Insights into the Pt (111) Surface Aid in Predicting the Selective Hydrogenation Catalyst


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
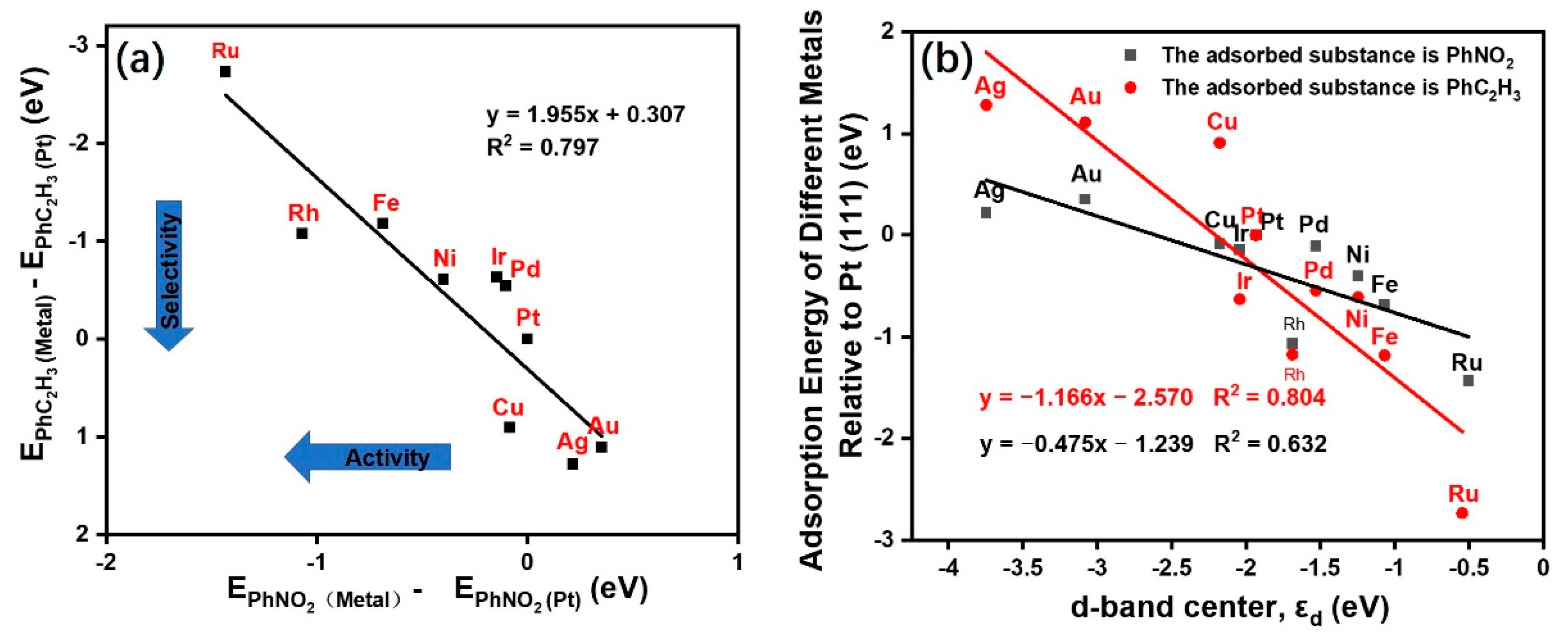
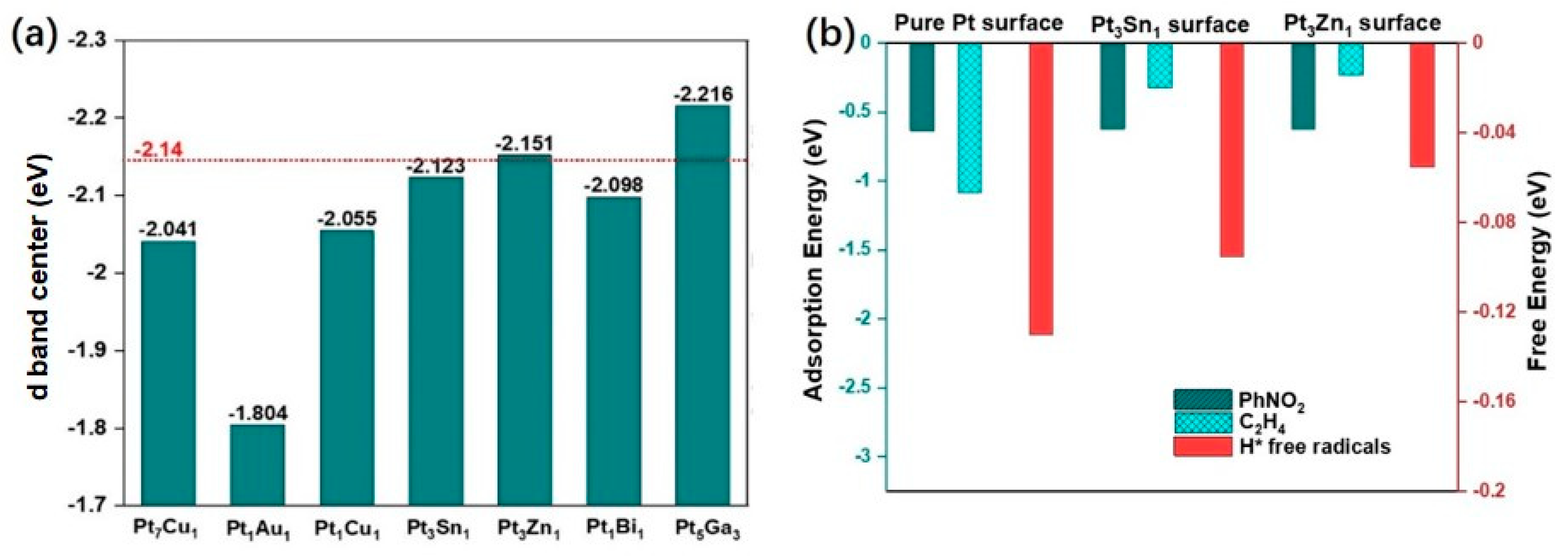
2.1. Screening of Transition Metals
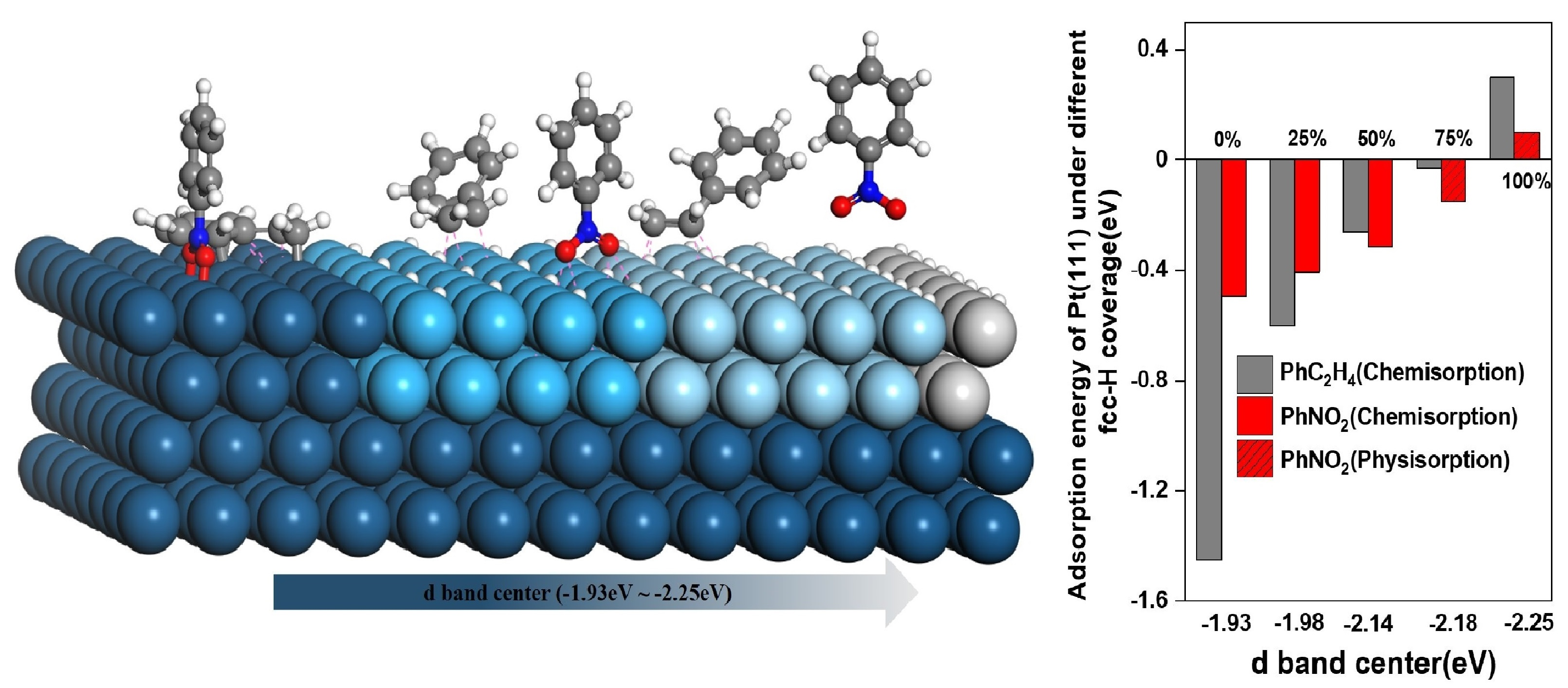
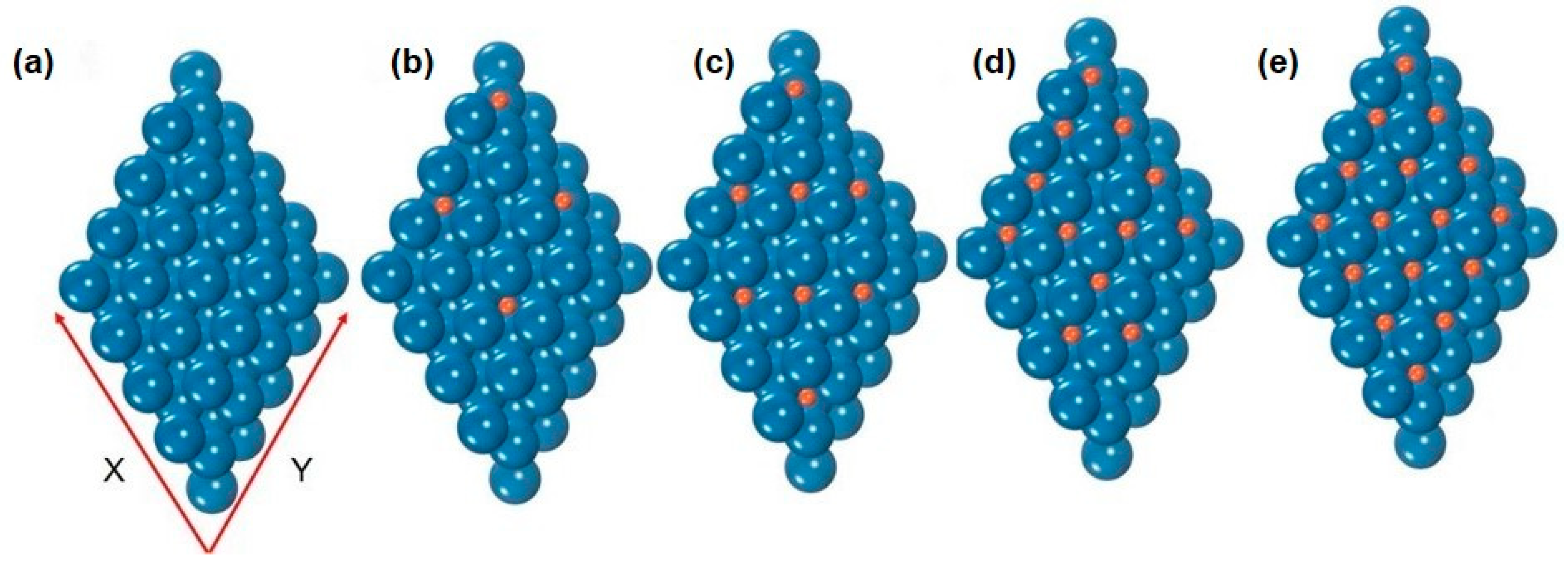
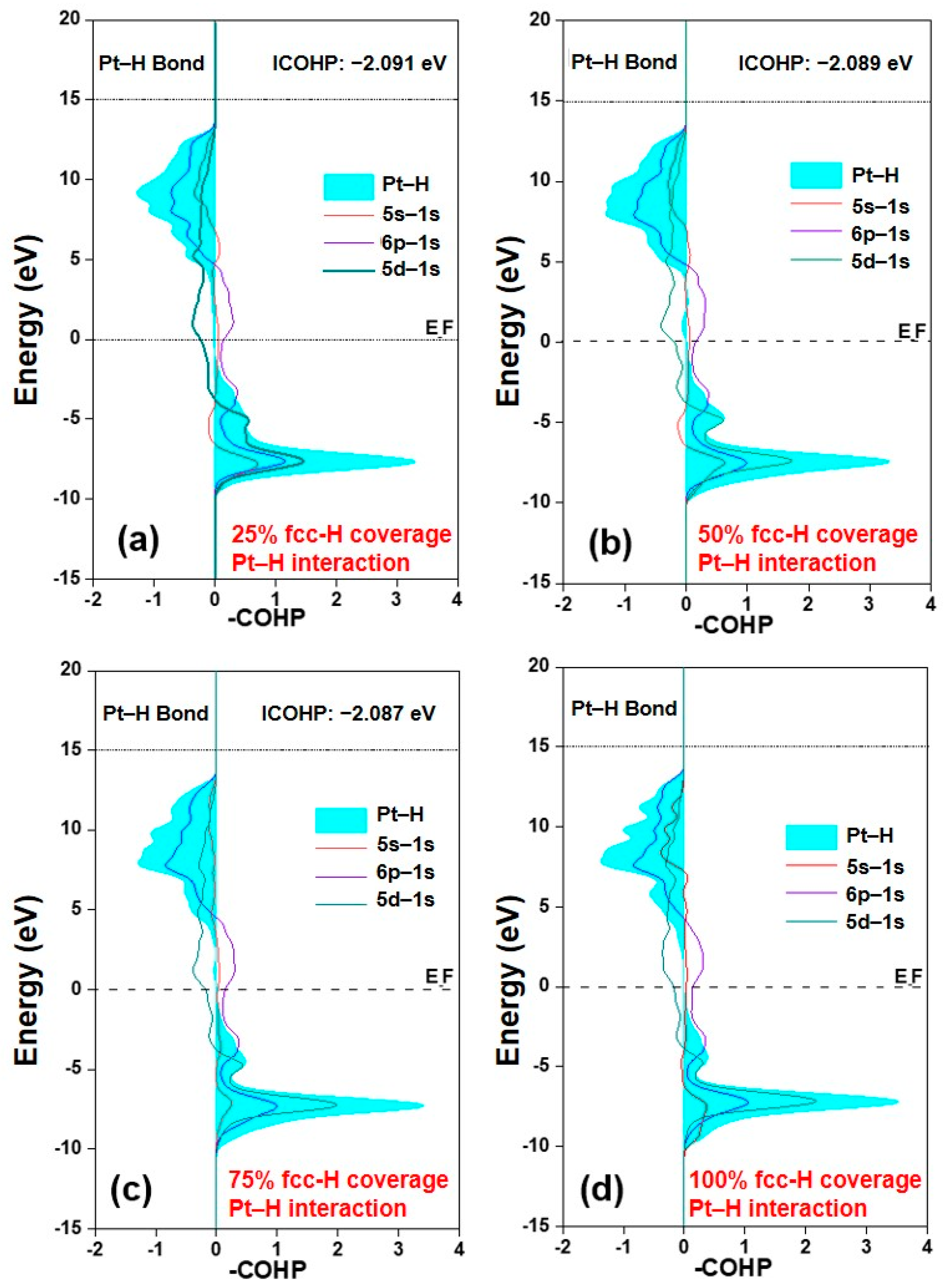
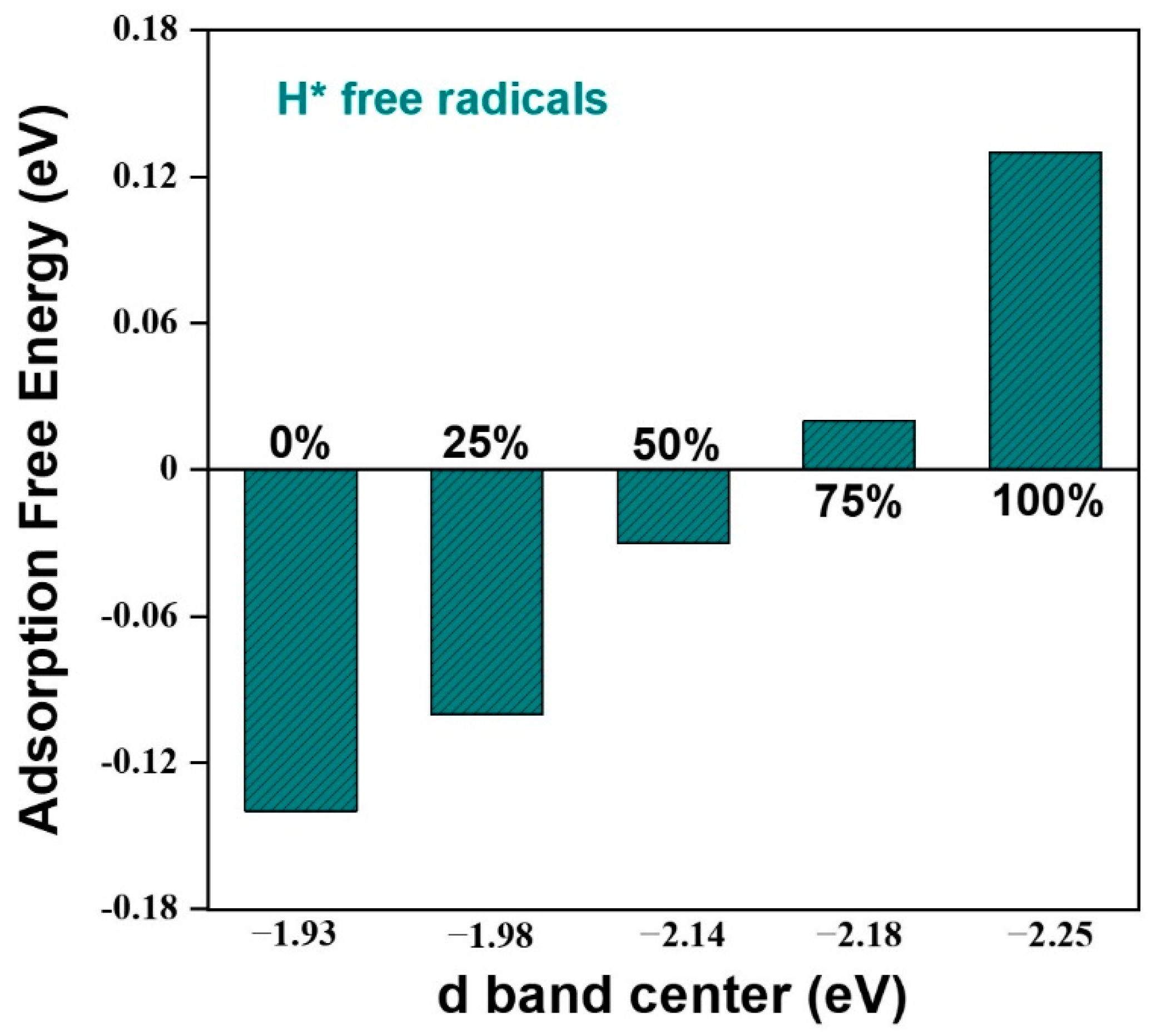
2.2. Regulation of the Electronic Properties of the Pt Surface by Hydrogen Coverage
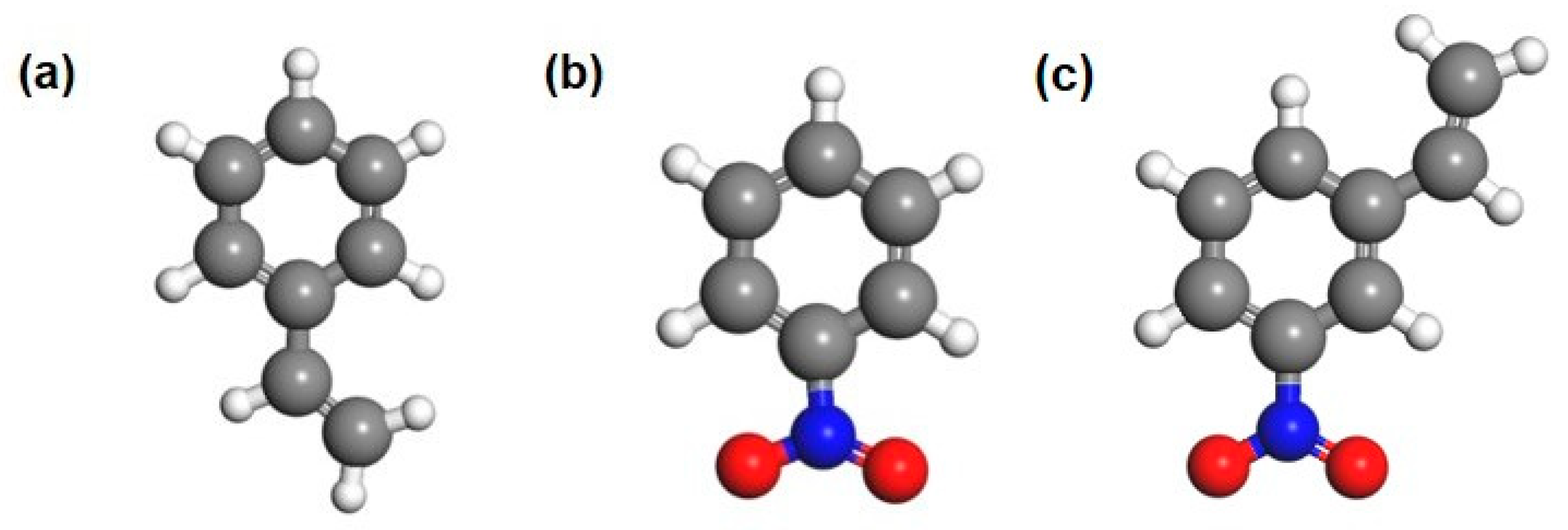
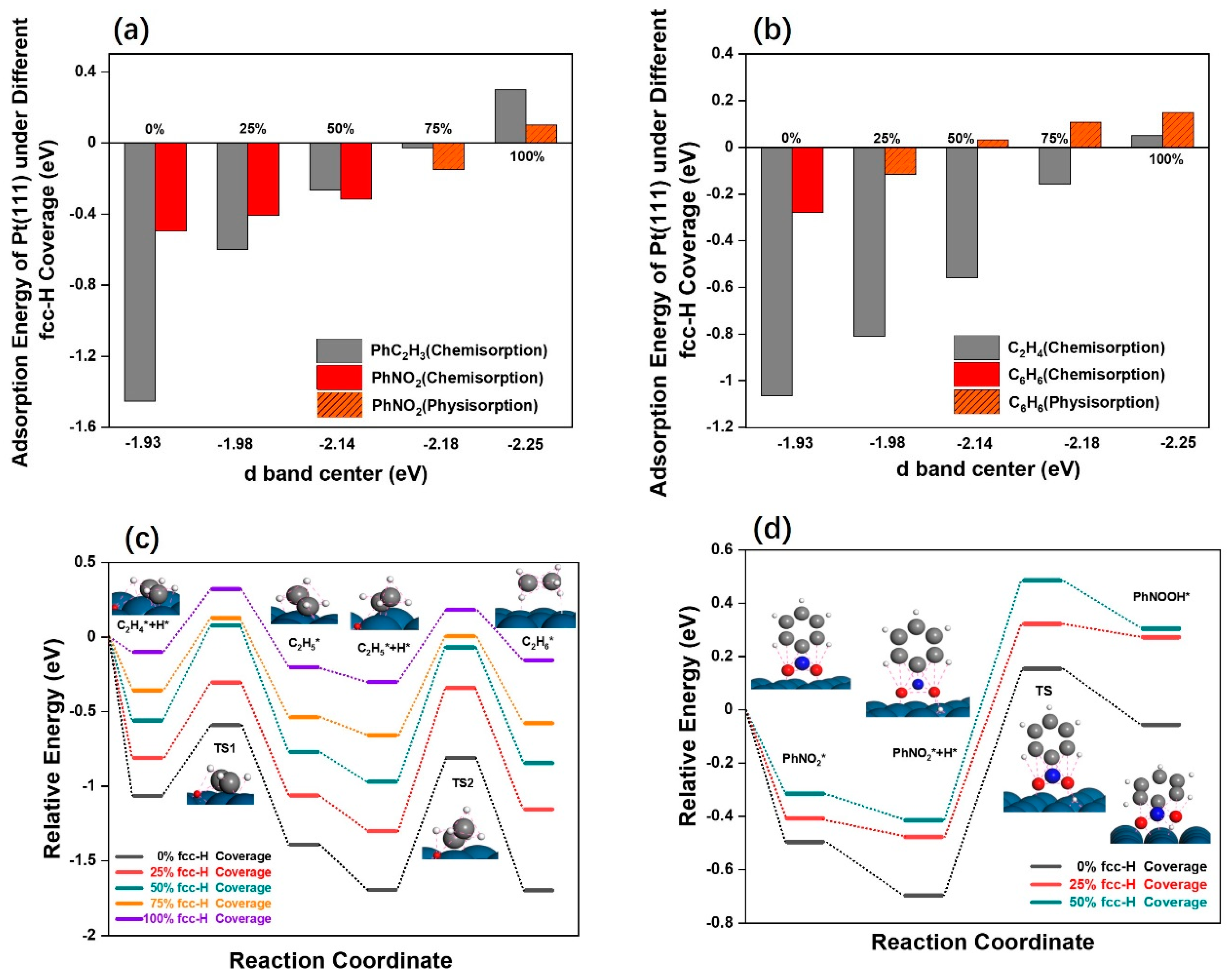
2.3. The Adsorption Energy and Reaction Path
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dzade, N.Y. CO2 and H2O Coadsorption and Reaction on the Low-Index Surfaces of Tantalum Nitride: A First-Principles DFT-D3 Investigation. Catalysts 2020, 10, 1217. [Google Scholar] [CrossRef]
- Lou, Y.; Zheng, Y.; Li, X.; Ta, N.; Xu, J.; Nie, Y.; Cho, K.; Liu, J. Pocketlike Active Site of Rh1/MoS2 Single-Atom Catalyst for Selective Crotonaldehyde Hydrogenation. J. Am. Chem. Soc. 2019, 141, 19289–19295. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Guo, Q.; Sun, Y.; Chen, S.; Wang, J.