Effects of Modifying Acidity and Reducibility on the Activity of NaY Zeolite in the Oxidative Dehydrogenation of n-Octane


Abstract
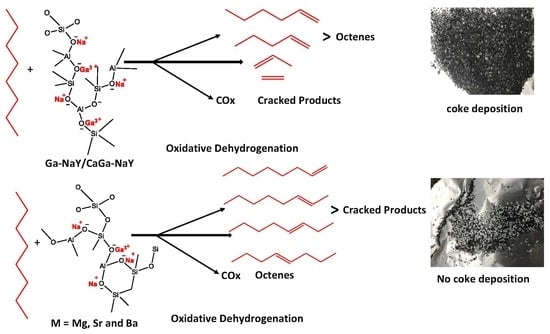
:
1. Introduction
2. Results and Discussion
2.1. Catalysts Characterization
2.1.1. Powder XRD
2.1.2. FT-IR
2.1.3. ICP-OES Studies
2.1.4. Temperature Programmed Desorption with NH3 and CO2
2.1.5. Pyridine FT-IR
2.1.6. Temperature Programmed Reduction
2.2. Catalytic Performance
2.2.1. Ionic Exchange Capacity Study
2.2.2. Product Selectivity at iso-Conversion
3. Materials and Experimental Methods
3.1. Catalysts Preparation
3.2. Catalysts Characterization
3.3. Catalytic Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Farahani, M.D.; Dasireddy, V.D.B.C.; Friedrich, H.B. Oxidative Dehydrogenation of n-Octane over Niobium-Doped NiAl2O4: An Example of Beneficial Coking in Catalysis over Spinel. ChemCatChem 2018, 10, 2059–2069. [Google Scholar] [CrossRef]
- Bandaru, H.; Mahomed, A.S.; Singh, S.; Friedrich, H.B. The Effect of Varying the Metal Ratio in Chromium Molybdate Catalysts for the Oxidative Dehydrogenation of n-Octane. Mol. Catal. 2018, 460, 74–82. [Google Scholar] [CrossRef]
- Fadlalla, M.I.; Farahani, M.D.; Friedrich, H.B. Three Inter-linked Active Sites in the Dehydrogenation of n-Octane over Magnesium Molybdate Based Catalysts and their Influence on Coking and Cracking Side Reactions. Mol. Catal. 2018, 461, 86–96. [Google Scholar] [CrossRef]
- Elkhalifa, E.A.; Friedrich, H.B. Effects of Boron and Barium Dopants on VMgO Catalysts Employed in the Oxidative Dehydrogenation of n-Octane. Kinet. Catal. 2015, 56, 212–221. [Google Scholar] [CrossRef]
- Obligacion, J.V.; Chirik, P.J. Earth-abundant Transition Metal Catalysts for Alkene Hydrosilylation and Hydroboration. Nat. Rev. Chem. 2018, 2, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Maleki, A. Green Oxidation Protocol: Selective Conversions of Alcohols and Alkenes to Al- dehydes, Ketones and Epoxides by Using a New Multiwall Carbon Nanotube-based Hybrid Nanocatalyst via Ultrasound Irradiation. Ultrason. Sonochem. 2017, 40, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Franke, R.; Selent, D.; Börner, A. Applied Hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef]
- Sattler, J.J.H.B.; Ruiz-Martinez, J.; Santillan-Jimenez, E.; Weckhuysen, B.M. Catalytic Dehydrogenation of Light Alkanes on Metals and Metal Oxides. Chem. Rev. 2014, 114, 10613–10653. [Google Scholar] [CrossRef]
- Shi, L.; Wang, Y.; Yan, B.; Song, W.; Shao, D.; Lu, A. Progress in Selective Oxidative Dehydrogenation of Light Alkanes to Olefins Promoted by Boron Nitride Catalysts. Chem. Commun. 2018, 54, 10936. [Google Scholar] [CrossRef]
- Yun, Y.S.; Lee, M.; Sung, J.; Yun, D.; Kim, T.Y.; Park, H.; Lee, K.R.; Song, C.K.; Kim, Y.; Lee, J.; et al. Promoting Effect of Cerium on MoVTeNb Mixed Oxide Catalyst for Oxidative Dehydrogenation of Ethane to Ethylene. Appl. Catal. B. 2018, 237, 554–562. [Google Scholar] [CrossRef]
- Grabowski, R. Kinetics of oxidative dehydrogenation of C2-C3 alkanes on oxide catalysts. Catal. Rev. 2007, 48, 199–268. [Google Scholar] [CrossRef]
