | t-BuOK | PhCl | Zn | - | 0 |
| 12 | t-BuOK | PhCl | Zn | LiI | 0 |
| 13 | NEt3 | PhCl | Zn | NaI | 0 |
| 14 | NEt3 | toluene | Zn | LiI | 54 |
| 15 | NEt3 | anisole | Zn | LiI | 416 |
| 16 | NEt3 | benzene | Zn | LiI | 85 |
| 17 | NEt3 | 2-Cl-Tol | Zn | LiI | 799 |
| 18 | NEt3 | THF | Zn | LiI | 0 |
| 19 | NEt3 | CH2Cl2 | Zn | LiI | 0 |
| 20 | NEt3 | DMF | Zn | LiI | 0 |
| 21 | NEt3 | 1,4-dioxane | Zn | LiI | 0 |
| 22 | NEt3 | PhCl | - | LiI | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Hahm, H.; Ryu, J.Y.; Byun, S.; Park, D.-A.; Lee, S.H.; Lim, H.; Lee, J.; Hong, S. Pyridine-Chelated Imidazo[1,5-a]Pyridine N-Heterocyclic Carbene Nickel(II) Complexes for Acrylate Synthesis from Ethylene and CO2. Catalysts 2020, 10, 758. https://doi.org/10.3390/catal10070758
Kim J, Hahm H, Ryu JY, Byun S, Park D-A, Lee SH, Lim H, Lee J, Hong S. Pyridine-Chelated Imidazo[1,5-a]Pyridine N-Heterocyclic Carbene Nickel(II) Complexes for Acrylate Synthesis from Ethylene and CO2. Catalysts. 2020; 10(7):758. https://doi.org/10.3390/catal10070758
Chicago/Turabian StyleKim, Jiyun, Hyungwoo Hahm, Ji Yeon Ryu, Seunghwan Byun, Da-Ae Park, Seoung Ho Lee, Hyunseob Lim, Junseong Lee, and Sukwon Hong. 2020. "Pyridine-Chelated Imidazo[1,5-a]Pyridine N-Heterocyclic Carbene Nickel(II) Complexes for Acrylate Synthesis from Ethylene and CO2" Catalysts 10, no. 7: 758. https://doi.org/10.3390/catal10070758