Amino Acid Replacement at Position 228 Induces Fluctuation in the Ω-Loop of KPC-3 and Reduces the Affinity against Oxyimino Cephalosporins: Kinetic and Molecular Dynamics Studies

 , ,
, ,
Abstract
:1. Introduction
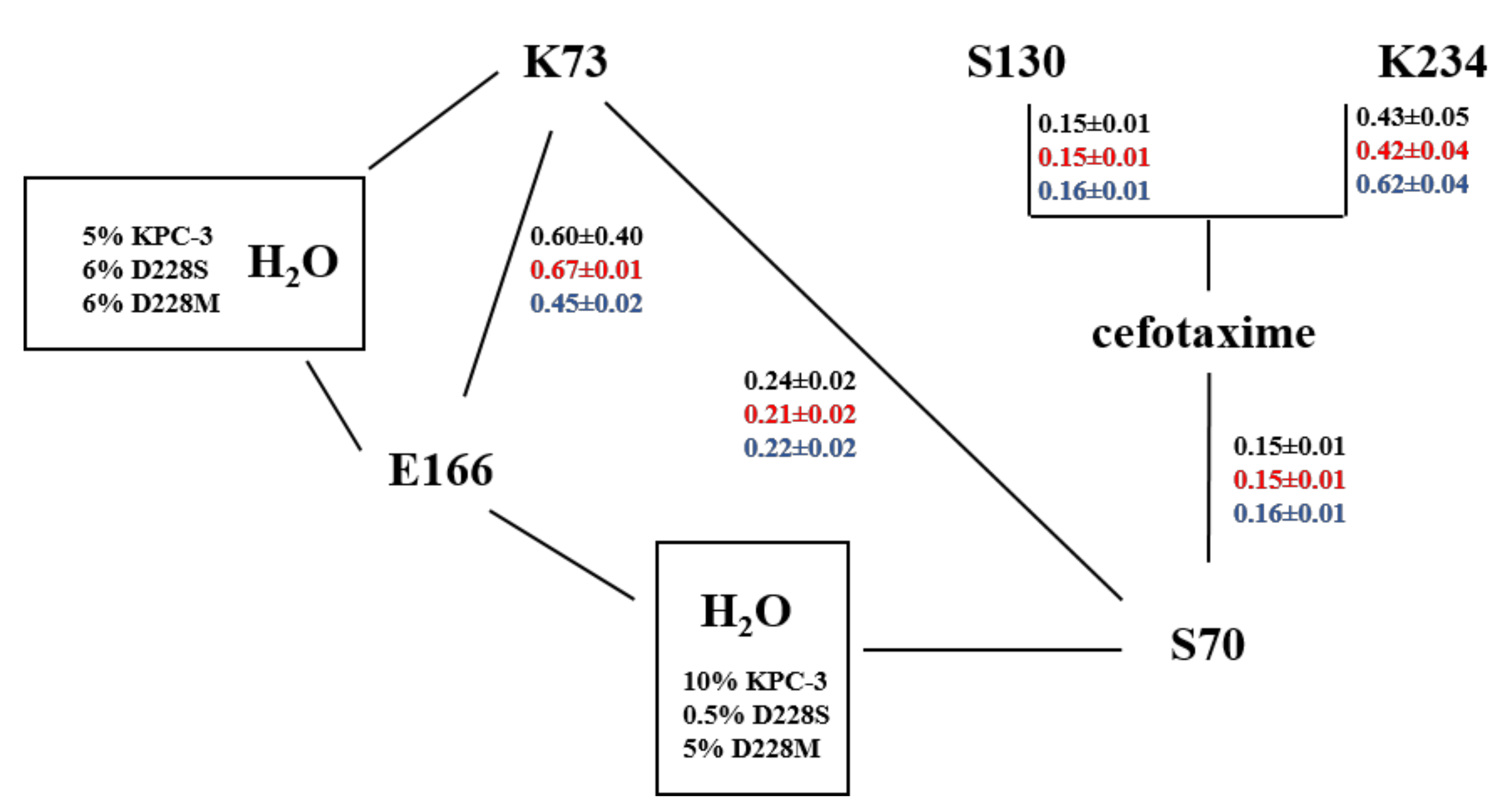
2. Results
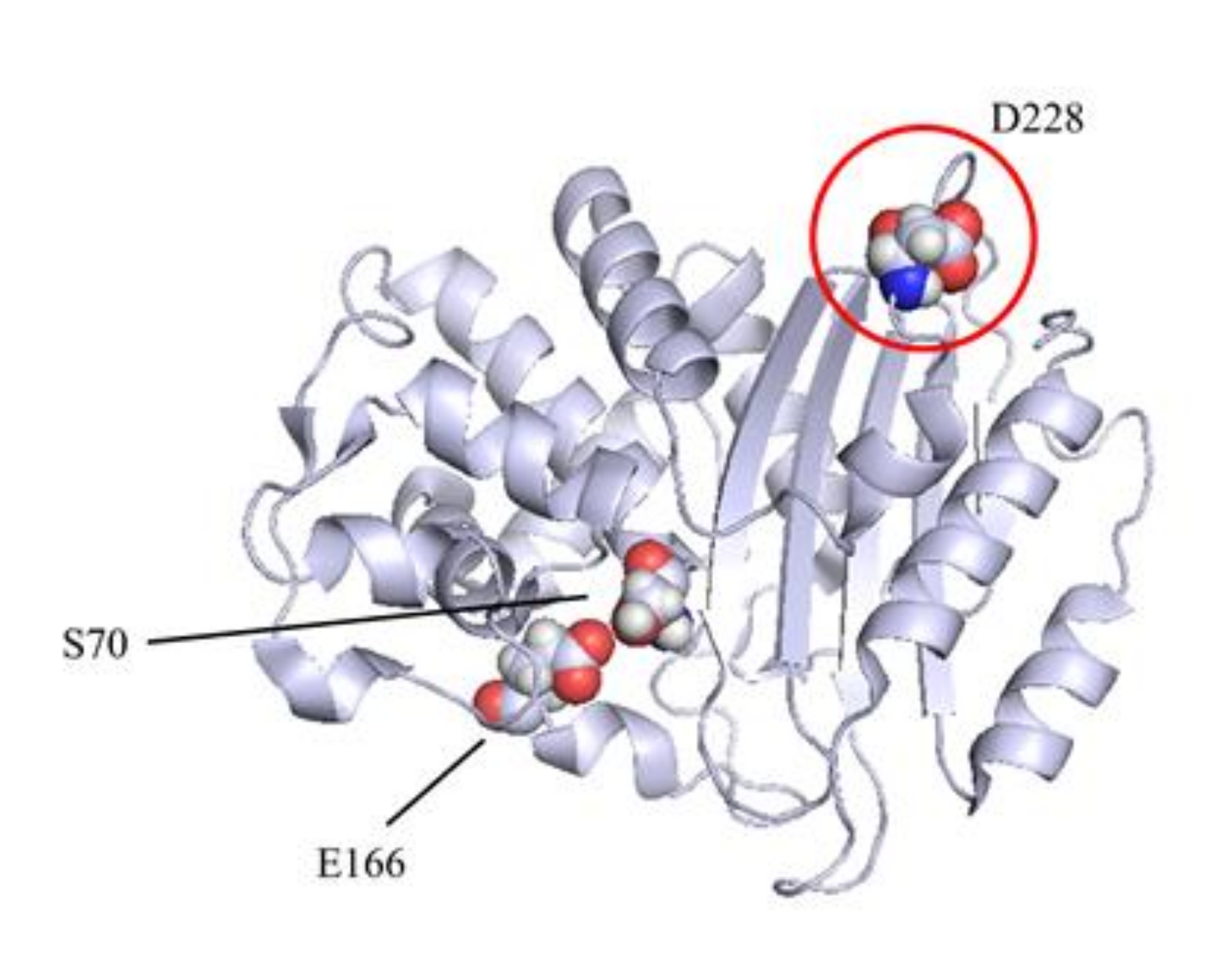
2.1. KPC-3 Mutants
2.2. Cloning and β-Lactamase Purification
2.3. Steady-State Kinetic Parameters
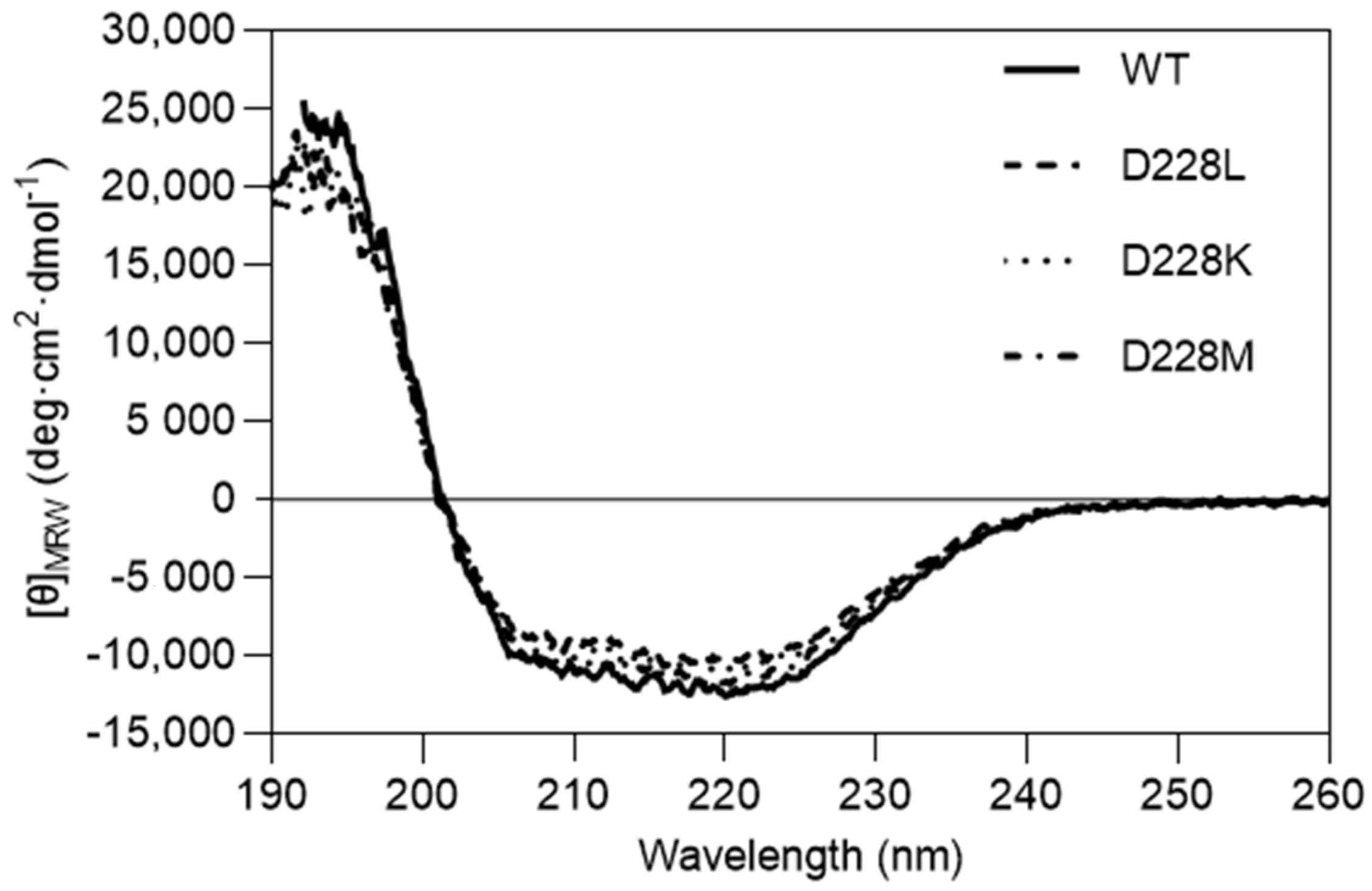
2.4. Circular Dichroism
2.5. Fluorescence Assay
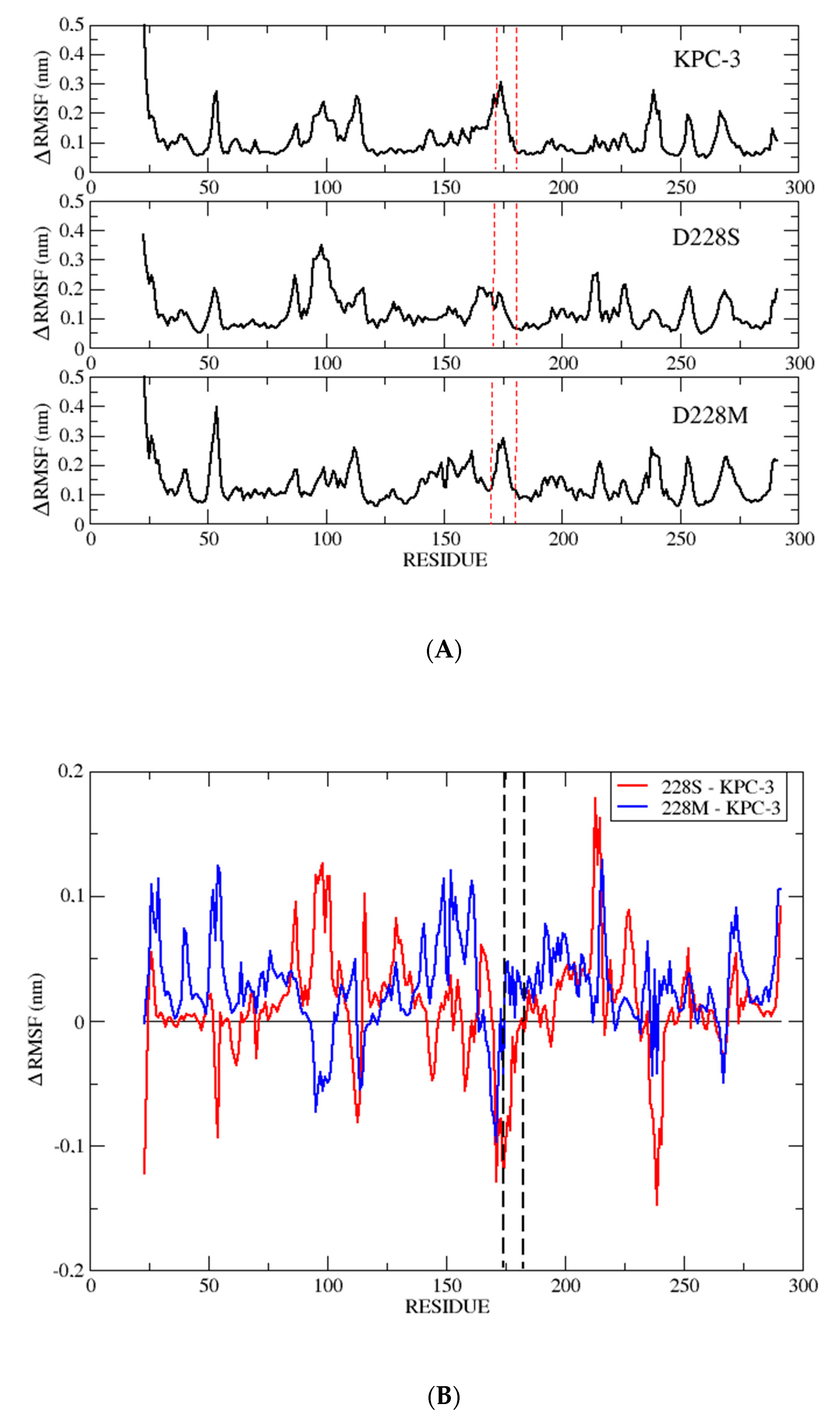
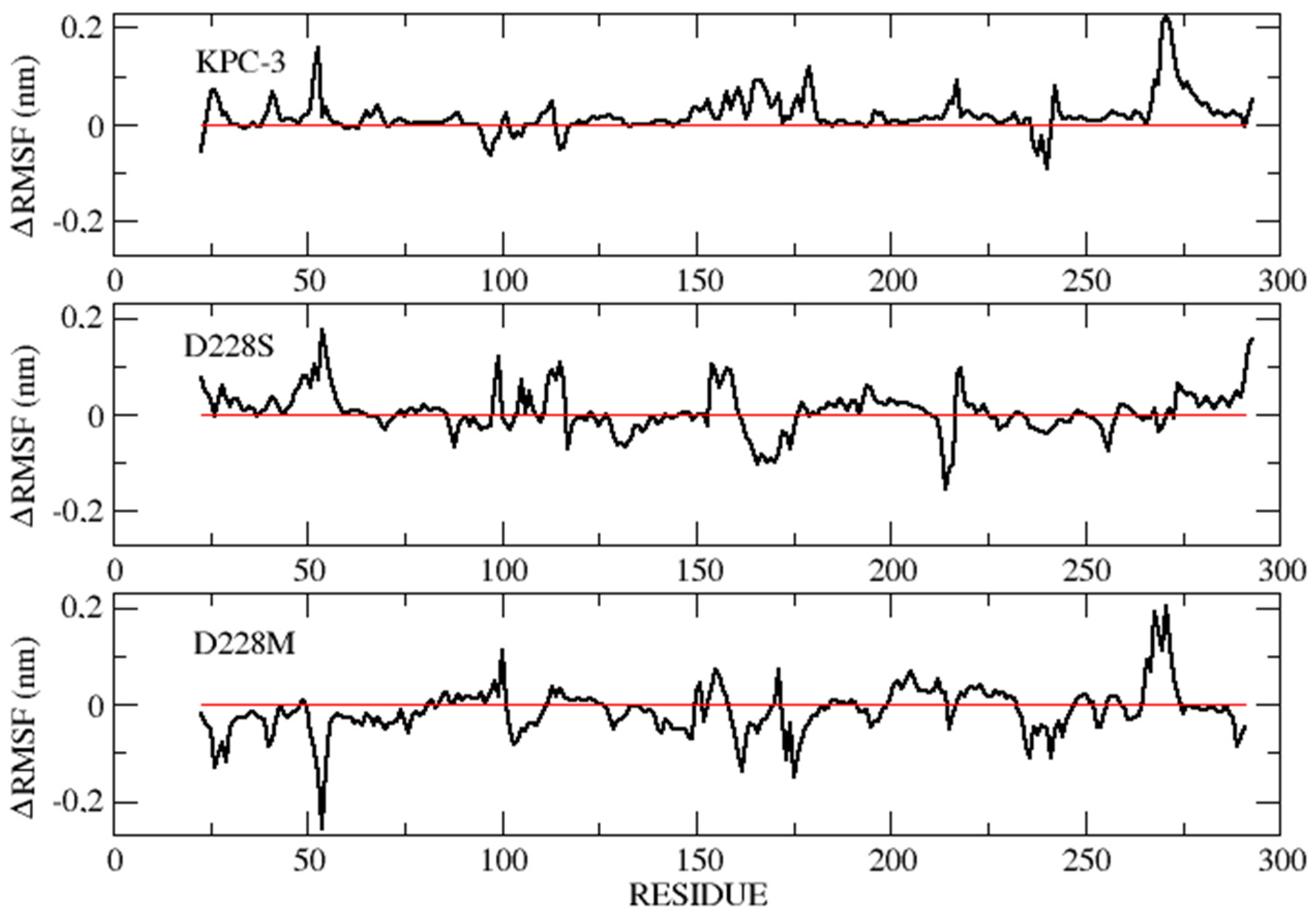
2.6. MD Simulations
3. Discussion
4. Materials and Methods
4.1. Cloning and Site-Saturation Mutagenesis
4.2. Overexpression and Purification of KPC-3 and D228X Mutants
4.3. Determination of Kinetic Parameters
4.4. Fluorescence Emission Spectroscopy
4.5. Circular Dichroism
4.6. Molecular Dynamics (MD) Simulations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bush, K.; Bradford, P.A. Epidemiology of β-lactamase-producing pathogens. Clin. Microbiol. Rev. 2020, 33, e00047-19. [Google Scholar] [CrossRef] [PubMed]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef] [PubMed]
- Bush, K. Past and present perspectives on β-lactamases. Antimicrob. Agents Chemother. 2018, 62, e01076-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambler, R.P. The structure of β-lactamases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1980, 289, 321–331. [Google Scholar]
