1. Introduction
Molecular hydrogen is fundamental for a large number of catalytic processes, involving chemical and petrochemical conversions of industrial interest. Included in these, ammonia and methanol syntheses, hydroformylation and Fischer-Tropsch reactions as well as catalytic reforming, hydrotreating (hydrodesulfurization), hydrocracking and either hydrogenation or dehydrogenation of hydrocarbons, oils and fats have to be mentioned [
1]. Among the catalytic transformations involving hydrogen, a significant amount of important catalysts contains supported transition metals [
2].
Recently, sp
-like carbonaceous species, i.e., styrene derivatives, pristine or defective graphene along with carbon nanotubes and graphitic materials, have increased significantly in attractiveness as catalytic material [
3,
4,
5,
6,
7] and, specifically, as supports [
8,
9,
10,
11,
12,
13].
One of the key phenomena concerning the hydrogen involvement is clearly its interaction with the supported metal systems [
1]. Therefore, the understanding at atomistic level of the mechanisms involving the interaction of hydrogen with such systems represents a substantial target in catalysis [
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24]. Typical elementary events considered as critical in investigating the surface dynamic phenomena are both the physical and chemical molecular H
2 sorption as well as the subsequent fragmentation/recombination, migration and desorption of any surface hydrogen species [
1,
14,
15,
16,
17,
18,
19,
20,
21].
A lot of information has been acquired both by experimental and theoretical studies [
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26], but the analysis of the causes, which dynamically originate the local surface coverage, is still poor [
24,
27]. In this context, as an example, hydrogen spillover is simply defined as the migration of H atoms to the support from the metal particles, following the dissociative adsorption of H
2 molecule on the metallic sites [
28]. However, the spillover mechanism, with the complementary reverse spillover, are still far from being fully understood and further investigations are needed to reach the whole grasp of these phenomena.
From a theoretical point of view, Density Functional Theory (DFT) was employed to compute the energy barrier characterizing the hydrogen migration through the Pt
4, Pd
6 and Pd
13 clusters’ sites and from these to the graphene surface. In all the evaluated cases, the computed energy barriers were 2.0–2.7 eV [
29,
30,
31,
32], suggesting that the hydrogen spillover is mostly an energetically hard process. Rangel et al. [
33], studying the hydrogen fragmentation, migration and diffusion on one Pd
4 cluster supported on defective graphene, however, observed that the dissociation of the molecular H
2 on the Pd clusters could already occur overcoming energy barriers lower than 0.6 eV and found that the atomic hydrogen migration from the metal to the support occurred overcoming energy barrier values equal to 0.8 eV (on pyridinic defects) and just 0.5 eV (on pyrrolic defects). As a consequence, it was possible to infer that hydrogen fragmentation and migration in these systems might occur almost spontaneously even at room temperature [
33].
Studying the energies of sequential H
2 molecules adsorption on Pd clusters at DFT level, Ramos-Castillo et al. [
34] suggested that one Pd
4 cluster is able to coordinate several H
2 molecules by shaping H
2 molecular complexes. In this case, the maximum number of H
2 molecules for one Pd
4 cluster supported on graphene resulted equal to four. Conversely, Ebrahimi et al. [
35] employed a series of accurate Molecular Dynamics (MD) simulations to characterize the phenomena involved in the H
2 adsorption on graphene nanoribbons, thus observing that the deformation of the graphene structure is closely related to the hydrogen surface coverage.
Recently, Prestianni et al. [
24], in an article from now on referred as paper
I, introduced a new, quite simple, approach in which the H
2 molecules were addressed “as projectiles” on a supported metal crystallite, namely on a Pd
4 cluster supported on graphene. At lower values of the H
2 kinetic energies, it was observed that the molecular sticking, hence the cluster site hitting was ruled by steering effects imputable to the Pd cluster, which easily reoriented the incoming H
2 molecule to the metallic surface sites, and in this way prompting the fragmentation thus atomic adsorption. Sticking decreased at higher molecular kinetic energies and it appeared that very fast molecules were able to acquire a kind of elusiveness toward the metallic surface which, because of this, was not able to steer and adsorb them.
To add new insights useful to unravel phenomena related to: (i) the surface ruling and hitting of H2 molecules on metal sites of both pristine and already adsorbed clusters; (ii) the sticking and fragmentation and/or diffusion of atomic hydrogen; and (iii) the existence of spillover and reverse-spillover phenomena, the simple theoretical protocol described in I was mostly employed in the simulations reported in the following.