-Q.; Wu, M.; Fu, W.; Tang, Y.; Duan, X.; Chen, D.; et al. Optimising surface d charge of AuPd nanoalloy catalysts for enhanced catalytic activity. Nat. Commun. 2019, 10, 1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferri, P.; Li, C.; Paris, C.; Vidal-Moya, A.; Moliner, M.; Boronat, M.; Corma, A. Chemical and Structural Parameter Connecting Cavity Architecture, Confined Hydrocarbon Pool Species, and MTO Product Selectivity in Small-Pore Cage-Based Zeolites. ACS Catal. 2019, 9, 11542–11551. [Google Scholar] [CrossRef]
- Blaser, H.-U. CHEMISTRY: A Golden Boost to an Old Reaction. Science 2006, 313, 312–313. [Google Scholar] [CrossRef] [PubMed]
- Hamm, G.; Schmidt, T.; Breitbach, J.; Franke, D.; Becker, C.; Wandelt, K. The Adsorption of Ethene on Pd(111) and Ordered Sn/Pd(111) Surface Alloys. Zeitschrift Phys. Chem. 2009, 223, 209–232. [Google Scholar] [CrossRef]
- Downing, R.; Kunkeler, P.; Van Bekkum, H. Catalytic syntheses of aromatic amines. Catal. Today 1997, 37, 121–136. [Google Scholar] [CrossRef]
- Blaser, H.-U.; Steiner, H.; Studer, M. Selective Catalytic Hydrogenation of Functionalized Nitroarenes: An Update. ChemCatChem 2009, 1, 210–221. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, M.; Wang, A.; Zhang, T. Selective Hydrogenation over Supported Metal Catalysts: From Nanoparticles to Single Atoms. Chem. Rev. 2020, 120, 683–733. [Google Scholar] [CrossRef]
