Atom-economical Synthesis and Characterization of Poly(oxindolidene thienylene vinylene) Based on Aldol Polycondensation Reaction

Abstract
:1. Introduction
2. Results and Discussion
2.1. Polymerizations
2.2. Optical Properties and Microstructures
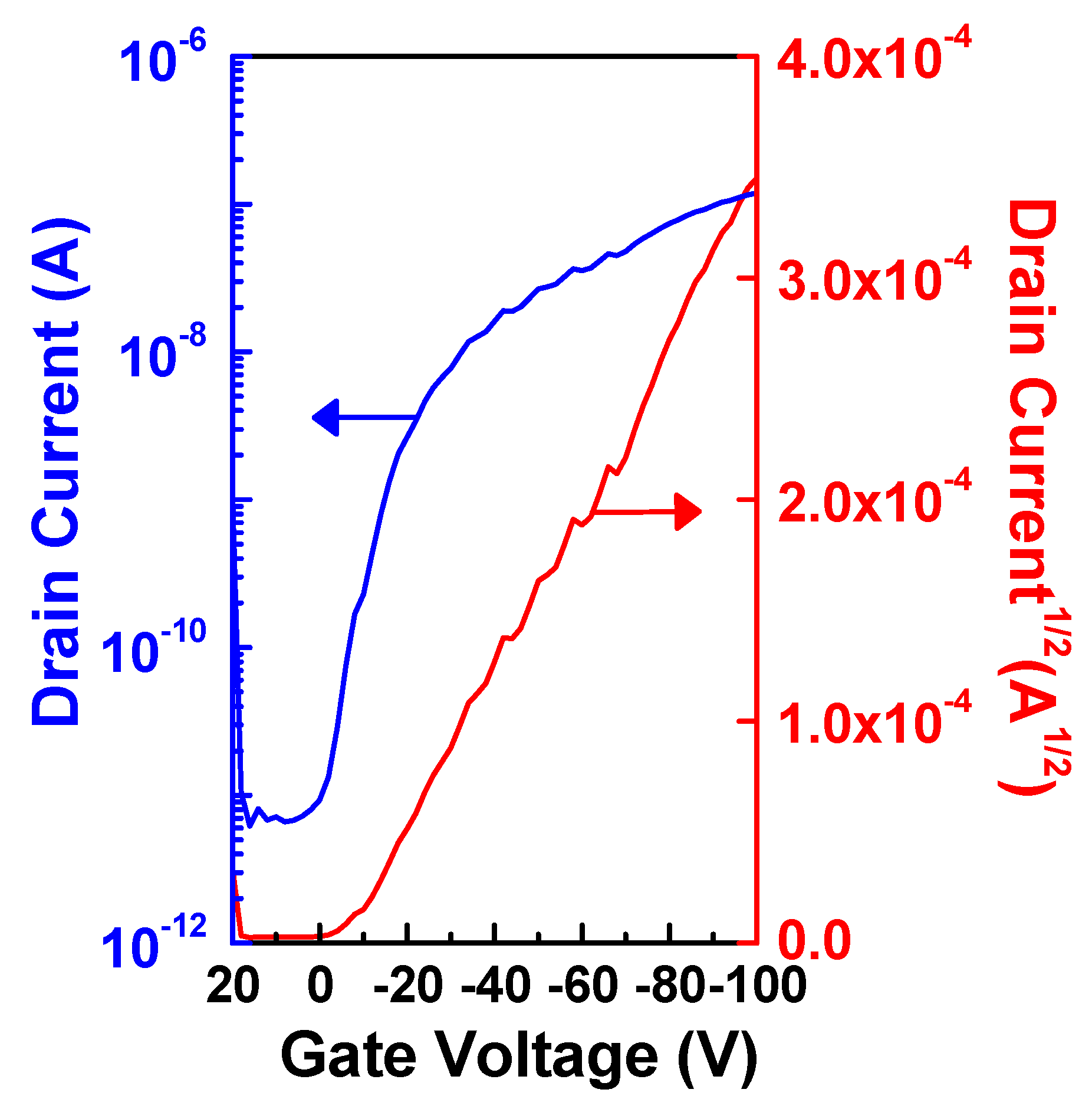
2.3. OFET Performances
3. Experimental
3.1. Materials
3.2. Synthesis of OTV
3.2.1. 1-Bromo-2-hexyldecane (1)
3.2.2. 6-Bromo-(N-2-hexyldecyl)isatin (2)
3.2.3. 6-Bromo-(N-2-hexyldecyl)oxindole (3)
3.2.4. 5-Iodothiophene-2-carbaldehyde (4)
3.2.5. 5-(Trimetyltin)thiophene-2-carbaldehyde (5)
3.2.6. N-(2-hexyldecyl)oxindolidene thienylene vinylene (OTV)
3.3. Synthesis of POTV
3.4. Measurements and Characterization
3.5. OFET Fabrication
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boudreault, P.L.T.; Najari, A.; Leclerc, M. Processable low-bandgap polymers for photovoltaic applications. Chem. Mater. 2011, 23, 456–469. [Google Scholar] [CrossRef]
- Beaujuge, P.M.; Fréchet, J.M.J. Molecular design and ordering effects in π-functional materials for transistor and solar cell applications. J. Am. Chem. Soc. 2011, 133, 20009–20029. [Google Scholar] [CrossRef]
- Dou, L.; Liu, Y.; Hong, Z.; Li, G.; Yang, Y. Low-bandgap near-IR conjugated polymers/molecules for organic electronics. Chem. Rev. 2015, 115, 12633–12665. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zheng, T.; Wu, Q.; Schneider, A.M.; Zhao, D.; Yu, L. Recent advances in bulk heterojunction polymer solar cells. Chem. Rev. 2015, 115, 12666–12731. [Google Scholar] [CrossRef] [PubMed]
- Holliday, S.; Li, Y.; Luscombe, C.K. Recent advances in high performance donor-acceptor polymers for organic photovoltaics. Prog. Polym. Sci. 2017, 70, 34–51. [Google Scholar] [CrossRef]
- Mizoroki, T.; Mori, K.; Ozaki, A. Arylation of olefin with aryl iodide catalyzed by palladium. Bull. Chem. Soc. Jpn. 1971, 44, 581. [Google Scholar] [CrossRef]
- Heck, R.F.; Nolley, J.P., Jr. Palladium-catalyzed vinylic hydrogen substitution reactions with aryl, benzyl, and styryl halides. J. Org. Chem. 1972, 37, 2320–2322. [Google Scholar] [CrossRef]
- Tamao, K.; Sumitani, K.; Kumada, M. Selective carbon-carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel-phosphine complexes. J. Am. Chem. Soc. 1972, 94, 4374–4376. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A convenient synthesis of acetylenes: Catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 1975, 50, 4467–4470. [Google Scholar] [CrossRef]