By these, the individual surface events on a very basic system—taking into consideration the collision of H2 molecules on a supported fragment, shaped in a periodic system characterized by one unit formed by a (pristine or hydrogen preadsorbed) Pd4 cluster placed on a C47 graphene model—were visually evaluated, by employing DFT Molecular Dynamics.
In details, the modeling routine implied the analysis of the local rearrangements following the single collisions of H2 molecules with a [2 mH+(n − − m)H2]/Pd4/C47 supported fragment, being . Hence, the effects of the fragment hydrogen coverage as well as the starting speed and the orientation of the H2 molecule were right correlated to the structural, mechanistic and energetic local changes induced on the same [2 mH+(n − − m)H2]/Pd4/C47 supported fragments.
2. Results and Discussion
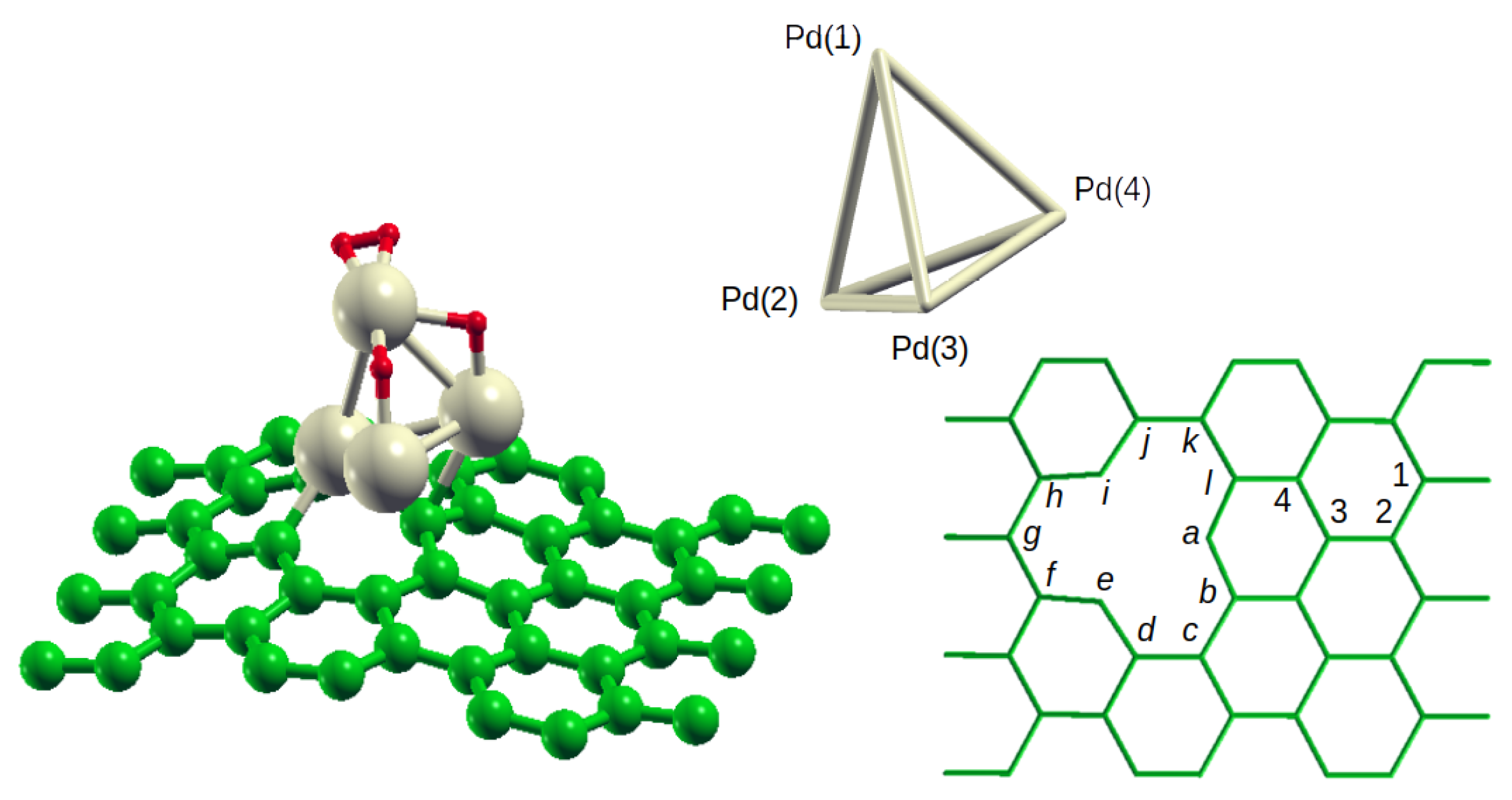
The model in this work is the same already used for the preliminary investigations reported in
I. Briefly, the studied system is a tetrahedral Pd
4 cluster anchored on the vancancy of a defective graphene sheet, simulated by a periodic system with a supercell of 47 carbon atoms, with cell vector (12.78, 9.84, 30.00 Å); the projectiles are H
2 molecules having predefined kinetic energies, k
, of 0.005, 0.01, 0.015 and 0.1 eV. In paper
I it is evidenced that the hitting of H
on Pd
/C
results in fragmentation only for the lowest values of projectile kinetic energy; the H
molecule is adsorbed for k
= 0.015 eV and it bounces at the higher energies. This behavior was ascribed to an energetic balance between the projectile k
and its interaction energy with the cluster. In agreement with the steering mechanism proposed by Gross and Scheffler [
36], low kinetic energy projectiles can undergo more easily to a process of steering and reorienting, which could promote their activation and fragmentation. On the other hand, when the second hitting was considered, i.e., H
shot either on H
Pd
/C
or on 2HPd
/C