- Corma, A. Chemoselective Hydrogenation of Nitro Compounds with Supported Gold Catalysts. Science 2006, 313, 332–334. [Google Scholar] [CrossRef]
- Shimizu, K.-I.; Miyamoto, Y.; Kawasaki, T.; Tanji, T.; Tai, Y.; Satsuma, A. Chemoselective Hydrogenation of Nitroaromatics by Supported Gold Catalysts: Mechanistic Reasons of Size- and Support-Dependent Activity and Selectivity. J. Phys. Chem. C 2009, 113, 17803–17810. [Google Scholar] [CrossRef]
- Berguerand, C.; Yarulin, A.; Cárdenas-Lizana, F.; Wärnå, J.; Sulman, E.; Murzin, D.Y.; Kiwi-Minsker, L. Chemoselective Liquid Phase Hydrogenation of 3-Nitrostyrene over Pt Nanoparticles: Synergy with ZnO Support. Ind. Eng. Chem. Res. 2015, 54, 8659–8669. [Google Scholar] [CrossRef]
- Furukawa, S.; Takahashi, K.; Komatsu, T. Well-structured bimetallic surface capable of molecular recognition for chemoselective nitroarene hydrogenation† †Electronic supplementary information (ESI) available: FT-IR spectra, recycling test, kinetic studies, and DFT calculations. Chem. Sci. 2016, 7, 4476–4484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serna, P.; Corma, A. Transforming Nano Metal Nonselective Particulates into Chemoselective Catalysts for Hydrogenation of Substituted Nitrobenzenes. ACS Catal. 