- Bush, K.; Jacoby, G.A. Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Garau, G.; García-Sáez, I.; Bebrone, C.; Anne, C.; Mercuri, P.; Galleni, M.; Frère, J.M.; Dideberg, O. Update of the standard numbering scheme for class B beta-lactamases. Antimicrob. Agents Chemother. 2004, 48, 2347–2349. [Google Scholar] [CrossRef] [Green Version]
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, present, and future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef] [Green Version]
- Palzkill, T. Structural and Mechanistic Basis for Extended-Spectrum Drug-Resistance Mutations in Altering the Specificity of TEM, CTX-M, and KPC β-lactamases. Front. Mol. Biosci. 2018, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Naas, T.; Cuzon, G.; Villegas, M.V.; Lartigue, M.F.; Quinn, J.P.; Nordmann, P. Genetic structures at the origin of acquisition of the β-lactamase blaKPC gene. Antimicrob. Agents Chemother. 2008, 52, 1257–1263. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Fernandez, S.; Villa, L.; Carta, C.; Venditti, C.; Giordano, A.; Venditti, M.; Mancini, C.; Carattoli, A. Klebsiella pneumoniae ST258 producing KPC-3 identified in Italy carries novel plasmids and OmpK36/OmpK35 porin variants. Antimicrob. Agents Chemother. 2012, 56, 2143–2145. [Google Scholar] [CrossRef] [Green Version]
- Pitout, J.D.; Nordmann, P.; Poirel, L. Carbapenemase-producing Klebsiella pneumoniae, a key pathogen set for global nosocomial dominance. Antimicrob. Agents Chemother. 2015, 59, 5873–5884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Chavda, K.D.; Melano, R.G.; Jacobs, M.R.; Levi, M.H.; Bonomo, R.A.; Kreiswirth, B.N. Complete sequence of a bla(KPC-2)-harboring IncFII(K1) plasmid from a Klebsiella pneumoniae sequence type 258 strain. Antimicrob. Agents Chemother. 