- Gaab, S.; Machli, M.; Grasselli, R.K.; Lercher, J.A. Oxidative Dehydrogenation of Ethane over Novel Li/Dy/Mg Mixed Oxides: Structure–activity Study. Top. Catal. 2003, 23, 151–158. [Google Scholar] [CrossRef]
- Madeira, L.M.; Martìn-Aranda, R.M.; Maldonado-Hódar, F.J.; Fierro, J.L.G.; Portela, M.F. Oxidative Dehydrogenation of n-Butane over Alkali and Alkaline Earth-Promoted α-NiMoO4 Catalysts. J. Catal. 1997, 169, 469–479. [Google Scholar] [CrossRef]
- Mulla, S.A.R.; Choudhary, V.R. Oxidative Conversion of Ethane to Ethylene over Supported SrO-Promoted Er2O3 Catalyst. J. Mol. Catal. A Chem. 2004, 223, 259–262. [Google Scholar] [CrossRef]
- Maiti, A.; Govind, N.; Kung p King-Smith, D.; Miller, J.E. Effect of Surface Phosphorus on the Oxidative Dehydrogenation of Ethane: A First-Principles Investigation. J. Chem. Phys. 2002, 117, 8080–8088. [Google Scholar] [CrossRef]
- Dai, H.X.; Ng, C.F.; Au, C.T. SrCl2-promoted REOx (RE= Ce, Pr, Tb) catalysts for the selective oxidation of ethane: A study on performance and defect structures for ethene formation. J. Catal. 2001, 199, 177–192. [Google Scholar] [CrossRef]
- Gounden, N.; Friedrich, H.B.; Mahadevaiah, N.; Fadlalla, M.I. Octenes and Aromatics from the Oxidative Dehydrogenation of n-Octane over Co/TiO2 Catalysts. Catal. Lett. 2014, 144, 2043–2051. [Google Scholar] [CrossRef]
- Elkhalifa, E.A.; Friedrich, H.B. Oxidative Dehydrogenation and Aromatization of n-Octane over VMgO Catalysts Obtained by Using Different MgO Precursors and Different Precursor Treatments. J. Mol. Catal. A Chem. 2014, 392, 22–30. [Google Scholar] [CrossRef]
- Vedrine, J.C. Heterogeneous Catalytic Partial Oxidation of Lower Alkanes (C1–C6) on Mixed Metal Oxides. J. Energy Chem. 2016, 25, 936–946. [Google Scholar] [CrossRef]
- Atanga, M.A.; Rezaei, F.; Jawad, A.; Fitch, M.; Rownaghi, A.A. Oxidative Dehydrogenation of Propane to Propylene with Carbon Dioxide. Appl. Catal. B. 2018, 220, 429–445. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, F.; Zhang, Y.; Miao, C.; Hua, W.; Yue, Y.; Gao, Z. Oxidative Dehydrogenation of Ethane With CO2 over Cr Supported on Submicron ZSM-5 Zeolite. Chin. J. Catal. 2015, 36, 1242–1248. [Google Scholar] [CrossRef]
- Lin, X.; Hoel, C.A.; Sachtler, W.M.H.; Poeppelmeier, K.R.; Weitz, E. Oxidative Dehydrogenation (ODH) of Ethane With O2 as Oxidant on Selected Transition Metal-Loaded Zeolites. J. Catal. 2009, 265, 54–62. [Google Scholar] [CrossRef]
- Cheng, Y.; Lei, T.; Miao, C.; Hua, W.; Yue, Y.; Gao, Z. Ga2O3/NaZSM-5 for C2H6 dehydrogenation in the presence of CO2: Conjugated effect of silanol. Micropor. Mesopor. Mater. 2018, 268, 235–242. [Google Scholar] [CrossRef]
- Richardson, J.T.; Scates, R.M.; Twigg, M.V. X-ray diffraction study of the hydrogen reduction of NiO/α-Al2O3 steam reforming catalysts. Appl. Catal. A Gen. 2004, 267, 35–46. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, K.; Dong, D.; Li, D.; Hill, M.R.; Hill, A.J.; Wang, H. Synthesis of Hierarchical Porous Zeolite NaY Particles with Controllable Particle Sizes. Micropor. Mesopor. Mater. 2010, 127, 167–175. [Google Scholar] [CrossRef]