- King, A.O.; Okukado, N.; Negishi, E. Highly general stereo-, regio-, and chemo-selective synthesis of terminal and internal conjugated enynes by the Pd-catalysed reaction of alkynylzinc reagents with alkenyl halides. J. Chem. Soc. Chem. Commun. 1977, 683–684. [Google Scholar] [CrossRef]
- Negishi, E.; King, A.O.; Okukado, N. Selective carbon-carbon bond formation via transition metal catalysis. 3. A highly selective synthesis of unsymmetrical biaryls and diarylmethanes by the nickel- or palladium-catalyzed reaction of aryl- and benzylzinc derivatives with aryl halides. J. Org. Chem. 1977, 42, 1821–1823. [Google Scholar] [CrossRef]
- Kosugi, M.; Sasazawa, K.; Shimizu, Y.; Migita, T. Reactions of allyltin compounds III. Allylation of aromatic halides with allyltributyltin in the presence of tetrakis(triphenylphosphine)palladium(0). Chem. Lett. 1977, 6, 301–302. [Google Scholar] [CrossRef]
- Milstein, D.; Stille, J.K. A general, selective, and facile method for ketone synthesis from acid chlorides and organotin compounds catalyzed by palladium. J. Am. Chem. Soc. 1978, 100, 3636–3638. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Stereoselective synthesis of arylated (E)-alkenes by the reaction of alk-1-enylboranes with aryl halides in the presence of palladium catalyst. J. Chem. Soc. Chem. Commun. 1979, 866–867. [Google Scholar] [CrossRef]
- Miyaura, N.; Yamada, K.; Suzuki, A. A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 20, 3437–3440. [Google Scholar] [CrossRef] [Green Version]
- Carsten, B.; He, F.; Son, H.J.; Xu, T.; Yu, L. Stille polycondensation for synthesis of functional materials. Chem. Rev. 2011, 111, 1493–1528. [Google Scholar] [CrossRef]
- Kuwabara, J.; Yasuda, T.; Takase, N.; Kanbara, T. Effects of the terminal structure, purity, and molecular weight of an amorphous conjugated polymer on its photovoltaic characteristics. ACS Appl. Mater. Interfaces 2016, 8, 1752–1758. [Google Scholar] [CrossRef]
- Zhang, G.; Dai, Y.; Liu, Y.; Liu, J.; Lu, H.; Qiu, L.; Cho, K. Facile green synthesis of isoindigo-based conjugated polymers using aldol polycondensation. Polym. Chem. 2017, 8, 3448–3456. [Google Scholar] [CrossRef]