2015, 5, 7114–7121. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J. Theoretical Surface Science and Catalysis—Calculations and Concepts; Elsevier: Amsterdam, The Netherlands, 2000; Volume 45, pp. 71–129. [Google Scholar]
- Zhang, Z.; Li, Y.-L.; Gu, J.; Ding, L.; Xue, N.; Peng, L.; Guo, X.; Zhu, Y.; Ma, J.; Ding, W. The effect of electrostatic field on the catalytic properties of platinum clusters confined in zeolite for hydrogenation. Catal. Sci. Technol. 2018, 8, 6384–6395. [Google Scholar] [CrossRef]
- Romero, A.H.; Cerecetto, H. A Common, Facile and Eco-Friendly Method for the Reduction of Nitroarenes, Selective Reduction of Poly-Nitroarenes and Deoxygenation of N -Oxide Containing Heteroarenes Using Elemental Sulfur. Eur. J. Org. Chem. 2020, 2020, 1853–1865. [Google Scholar] [CrossRef]
- He, D.; Song, X.; Li, W.; Tang, C.; Liu, J.; Ke, Z.; Jiang, C.; Xiao, X. Inside Back Cover: Active Electron Density Modulation of Co3O4-Based Catalysts Enhances their Oxygen Evolution Performance (Angew. Chem. Int. Ed. 17/2020). Angew. Chem. Int. Ed. 2020, 59, 6936. [Google Scholar] [CrossRef]