2013, 57, 1542–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerdeira, L.T.; Cunha, M.P.V.; Francisco, G.R.; Bueno, M.F.C.; Araujo, B.F.; Ribas, R.M.; Gontijo-Filho, P.P.; Knöbl, T.; de Oliveira Garcia, D.; Lincopan, N. IncX3 plasmid harboring a non-Tn4401 genetic element (NTEKPC) in a hospital-associated clone of KPC-2-producing Klebsiella pneumoniae ST340/CG258. Diagn. Microbiol. Infect. Dis. 2017, 89, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Alba, J.; Ishii, Y.; Thomson, K.; Moland, E.S.; Yamaguchi, K. Kinetics study of KPC-3, a plasmid-encoded class A carbapenem-hydrolyzing β-lactamase. Antimicrob. Agents Chemother. 2005, 49, 4760–4762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, W.; Bethel, C.R.; Thomson, J.M.; Bonomo, R.A.; van den Akker, F. Crystal structure of KPC-2: Insights into carbapenemase activity in class A β-lactamases. Biochemistry 2007, 46, 5732–5740. [Google Scholar] [CrossRef] [Green Version]
- Papp-Wallace, K.M.; Taracila, M.; Wallace, C.J.; Hujer, K.M.; Bethel, C.R.; Hornick, J.M.; Bonomo, R.A. Elucidating the role of Trp105 in the KPC-2 β-lactamase. Protein Sci. 2010, 19, 1714–1727. [Google Scholar] [CrossRef]
- Papp-Wallace, K.M.; Taracila, M.A.; Smith, K.M.; Xu, Y.; Bonomo, R.A. Understanding the molecular determinants of substrate and inhibitor specificities in the carbapenemase KPC-2: Exploring the roles of Arg220 and Glu276. Antimicrob. Agents Chemother. 2012, 56, 4428–4438. [Google Scholar] [CrossRef] [Green Version]