, it was revealed how the presence of already adsorbed species on the cluster affects the fate of the incoming molecule. In particular, it was evident that, due to repulsive effects from the preadsorbed not-dissociated H
, steering effects occur at higher projectile kinetic energies.
The target from which the investigation here reported starts is the system where one H
2 molecule is adsorbed atop the Pd(1) atom (with Pd–H distances of 1.853 and 1.887 Å, and H–H bond length equal to 0.849 Å), one H atom is located on the Pd(1)Pd(3) edge (being 1.887 and 1.738 the H–Pd(1) and H–Pd(3) distances, respectively) and another H atom on the Pd(1)Pd(4) edge (with H–Pd(1) and H–Pd(4) equal to 1.862 and 1.724 Å) of a Pd
4 tetrahedron anchored on defective graphene (see
Figure 1 for atoms numbering). To simplify the discussion, the following notation is used throughout: (
i) H
is a hydrogen molecule adsorbed on the Pd(n) site of Pd
4; (
ii) H
is a H atom
[
][
]-bonded to the Pd(n
)[Pd(n
)][Pd(n
)] site(s) of the cluster; and (
iii) the Pd
/C
cluster/graphene support is implied. With this notation, the target system above is indicated as H
H
H
. Clearly,
,
and
hydrogen atoms are characterized by 1, 2 and 3 apices, respectively. Furthermore, it is worth investigating the effect of H
2 adsorption on the Pd
4 configuration with respect to the graphene defect. The C atoms defining the defect contour in C
47 are labeled with letters ranging from
a to
l (
Figure 1), and the anchoring of Pd
4 can be defined by the bonds occurring between the Pd centers of the cluster and those C atoms. Thus, in the H
H
H
system, Pd(2) is bonded to the contour atoms
e and
i (Pd–C = 1.989 and 2.002 Å), Pd(3) is bonded to the contour atom
c (Pd–C = 2.383 Å) and Pd(4) to
a and
l (Pd–C = 2.258 and 2.424 Å); the shorthand notation (2
ei3
c4
al) will be used for this anchoring configuration. For comparison, the one in Pd
4/C
47, without adsorbed hydrogen, is (2
aei3
abc4
al), with Pd–C distances equal to 1.937, 2.017, 2.053 Å (2aei), 2.523, 2.396, 2.536 Å (3abc) and 2.027, 2.218 Å (4al), so that in our initial target some bonds between the cluster and the graphene sheet are elongated/broken.