- Formenti, D.; Ferretti, F.; Scharnagl, F.K.; Beller, M. Reduction of Nitro Compounds Using 3d-Non-Noble Metal Catalysts. Chem. Rev. 2019, 119, 2611–2680. [Google Scholar] [CrossRef]
- Mao, J.; Chen, W.; Sun, W.; Chen, Z.; Pei, J.; He, D.; Lv, C.; Wang, D.; Li, Y. Rational Control of the Selectivity of a Ruthenium Catalyst for Hydrogenation of 4-Nitrostyrene by Strain Regulation. Angew. Chem. Int. Ed. 2017, 56, 11971–11975. [Google Scholar] [CrossRef]
- Trandafir, M.M.; Pop, L.; Hădade, N.D.; Florea, M.; Neatu, F.; Teodorescu, C.M.; Duraki, B.; Van Bokhoven, J.A.; Grosu, I.; Parvulescu, V.; et al. An adamantane-based COF: Stability, adsorption capability, and behaviour as a catalyst and support for Pd and Au for the hydrogenation of nitrostyrene. Catal. Sci. Technol. 2016, 6, 8344–8354. [Google Scholar] [CrossRef]
- Wei, C.; Sun, Y.; Scherer, G.G.; Fisher, A.C.; Sherburne, M.P.; Ager, J.W.; Xu, Z.J. Surface Composition Dependent Ligand Effect in Tuning the Activity of Nickel–Copper Bimetallic Electrocatalysts toward Hydrogen Evolution in Alkaline. J. Am. Chem. Soc. 2020, 142, 7765–7775. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gao, Z.; Wang, S.; Wang, G.; Gao, X.; Zhang, B.; Xing, S.; Zhao, S.; Qin, Y. Origin of synergistic effects in bicomponent cobalt oxide-platinum catalysts for selective hydrogenation reaction. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Macino, M.; Barnes, A.J.; Althahban, S.M.; Qu, R.; Gibson, E.K.; Morgan, D.J.; Freakley, S.J.; Dimitratos, N.; Kiely, C.J.; Gao, X.; et al. Tuning of catalytic sites in Pt/TiO2 catalysts for the chemoselective hydrogenation of 3-nitrostyrene. Nat. Catal. 2019, 2, 873–881. [Google Scholar] [CrossRef]
- Zhu, J.; Hu, L.; Zhao, P.; Lee, L.Y.S.; Wong, K.-Y. Recent Advances in Electrocatalytic Hydrogen Evolution Using Nanoparticles. Chem. Rev. 2020, 120, 851–918. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.-L.; Zou, J.; Chen, Z. Advanced Thermoelectric Design: From Materials and Structures to Devices. Chem. Rev. 2020, 120, 7399–7515. [Google Scholar] [CrossRef] [PubMed]
- Mu, R.; Fu, Q.; Xu, H.; Zhang, H.; Huang, Y.; Jiang, Z.; Zhang, S.; Tan, D.; Bao, X. Synergetic Effect of Surface and Subsurface Ni Species at Pt−Ni Bimetallic Catalysts for CO Oxidation. J. Am. Chem. Soc. 2011, 133, 1978–1986. [Google Scholar] [CrossRef]