- Mehta, S.G.; Rice, K.; Palzkill, T. Natural variants of the KPC-2 carbapenemase have evolved increased catalytic efficiency for ceftazidime hydrolysis at the cost of enzyme stability. PLoS Pathog. 2015, 11, e1004949. [Google Scholar] [CrossRef]
- Matagne, A.; Lamotte-Brasseur, J.; Frère, J.M. Catalytic properties of class A β-lactamases: Efficiency and diversity. Biochem. J. 1998, 330, 581–598. [Google Scholar] [CrossRef]
- Munoz-Price, L.S.; Poirel, L.; Bonomo, R.A.; Schwaber, M.J.; Daikos, G.L.; Cormican, M.; Cornaglia, G.; Garau, J.; Gniadkowski, M.; Hayden, M.K.; et al. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect. Dis. 2013, 13, 785–796. [Google Scholar] [CrossRef] [Green Version]
- Perilli, M.; Bottoni, C.; Grimaldi, A.; Segatore, B.; Celenza, G.; Mariani, M.; Bellio, P.; Frascaria, P.; Amicosante, G. Carbapenem-resistant Klebsiella pneumoniae harbouring bla(KPC-3) and bla(VIM-2) from central Italy. Diagn. Microbiol. Infect. Dis. 2013, 75, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Piccirilli, A.; Mercuri, P.S.; Galleni, M.; Aschi, M.; Matagne, A.; Amicosante, G.; Perilli, M. P174E substitution in GES-1 and GES-5 β-lactamases improves catalytic efficiency towards carbapenems. Antimicrob. Agents Chemother. 2018, 62, e01851-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segel, I.H. Biochemical Calculations, 2nd ed.; John Wiley & Sons: New York, NY, USA, 1976; pp. 236–241. [Google Scholar]