- Onwubiko, A.; Yue, W.; Jellett, C.; Xiao, M.; Chen, H.Y.; Ravva, M.K.; Hanifi, D.A.; Knall, A.C.; Purushothaman, B.; Nikolka, M.; et al. Fused electron deficient semiconducting polymers for air stable electron transport. Nat. Commun. 2018, 9, 416. [Google Scholar] [CrossRef]
- Himmelberger, S.; Vandewal, K.; Fei, Z.; Heeney, M.; Salleo, A. Role of molecular weight distribution on charge transport in semiconducting polymers. Macromolecules 2014, 47, 7151–7157. [Google Scholar] [CrossRef]
- Lu, L.; Zheng, T.; Xu, T.; Zhao, D.; Yu, L. Mechanistic studies of effect of dispersity on the photovoltaic performance of PTB7 polymer solar cells. Chem. Mater. 2015, 27, 537–543. [Google Scholar] [CrossRef]
- Yokoyama, A.; Miyakoshi, R.; Yokozawa, T. Chain-growth polymerization for poly(3-hexylthiophene) with a defined molecular weight and a low polydispersity. Macromolecules 2004, 37, 1169–1171. [Google Scholar] [CrossRef]
- Iovu, M.C.; Sheina, E.E.; Gil, R.R.; McCullough, R.D. Experimental evidence for the quasi-“living” nature of the grignard metathesis method for the synthesis of regioregular poly(3-alkylthiophenes). Macromolecules 2005, 38, 8649–8656. [Google Scholar] [CrossRef]
- Sheina, E.E.; Liu, J.; Lovu, M.C.; Laird, D.W.; McCullough, R.D. Chain growth mechanism for regioregular nickel-initiated cross-coupling polymerizations. Macromolecules 2004, 37, 3526–3528. [Google Scholar] [CrossRef]
- Higashihara, T.; Goto, E.; Ueda, M. Purification-free and protection-free synthesis of regioregular poly(3-hexylthiophene) and poly(3-(6-hydroxyhexyl)thiophene) using a zincate complex of tBu4ZnLi2. ACS Macro Lett. 2012, 1, 167–170. [Google Scholar] [CrossRef]
- Higashihara, T.; Goto, E. Controlled synthesis of low-polydisperse regioregular poly(3- hexylthiophene) and related materials by zincate-complex metathesis polymerization. Polym. J. 2014, 46, 381–390. [Google Scholar] [CrossRef]
- Goto, E.; Mori, H.; Ueda, M.; Higashihara, T. Controlled polymerization of electron-deficient naphthalene-diimide containing monomer by negishi-type catalyst-transfer polymerization. J. Photopolym. Sci. Technol. 2015, 28, 279–283. [Google Scholar] [CrossRef] [Green Version]
- Goto, E.; Ochiai, Y.; Ueda, M.; Higashihara, T. Transition-metal-free and halogen-free controlled synthesis of poly(3-alkylthienylene vinylene): Via the Horner-Wadsworth-Emmons condensation reaction. Polym. Chem. 2018, 9, 1996–2001. [Google Scholar] [CrossRef]