- She, W.; Qi, T.; Cui, M.; Yan, P.; Ng, S.W.; Li, W.; Li, G.; Weng, N.S. High Catalytic Performance of a CeO2-Supported Ni Catalyst for Hydrogenation of Nitroarenes, Fabricated via Coordination-Assisted Strategy. ACS Appl. Mater. Interfaces 2018, 10, 14698–14707. [Google Scholar] [CrossRef]
- Liu, L.; Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Geng, Z.; Zhao, S.; Wang, L.; Li, H.; Wang, X.; Zheng, X.; Zhu, J.; Li, Z.; Si, R.; et al. Pt Single Atoms Embedded in the Surface of Ni Nanocrystals as Highly Active Catalysts for Selective Hydrogenation of Nitro Compounds. Nano Lett. 2018, 18, 3785–3791. [Google Scholar] [CrossRef]
- Takigawa, I.; Shimizu, K.-I.; Tsuda, K.; Takakusagi, S. Machine-learning prediction of the d-band center for metals and bimetals. RSC Adv. 2016, 6, 52587–52595. [Google Scholar] [CrossRef] [Green Version]
- Vandichel, M.; Grönbeck, H. A dimer path for CO dissociation on PtSn. Catal. Sci. Technol. 2019, 9, 695–701. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Sun, M.; Hu, Z.-P.; Ren, J.-T.; Zhang, S.; Yuan, Z.-Y. New insight into the enhanced catalytic performance of ZnPt/HZSM-5 catalysts for direct dehydrogenation of propane to propylene. Catal. Sci. Technol. 2019, 9, 1979–1988. [Google Scholar] [CrossRef]