2.1. Hitting of H2 on the Hydrogenated Pd4 Cluster
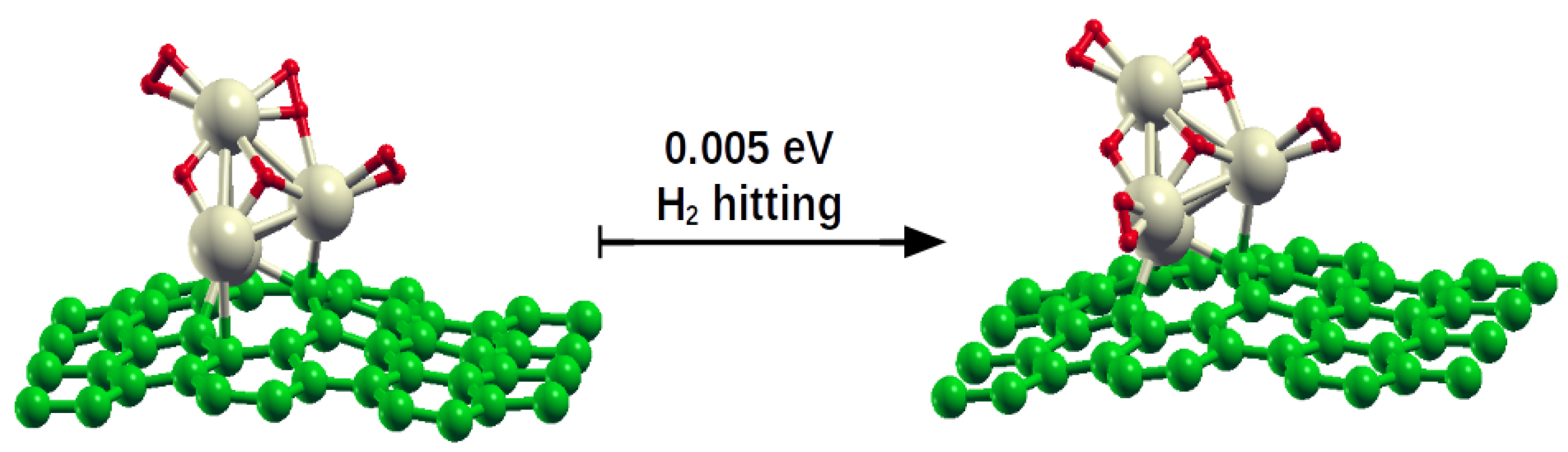
H2 on HHH: The hitting of H2 on the HHH system results in adsorption on Pd(4) if the projectile kinetic energy is 0.01 or 0.015 eV; this gives raise to the HHHH system, of which, in the following, NVT equilibration is analyzed. In this system, the anchoring configuration of Pd4 on the graphene defect slightly changes, becoming now (2ei3bc4a). At lower energy, 0.005 eV, the H2 molecule interacts with the cluster but cannot bind to it, hence it steps away. At higher energy, 0.1 eV, the projectile undergoes a sort of reflection: it hits the cluster after 97 fs from the beginning of simulation and its interaction with Pd(1) lasts only 22 fs, and then it goes rotating in the direction opposite to the incoming one.
H2 on HHHH: After NVT equilibration, the H
H
H
H
system maintains its configuration. Every simulation of H
2 hitting, beginning with a different projectile initial orientation, invariably resulted in a H
2 molecule adsorbed on the cluster. At k
= 0.005 eV, the projectile bounces on Pd(3), turns around the cluster and interacts with it in proximity of Pd(4). The palladium cluster, after the hitting, is anchored on graphene only through the Pd(2) and Pd(3) centers. Following equilibration, the H
2 molecule (former the projectile) locates between Pd(1) and Pd(4), and the cluster adopts a (2
aei3
d4
a) position with respect to the defect. Essentially the same result is obtained at k
= 0.01 eV, even if in this case the projectile firstly interacts with Pd(1), and also at k
= 0.015 eV, where following the hit H
2 weakly interacts with Pd(3) and only after equilibration it shifts on the Pd(1)–Pd(4) edge. Finally, at k
= 0.1 eV, the H
2 molecule adsorbs on Pd(1) after bouncing on Pd(3) and again moves to the Pd(1)–Pd(4) edge after NVT. It is then clear that the result of the H
2 hitting on H
H
H
H
is the new system H
H
H
H
H
, where the apex 1(4) underlines that the H
2 molecule is adsorbed atop Pd(1) but one of its atoms interact also with Pd(4). Presumably, this occurrence is observed because both Pd(1) and Pd(4) are already occupied by another H
2 molecule in atop position. Some shapshots of these simulations are collected in
Figure 2.
H2 on HHHHH: In the H
H
H
H
H
system, only the Pd(3) position is free to adsorb another H
2 molecule, being Pd(2) the main site that anchors the Pd
4 cluster to graphene. As a matter of fact, the adsorption on Pd(3) is just what happens when a k
= 0.005 eV H
2 projectile is shot on the target, giving rise to a seemingly hydrogen-saturated H
H
H
H
H
H
palladium cluster, with this one in (2
aei4
a) configuration with respect to the defect arrangement (see
Figure 3).