- Alwash, A.H.; Abdullah, A.Z.; Ismail, N. Elucidation of Reaction Behaviors in Sonocatalytic Decolorization of Amaranth Dye in Water Using Zeolite Y Co-incorporated with Fe and TiO2. Adv. Chem. Eng. Sci. 2013, 3, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Chanthaanont, P.; Sirivat, A. Effect of Transition Metal Ion-Exchanged into the Zeolite Y on Electrical Conductivity and Response of PEDOT-PSS/MY Composites toward SO2. Adv. Polym. Tech. 2013, 32, 21367–21372. [Google Scholar] [CrossRef]
- Zhao, J.; Yin, Y.; Li, Y.; Chen, W.; Liu, B. Synthesis and Characterization of Mesoporous Zeolite Y by Using Block Copolymers as Templates. Chem. Eng. J. 2016, 284, 405–411. [Google Scholar] [CrossRef]
- Pal, N.; Pramanik, M.; Bhaumik, A.; Ali, M. Highly Selective and Direct Oxidation of Cyclohexane to Cyclohexanone Over Vanadium Exchanged NaY at Room Temperature Under Solvent-Free Conditions. J. Mol. Catal. A Chem. 2014, 392, 299–307. [Google Scholar] [CrossRef]
- Król, M.; Mozgawa, W.; Jastrzębski, W.; Barczyk, K. Application of IR Spectra in the Studies of Zeolites from D4R and D6R Structural Groups. Micropor. Mesopor. Mater. 2012, 156, 181–188. [Google Scholar] [CrossRef]
- Shariatinia, Z.; Bagherpour, A. Synthesis of Zeolite NaY and its Nanocomposites With Chitosan as Adsorbents for Lead(II) Removal From Aqueous Solution. Powder Technol. 2018, 388, 744–763. [Google Scholar] [CrossRef]
- Datka, J.; Gil, B.; Baran, P. Heterogeneity of OH groups in HZSM-5 Zeolites: Splitting of OH and OD Bands in Low-Temperature IR Spectra. Micropor. Mesopor. Mater. 2003, 58, 291–294. [Google Scholar] [CrossRef]
- Niwa, M.; Katada, N. New Method for the Temperature- Programmed Desorption (TPD) of Ammonia Experiment for Characterization of Zeolite Acidity: A Review. Chem. Rec. 2013, 13, 432–455. [Google Scholar] [CrossRef] [PubMed]
- Brueva, T.R.; Mishin, I.V.; Kapustin, G.I. Distribution of Acid-site Strengths in Hydrogen Zeolites and Relationship Between Acidity and Aatalytic Activity. Thermochim. Acta 2001, 379, 15–23. [Google Scholar] [CrossRef]
- Almutairi, S.M.T.; Mezari, B.; Filonenko, G.A.; Magusin, P.C.M.M.; Rigutto, M.S.; Pidko, E.A.; Hensen, E.J.M. Influence of Extraframework Aluminum on the Brønsted Acidity and Catalytic Reactivity of Faujasite Zeolite. ChemCatChem 2013, 5, 452–466. [Google Scholar] [CrossRef]
- Jie, Y.; Dinghua, Y.; Peng, S.; He, H. Alkaline Earth Metal Modified NaY for Lactic Acid Dehydration to Acrylic Acid: Effect of Basic Sites on the Catalytic Performance. Chin. J. Catal. 2011, 32, 405–411. [Google Scholar]
- Liu, C.; Aika, K. Ammonia Adsorption on Ion Exchanged Y-zeolites as Ammonia Storage Material. J. Jpn. Petrol. Inst. 2003, 46, 301–307. [Google Scholar] [CrossRef]
- Choi, S.W.; Kim, W.G.; So, J.S.; Moore, J.S.; Liu, Y.J.; Dixit, R.S.; Pendergast, J.G.; Sievers, C.; Sholl, D.S.; Nair, S.; et al. Propane dehydrogenation catalyzed by gallosilicate MFI zeolites with perturbed acidity. J. Catal. 2017, 345, 113–123. [Google Scholar] [CrossRef]
- Ates, A.; Akgül, G. Modification of natural zeolite with NaOH for removal of manganese in drinking water. Powder Technol. 2016, 287, 285–291. [Google Scholar] [CrossRef]