- Yoo, S.J.; Kim, S.-K.; Jeon, T.-Y.; Hwang, S.J.; Lee, J.-G.; Lee, S.-C.; Lee, K.-S.; Cho, Y.-H.; Sung, Y.-E.; Lim, T.-H. Enhanced stability and activity of Pt–Y alloy catalysts for electrocatalytic oxygen reduction. Chem. Commun. 2011, 47, 11414–11416. [Google Scholar] [CrossRef]
- Han, A.; Zhang, J.; Sun, W.; Chen, W.; Zhang, S.; Han, Y.; Feng, Q.; Zheng, L.; Gu, L.; Chen, C.; et al. Isolating contiguous Pt atoms and forming Pt-Zn intermetallic nanoparticles to regulate selectivity in 4-nitrophenylacetylene hydrogenation. Nat. Commun. 2019, 10, 1–7. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Pan, L.; Zhang, X.; Zou, J.-J. Insights into the Pt (111) Surface Aid in Predicting the Selective Hydrogenation Catalyst. Catalysts 2020, 10, 1473. https://doi.org/10.3390/catal10121473
Wang T, Pan L, Zhang X, Zou J-J. Insights into the Pt (111) Surface Aid in Predicting the Selective Hydrogenation Catalyst. Catalysts. 2020; 10(12):1473. https://doi.org/10.3390/catal10121473
Chicago/Turabian StyleWang, Tianzuo, Lun Pan, Xiangwen Zhang, and Ji-Jun Zou. 2020. "Insights into the Pt (111) Surface Aid in Predicting the Selective Hydrogenation Catalyst" Catalysts 10, no. 12: 1473. https://doi.org/10.3390/catal10121473