At higher energy (0.01, 0.015 and 0.1 eV), only almost perfect reflections occur. In this case, we also investigated the effect of very high energy particles, namely 0.5 and 1 eV, to see if spillover would take place. Instead, the high energy H2 always causes the desorption of molecules from the cluster. In particular, the k = 0.5 eV projectile adsorbs on Pd(1), substituting itself to the two H2 molecules locating atop it, which at once desorb, while in the k = 1 eV case the projectile reflects and one H2 from Pd(1) moves away from the cluster.
2.2. Hydrogen Hopping to Graphene
The hopping of one hydrogen atom from the Pd
4 cluster to position C(4) of C
47 (see
Figure 1), as well as the diffusion of H along the 4→3→2→1 path in the graphene sheet, was investigated by means of NEB calculations. To evaluate the effect on the hopping due to the presence of hydrogen adsorbed on the cluster, the H
H
, H
H
H
and H
H
H
H
H
systems (from now on indicated as R1, R2 and R3, respectively) were selected. The first of these systems has only one H
2 molecule fragmented on the palladium cluster while the second is just the starting target system of the molecular dynamics simulations. The third system is similar to the result of hydrogen hitting on H
H
H
H
, but with the adsorbed H
2 molecule shifted from Pd(4) to Pd(3); this was necessary because H
H
H
H
H
does not show any fragmented H atom that could hop onto graphene in any direction coherent to our sheet model. The change is legitimated by the fact that the result of hitting by MD simulations on a given H–Pd
4/C
47 system is to be considered only as one among a multitude of possible outcomes of the real hydrogen hitting, and the adsorption of H
2 on Pd(3) instead of Pd(4) can surely be included amid these outcomes. Further, with this choice, in all of the three systems the H-hopping could occur with somehow the same sequence of steps: the breaking of H–Pd(1) bond, the revolution around Pd(4), the breaking of H–Pd(4) bond and the formation of H–C(4). As a matter of fact, this allows to compare the hopping energetics in the three systems. In the final products the configuration of hydrogen species on the Pd