- Manavalan, P.; Johnson, W.C., Jr. Variable selection method improves the prediction of protein secondary structure from circular dichroism spectra. Anal. Biochem. 1987, 167, 76–85. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef]
- Provencher, S.W.; Glöckner, J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry 1981, 20, 33–37. [Google Scholar] [CrossRef]
- Van Stokkum, I.H.; Spoelder, H.J.; Bloemendal, M.; van Grondelle, R.; Groen, F.C. Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal. Biochem. 1990, 191, 110–118. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal. Biochem. 1993, 209, 32–44. [Google Scholar] [CrossRef]
- Sreerama, N.; Venyaminov, S.Y.; Woody, R.W. Estimation of the number of alpha-helical and beta-strand segments in proteins using circular dichroism spectroscopy. Protein Sci. 1999, 8, 370–380. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Piccirilli, A.; Brisdelli, F.; Aschi, M.; Celenza, G.; Amicosante, G.; Perilli, M. Kinetic Profile and Molecular Dynamic Studies Show that Y229W Substitution in an NDM-1/L209F Variant Restores the Hydrolytic Activity of the Enzyme toward Penicillins, Cephalosporins, and Carbapenems. Antimicrob. Agents Chemother. 2019, 63, e02270-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aschi, M.; D’Abramo, M.; Amadei, A. Photoindiced Electron Transfer in a Dichromophoric Peptide: A numerical experiment. Theor. Chem. Acc. 2016, 135, 132–142. [Google Scholar] [CrossRef]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput.-Aided Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrates (β-Lactams) | KPC-3 | D228S | D228W | D228K | D228M | D228L | D228I | D228G | |