- Yokozawa, T.; Muroya, D.; Sugi, R.; Yokoyama, A. Convenient method of chain-growth polycondensation for well-defined aromatic polyamides. Macromol. Rapid Commun. 2005, 26, 979–981. [Google Scholar] [CrossRef]
- Shao, M.; Keum, J.; Chen, J.; He, Y.; Chen, W.; Browning, J.F.; Jakowski, J.; Sumpter, B.G.; Ivanov, I.N.; Ma, Y.-Z.; et al. The isotopic effects of deuteration on optoelectronic properties of conducting polymers. Nat. Commum. 2014, 5, 3180. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run a | Base | Solvent | Temp. | Mnb | ÐM | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | sodium hexamethyldisilazide (NaHMDS)/15-crown-5 | Tetrahydrofuran (THF) | −40 °C | 1300 | 1.17 | 71 c |
| 2 | NaHMDS/15-crown-5 | THF | r. t. | 2100 | 1.97 | 58 c |
| 3 | NaHMDS/15-crown-5 | Toluene/THF f | r. t. | 2600 | 1.46 | ~100 d |
| 4 | lithium hexamethyldisilazide (LiHMDS)/12-crown-4 | Toluene/THF g | r. t. | 2500 | 1.37 | 66 c |
| 5 e | LiHMDS/12-crown-4 | Toluene/THF g | r. t. | 3000 | 1.45 | 73 c |
| 6 e | LiHMDS/12-crown-4 | Toluene/THF g | 30 °C | 3700 (5300) h | 1.47 (1.57) h | 65 c (40) h |
| Conditions | λmax (nm) | ε (L/mol/cm) | λonset (nm) | Eg (eV) |
|---|---|---|---|---|
| Solution | 486 | 2.47 × 104 | 677 | 1.82 |
| As-cast | 433, 577 | - | 740 | 1.66 |
| Annealed at 200 °C | 431, 588 | - | 748 | 1.64 |
| ION/IOFF | Max mobility (cm V−1 s−1) | Threshold voltage (V) | W/L | |
|---|---|---|---|---|
| POTV (Run 6) | 1.92 × 104 | 2.24 × 10−4 | −21.6 | 25 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, S.; Lin, P.-S.; Wu, W.-N.; Liu, C.-L.; Higashihara, T. Atom-economical Synthesis and Characterization of Poly(oxindolidene thienylene vinylene) Based on Aldol Polycondensation Reaction. Catalysts 2020, 10, 364. https://doi.org/10.3390/catal10040364
Sato S, Lin P-S, Wu W-N, Liu C-L, Higashihara T. Atom-economical Synthesis and Characterization of Poly(oxindolidene thienylene vinylene) Based on Aldol Polycondensation Reaction. Catalysts. 2020; 10(4):364. https://doi.org/10.3390/catal10040364
Chicago/Turabian StyleSato, Shingo, Po-Shen Lin, Wei-Ni Wu, Cheng-Liang Liu, and Tomoya Higashihara. 2020. "Atom-economical Synthesis and Characterization of Poly(oxindolidene thienylene vinylene) Based on Aldol Polycondensation Reaction" Catalysts 10, no. 4: 364. https://doi.org/10.3390/catal10040364