4 cluster slightly changes, giving rise to the systems H
H
, H
H
H
and H
H
H
H
H
, whose energies show that the spillover reactions always occurs with high endoergicity. Here, the apex C(4) underlines the displacement of the corresponding H atom from the cluster to the graphene site indexed as 4 (see
Figure 1), where it binds at a distance of 1.13–1.14 Å.
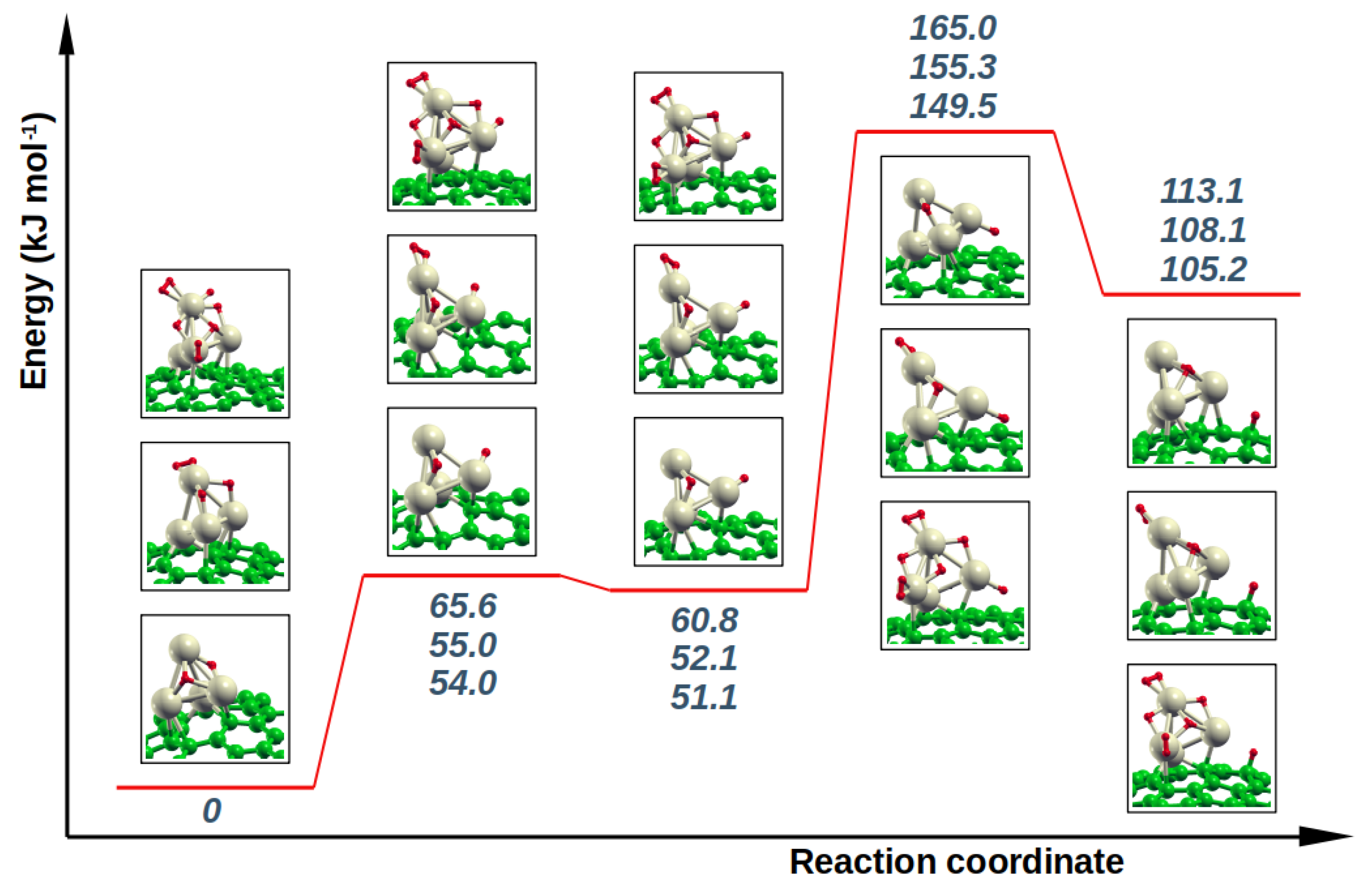
The results of the NEB calculations from reactants to products are collected in
Figure 4. As can be noticed, no matter the starting system, a metastable intermediate locates along the hopping path, where an H atom is located atop the Pd(4) center; the energy of the transition state from the reactant to this intermediate species slightly exceeds 65 kJ mol
in the least favorable case, but is always only a few kJ mol
above the energy of the intermediate itself. The step from intermediate to product would occur by overcoming an energy barrier of 104.3, 103.2 and 98.4 kJ mol
for the paths starting from R1, R2 and R3, respectively. This would suggest that the presence of hydrogen species on the Pd
4 cluster has only a small influence on the H-hopping. On the other hand, the intermediate-to-product step only reflects the energetics of H–Pd(4) bond breaking, while the effect of other H-species on the cluster should be better characterized by taking into account the highest energy barrier with respect to the reactant. If this is done, the difference between the three systems is clearly magnified, indicating that, even if the activation toward H-hopping is always quite difficult, the energy barrier sensibly decreases when the number of H-species on the cluster increases. As a matter of fact, the intermediate can be seen as a small ripple in the minimum energy path from reactant to product and it is possible to think that, in order for the hopping to occur, the reactant has to acquire all in one the energy to overcome the largest barrier.