|---|---|---|---|---|---|---|---|---|---|
| Benzylpenicillin | Km (µM) | 144 ± 22 | 97 ± 8 | 187 ± 1 | 75 ± 5 | 248 ± 14 | 20 ± 2 | 25 ± 2 | 70 ± 6 |
| kcat (s−1) | 708 ± 15 | 48 ± 2 | 157 ± 4 | 136 ± 6 | 131 ± 5 | 37 ± 1 | 29 ± 1 | 35 ± 2 | |
| kcat/Km (µM−1 s−1) | 4.92 | 0.49 | 0.84 | 1.81 | 0.53 | 1.85 | 1.16 | 0.5 | |
| Carbenicillin | Km (µM) | 108 ± 10 | 281 ± 12 | 179 ± 10 | 494 ± 18 | 240 ± 15 | 280 ± 30 | 182 ± 8 | 100 ± 5 |
| kcat (s−1) | 18 ± 1 | 17 ± 1 | 27 ± 1 | 36 ± 2 | 25 ± 2 | 14 ± 1 | 21 ± 2 | 70 ± 3 | |
| kcat/Km (µM−1 s−1) | 0.17 | 0.06 | 0.15 | 0.07 | 0.10 | 0.05 | 0.12 | 0.7 | |
| Cefotaxime | Km (µM) | 57 ± 3 | ≥1000 | 373 ± 21 | 629 ± 50 | 238 ± 15 | 390 ± 28 | 430 ± 30 | 950 ± 48 |
| kcat (s−1) | 333 ± 12 | N.D. | 108 ± 5 | 68 ± 2 | 931 ± 21 | 34 ± 2 | 41 ± 2 | 244 ± 11 | |
| kcat/Km (µM−1 s−1) | 5.84 | N.D. | 0.29 | 0.11 | 3.91 | 0.09 | 0.09 | 0.21 | |
| Cefazolin | Km (µM) | 189 ± 25 | 150 ± 8 | 119 ± 11 | 126 ± 14 | 141 ± 18 | 76 ± 6 | 121 ± 15 | 205 ± 17 |
| kcat (s−1) | 351 ± 9 | 11 ± 0.4 | 413 ± 3 | 644 ± 8 | 519 ± 14 | 64 ± 4 | 93 ± 4 | 271 ± 8 | |
| kcat/Km (µM−1 s−1) | 1.86 | 0.07 | 3.47 | 5.11 | 3.68 | 0.84 | 0.77 | 1.32 | |
| Ceftazidime | Km (µM) | 100 ± 15 | >1000 | 400 ± 18 | 506 ± 25 | 300 ± 20 | 321 ± 38 | 960 ± 23 | >1000 |
| kcat (s−1) | 1.4 ± 0.1 | N.D. | 3.2 ± 0.1 | 1.8 ± 0.1 | 2.2 ± 0.1 | 0.5 ± 0.1 | 3.7 ± 0.5 | N.D. | |
| kcat/Km (µM−1 s−1) | 0.01 | N.D. | 0.01 | 0.004 | 0.007 | 0.002 | 0.004 | N.D. | |
| Imipenem | Km (µM) | 88 ± 6 | 83 ± 4 | 128 ± 23 | 88 ± 5 | 87 ± 8 | 110 ± 10 | 104 ± 8 | 50 ± 2 |