- Xue, L.; He, H.; Liu, C.; Zhang, C.; Zhang, B. Promotion Effects and Mechanism of Alkali Metals and Alkaline Earth Metals on Cobalt-Cerium Composite Oxide Catalysts for N2O Decomposition. Environ. Sci. Tech. 2009, 43, 890–895. [Google Scholar] [CrossRef]
- Zhong, C.; Guo, X.; Mao, D.; Wang, S.; Wu, G.; Lu, G. Effects of Alkaline-Earth Oxides on the Performance of a CuO–ZrO2 Catalyst for Methanol Synthesis via CO2 Hydrogenation. RSC Adv. 2015, 5, 52958–52965. [Google Scholar] [CrossRef]
- Shao, C.; Lang, W.; Yan, X.; Guo, Y. Catalytic Performance of Gallium Oxide Based-Catalysts for the Propane Dehydrogenation Reaction: Effects of Support and Loading Amount. RSC Adv. 2017, 7, 4710–4723. [Google Scholar] [CrossRef] [Green Version]
- El-Malki, E.l.-M.; van Santen, R.A.; Sachtler, W.M.H. Introduction of Zn, Ga, and Fe into HZSM-5 Cavities by Sublimation: Identification of Acid Sites. J. Phys. Chem. B 1999, 103, 4611–4622. [Google Scholar] [CrossRef]
- Price, G.L.; Kanazi, V. Ga2O3/HZSM-5 Propane Aromatization Catalysts: Formation of Active Centers via Solid-State Reaction. J. Catal. 1990, 126, 267–278. [Google Scholar] [CrossRef]
- Li, J.; Zhang, C.; Cheng, X.; Qing, M.; Xu, J.; Wu, B.; Yang, Y.; Li, Y. Effects of Alkaline-Earth Metals on the Structure, Adsorption and Catalytic Behaviour of Iron-based Fischer–Tropsch Synthesis Catalysts. Appl. Catal. A Gen. 2013, 464–465, 10–19. [Google Scholar]
- Homeyer, S.T.; Sachtler, W.M.H. Elementary Steps in the Formation of Highly Dispersed Palladium in NaY. J. Catal. 1989, 118, 266–274. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Ga (wt %) | Alkali Metal (wt %) |
|---|---|---|
| GaNaY | 1.4 | - |
| CaGa-NaY | 1.1 | 1.7 |
| MgGa-NaY | 0.88 | 1.5 |
| SrGa-NaY | 1.0 | 1.5 |
| BaGa-NaY | 1.2 | 1.4 |
| Acidity Amount (µmol g−1) | ||||
|---|---|---|---|---|
| Zeolite | W (200 °C–300 °C) | M (300 °C–400 °C) | S (400 °C–600 °C) | Total Acidity |
| NaY | 30.9 | 0 | 0 | 30.9 |
| Ga-NaY | 21.0 | 31.6 | 0 | 52.6 |
| BaGa-NaY | 10.2 | 3.7 | 1.3 | 15.2 |
| MgGa-NaY | 10.3 | 11.8 | 4.3 | 26.4 |
| SrGa-NaY | 4.8 | 8.8 | 10.2 | 23.8 |
| CaGa-NaY | 19.9 | 14.5 | 7.0 | 41.4 |
| Basicity Amount (µmol g−1) | ||||
|---|---|---|---|---|
| Zeolite | W (0–160 °C) | M (160–400 °C) | S (> 400 °C) | Total Basicity |
| Ga-NaY | 76.5 | 13 | 3 | 92.5 |
| BaGa-NaY | 39 | 6.5 | 74 | 119.5 |
| MgGa-NaY | 66.5 | 23 | 16 | 105.5 |
| SrGa-NaY | 57 | 7.4 | 43 | 107.4 |
| CaGa-NaY | 57 | 5 | 0.4 | 62.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ndlela, S.S.; Friedrich, H.B.; Cele, M.N. Effects of Modifying Acidity and Reducibility on the Activity of NaY Zeolite in the Oxidative Dehydrogenation of n-Octane. Catalysts 2020, 10, 363. https://doi.org/10.3390/catal10040363
Ndlela SS, Friedrich HB, Cele MN. Effects of Modifying Acidity and Reducibility on the Activity of NaY Zeolite in the Oxidative Dehydrogenation of n-Octane. Catalysts. 2020; 10(4):363. https://doi.org/10.3390/catal10040363
Chicago/Turabian StyleNdlela, Siyabonga S., Holger B. Friedrich, and Mduduzi N. Cele. 2020. "Effects of Modifying Acidity and Reducibility on the Activity of NaY Zeolite in the Oxidative Dehydrogenation of n-Octane" Catalysts 10, no. 4: 363. https://doi.org/10.3390/catal10040363