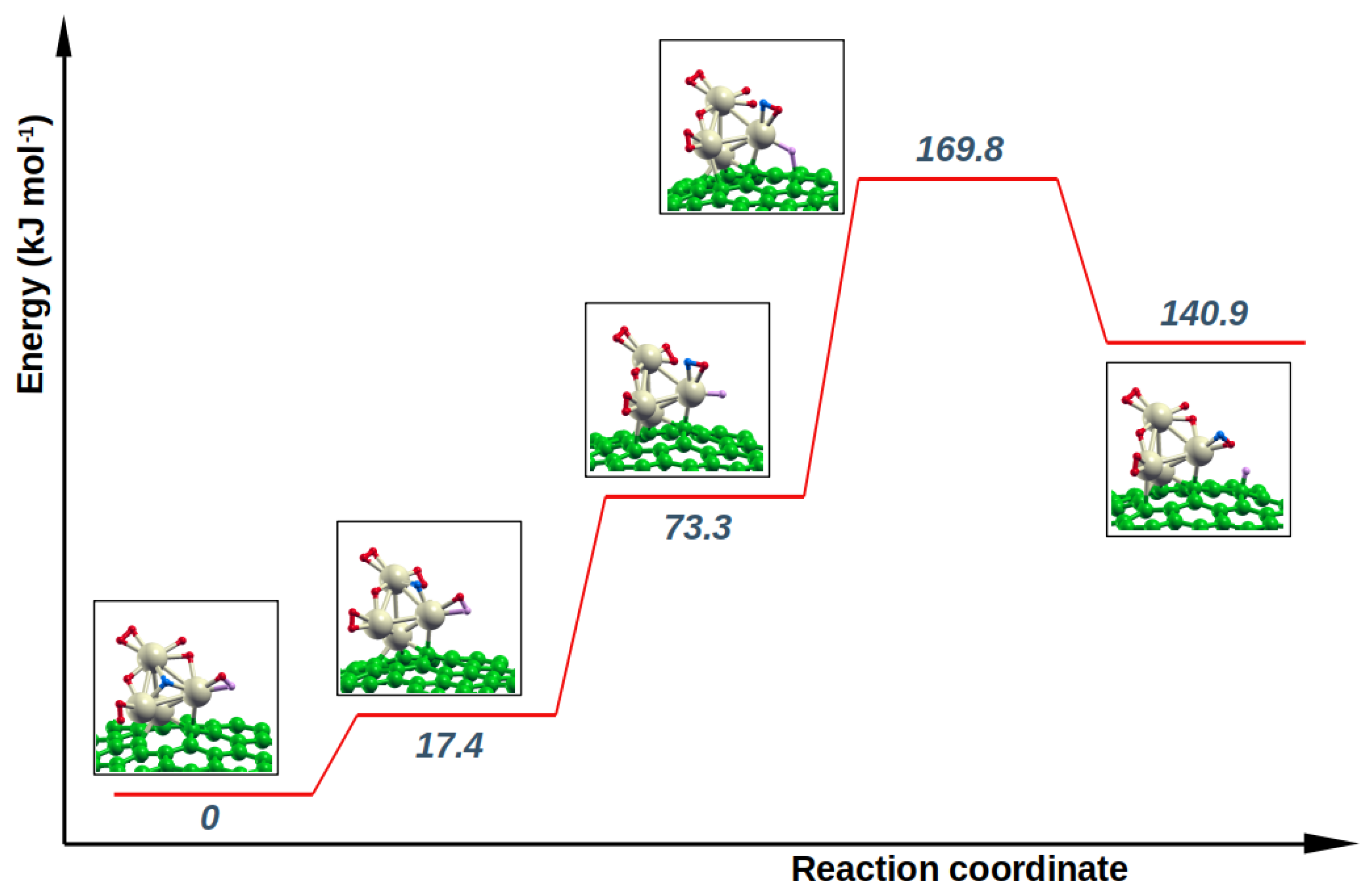
In the present whole saturated H
H
H
H
H
H
system, there is no hydrogen atoms that could spillover to graphene. In this case the possibility of a H-induced hopping was investigated. The H
atom could substitute a H atom of H
, which in turn let migrate the other of its H atom onto the graphene surface. In more details, the investigated pathway, represented schematically in
Figure 5, shows the essentially barrierless formation of two intermediate species, H
H
H
H
H
H
and H
H
H
H
H
H
, and an energy barrier of 169.8 kJ mol
, to end up with the H
H
H
H
H
H
final product. This last is 140.9 kJ mol
less stable than the initial reactant; the higher calculated endoergicity, with respect to the other spillover products, is presumably due to the fact that in this case, if the net process is considered, the H atom hopped on graphene was removed from a stable
position. Hence, it is possible to argue that for the same reason the energy barrier for this process is the highest of all the investigated ones, which suggests that H-spillover from a fully-saturated palladium cluster could be more difficult due to a necessary rearrangement of the hydrogen atomic species that need to get a right constellation, close to the receiving carbonaceous surface.
Finally, NEB calculations were performed to investigate the ease of diffusion of hydrogen atom far away from the anchored cluster, i.e., the hydrogen hopping along the 4→3→2→1 path of graphene (see
Figure 1). This was done by considering the spillover product H
H
H
as representative, which has energy of 108.1 kJ mol
higher than the R2 reactant. The results, reported in
Table 1, show that not all C position in C
47 are equivalent, as could be reasonably expected since the bonding of one H atom on graphene site causes a curvature of the sheet, and the ease with which this occurs depends on the position with respect to the defect. Indeed, in the present case, the properties of the graphene curvature are also heavily affected by the palladium cluster anchored on the defect and by the size of the model used. The same arguments apply to the energies of the transition states for the shifting from one C position to the next, whose trend reflects somehow the stability of the corresponding product and is quantified around 100 ± 15 kJ mol
.