| kcat(s−1) | 41 ± 2 | 34 ± 1 | 115 ± 9 | 116 ± 5 | 118 ± 2 | 19 ± 1.5 | 30 ± 1 | 26 ± 1 | |
| kcat/Km (µM−1 s−1) | 0.46 | 0.41 | 0.89 | 1.32 | 1.36 | 0.17 | 0.29 | 0.52 | |
| Meropenem | Km (µM) | 68 ± 3 | 19 ± 2 | 46 ± 1 | 99 ± 5 | 48 ± 3 | 34 ± 1 | 62 ± 2 | 36 ± 1 |
| kcat (s−1) | 51 ± 2 | 3 ± 0.1 | 7 ± 0.3 | 8 ± 0.5 | 5 ± 0.3 | 4 ± 0.2 | 5 ± 0.1 | 11 ± 1 | |
| kcat/Km(µM−1 s−1) | 0.75 | 0.16 | 0.15 | 0.08 | 0.10 | 0.12 | 0.08 | 0.31 | |
| Ertapenem | Km (µM) | 30 ± 1 | 63 ± 4 | 40 ± 3 | 26 ± 1 | 30 ± 2 | 21 ± 1 | 29 ± 2 | 18 ± 1 |
| kcat (s−1) | 5 ± 0.5 | 23 ± 2 | 25 ± 1 | 10 ± 1 | 82 ± 1 | 7 ± 0.4 | 11 ± 0.5 | 7 ± 0.2 | |
| kcat/Km (µM−1 s−1) | 0.17 | 0.37 | 0.62 | 0.38 | 2.73 | 0.33 | 0.38 | 0.39 |
| Enzymes | Alpha | Beta | Turn | Unordered | Total | NRMSD (a) |
|---|---|---|---|---|---|---|
| KPC-3 | ||||||
| CDSSTR | 39 | 17 | 18 | 26 | 100 | 0.055 |
| CONTIN | 36 | 16 | 19 | 29 | 100 | 0.036 |
| SELCON | 37 | 15 | 19 | 30 | 101 | 0.103 |
| AVERAGE | 37 ± 1.5 | 16 ± 1 | 18.7 ± 0.6 | 28 ± 2 | 100 ± 5 | |
| D228L | ||||||
| CDSSTR | 33 | 21 | 19 | 27 | 100 | 0.061 |
| CONTIN | 29 | 20 | 20 | 30 | 99 | 0.049 |
| SELCON | 32 | 17 | 20 | 31 | 100 | 0.130 |
| AVERAGE | 31 ± 2 | 19 ± 2 | 19.7 ± 0.6 | 29 ± 2 | 100 ± 7 | |
| D228K | ||||||
| CDSSTR | 34 | 18 | 19 | 28 | 99 | 0.059 |
| CONTIN | 31 | 17 | 20 | 31 | 99 | 0.043 |
| SELCON | 34 | 17 | 20 | 30 | 101 | 0.120 |
| AVERAGE | 33 ± 2 | 17.3 ± 0.6 | 19.7 ± 0.6 | 30 ± 1.5 | 100 ± 5 | |
| D228M | ||||||
| CDSSTR | 37 | 18 | 18 | 27 | 100 | 0.055 |
| CONTIN | 34 | 18 | 19 | 30 | 101 | 0.046 |
| SELCON | 35 | 16 | 20 | 30 | 101 | 0.123 |
| AVERAGE | 35 ± 1.5 | 17.3 ± 1 | 19 ± 1 | 29 ± 2 | 100 ± 5 | |
| GLOBAL AVERAGE | 34 ± 3 | 18 ± 2 | 19 ± 1 | 29 ± 2 | 100 ± 8 |
| Specie | Uncomplexed Species | Complexed Species |
|---|---|---|
| KPC-3 | 29.1 nm | 34.0 nm |
| D228S | 32.2 nm | 34.0 nm |
| D228M | 36.2 nm | 32.0 nm |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piccirilli, A.; Brisdelli, F.; Docquier, J.D.; Aschi, M.; Cherubini, S.; De Luca, F.; Matagne, A.; Amicosante, G.; Perilli, M. Amino Acid Replacement at Position 228 Induces Fluctuation in the Ω-Loop of KPC-3 and Reduces the Affinity against Oxyimino Cephalosporins: Kinetic and Molecular Dynamics Studies. Catalysts 2020, 10, 1474. https://doi.org/10.3390/catal10121474
Piccirilli A, Brisdelli F, Docquier JD, Aschi M, Cherubini S, De Luca F, Matagne A, Amicosante G, Perilli M. Amino Acid Replacement at Position 228 Induces Fluctuation in the Ω-Loop of KPC-3 and Reduces the Affinity against Oxyimino Cephalosporins: Kinetic and Molecular Dynamics Studies. Catalysts. 2020; 10(12):1474. https://doi.org/10.3390/catal10121474
Chicago/Turabian StylePiccirilli, Alessandra, Fabrizia Brisdelli, Jean Denis Docquier, Massimiliano Aschi, Sabrina Cherubini, Filomena De Luca, André Matagne, Gianfranco Amicosante, and Mariagrazia Perilli. 2020. "Amino Acid Replacement at Position 228 Induces Fluctuation in the Ω-Loop of KPC-3 and Reduces the Affinity against Oxyimino Cephalosporins: Kinetic and Molecular Dynamics Studies" Catalysts 10, no. 12: 1474. https://doi.org/10.3390/catal10121474