3. Models and Computational Details
Molecular dynamics simulations were performed within the density functional theory framework by using the SIESTA approach as implemented in the homonym code, version 4.2.1 [
37]. The employed relativistic norm-conserving Troullier–Martins pseudopotentials and the double zeta plus polarization quality numerical basis sets, on which the SIESTA method is based, were generated and tested as described elsewhere [
13]. The PBE exchange-correlation functional in the spin-polarized form was chosen for all calculations, along with a
sampling of the Monkhorst-Pack grid and a value of 450 Ry for the mesh cutoff.
The algorithm devised to simulate the hitting of H2 on the target can be summarized as follows. Start with the optimized geometry of Pd4/C47 (the HHH system, in the present work) and perform a 10 ps MD simulation (timestep of 0.5 fs) in the NVT (298 K) ensemble. (A) Once the system is equilibrated, take the atomic positions, velocities and forces of the last snapshot and add to the system a H2 molecule with the following characteristics: the first H atom is placed at a distance of 7 Å from the center of mass (CM) of the palladium cluster, with a given angle with respect to the normal to surface; the second H atom is located in the same direction at a distance of 0.75 Å from the first; the velocity vectors of each H atom have magnitudes according to the initial kinetic energy of the projectile and are oriented toward the CM of Pd4; and, finally, the forces acting on the two H centers can be safely assumed to be negligible (hence set to zero) due to the distance of H2 from the target. Restart the MD simulation for a duration of 2 ps, now using the NVE ensemble in order to not redistribute the energy possessed by the projectile, and analyze the product of the hitting. Do this for projectiles having initial kinetic energies of 0.005, 0.01, 0.015 and 0.1 eV. Since the aim is to investigate the adsorption and/or the fragmentation of H2 on Pd4/C47, the simulations ended with simple reflection or bouncing of the projectile were discarded, while those ended with an effective hitting, i.e., an incipient sticking, were retained, analyzed and then subjected to equilibration in the NVT ensemble for 10 ps. The same procedure is repeated starting from (A) to simulate the hitting of a new H2 molecule and continue to shoot H2 until no effective hit occurs, i.e., the Pd4 cluster appears to be H-saturated.
Hopping of hydrogen species from the metal cluster to the graphene surface sites (spillover) was studied by means of a home-made code, which implements the Climbing Image Nudged Elastic Band method (CI-NEB) [
38,
39] and is interfaced with the SIESTA program. Reactants and products were fully optimized by SIESTA periodic calculations and NEB images were generated using the image depending pair potential [
40]. Every NEB calculation used 13 images connected each other by dynamic springs and a convergence threshold of
E
/Å on the norm of atomic forces was employed.
4. Conclusions
DFT-based molecular dynamics simulations show that a small palladium cluster anchored on a defective site of graphene can be saturated with hydrogen by means of a flux of low-energy H2 molecules. Cluster H-saturation is reached when five hydrogen molecules are adsorbed/fragmented, taking into account that some cluster sites are not available for the interaction with H2. In fact, even if the cluster anchoring configuration slightly changes and/or weakens according to the degree of H-adsorption, one face of the cluster is always firmly interacting with the graphene defect region. As the number of hydrogen species on palladium increases, the energy barrier that rules the H-spillover onto graphene slightly decreases. H-spillover remains an onerous process, which is not surprising given the high endoergicity of the reaction, but the present results indicate that, in the here investigated system, a small palladium cluster on defective graphene, the energy barriers are reduced by 0.5–1.0 eV with respect to other systems (excluding of course those where H-spillover occurs toward a graphene defect). This appears true also if the spillover requires some sort of redistribution/rearrangement of atomic hydrogen on the cluster.
Finally, by considering the whole web of the different pathways presented in this study, straightforwardly it emerges that the reverse-spillover (i.e., hopping from graphene to Pd4, rearrangement of hydrogen species on the cluster, and formation and desorption of H2) is supposed to be an easy process, when occurring on the graphene-anchored palladium clusters. This should encourage experimental investigations to deeper probe this kind of systems, hence to give them a chance as reversible hydrogen storage device.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}