One-Pot Biocatalytic Preparation of Enantiopure Unusual α-Amino Acids from α-Hydroxy Acids via a Hydrogen-Borrowing Dual-Enzyme Cascade



Abstract
:
1. Introduction
2. Results and Discussion
2.1. Expression and Comparative Enzyme Activity of D-Hydroxy Acid Dehydrogenases
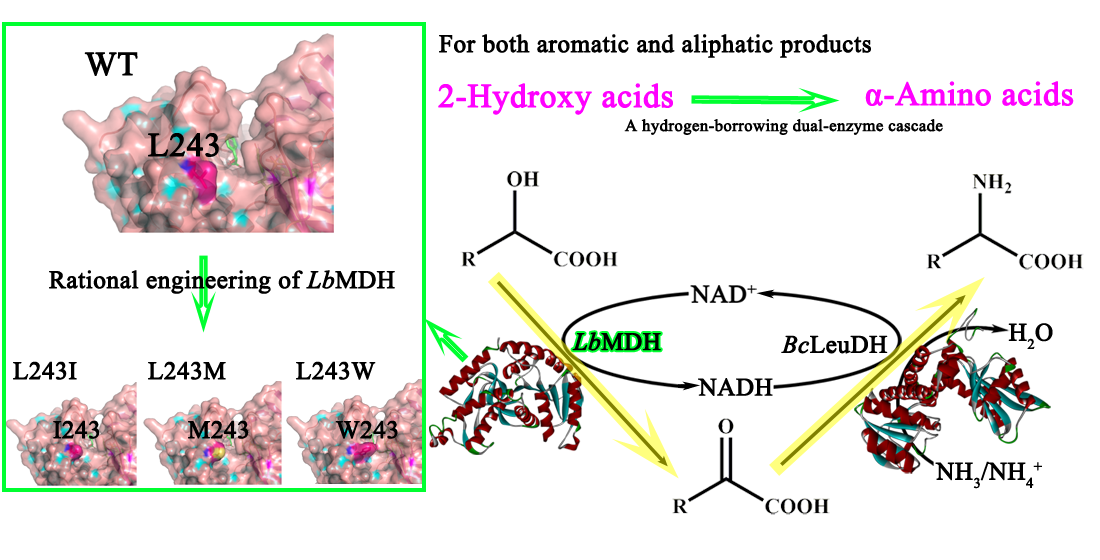
2.2. Rational Engineering of LbMDH
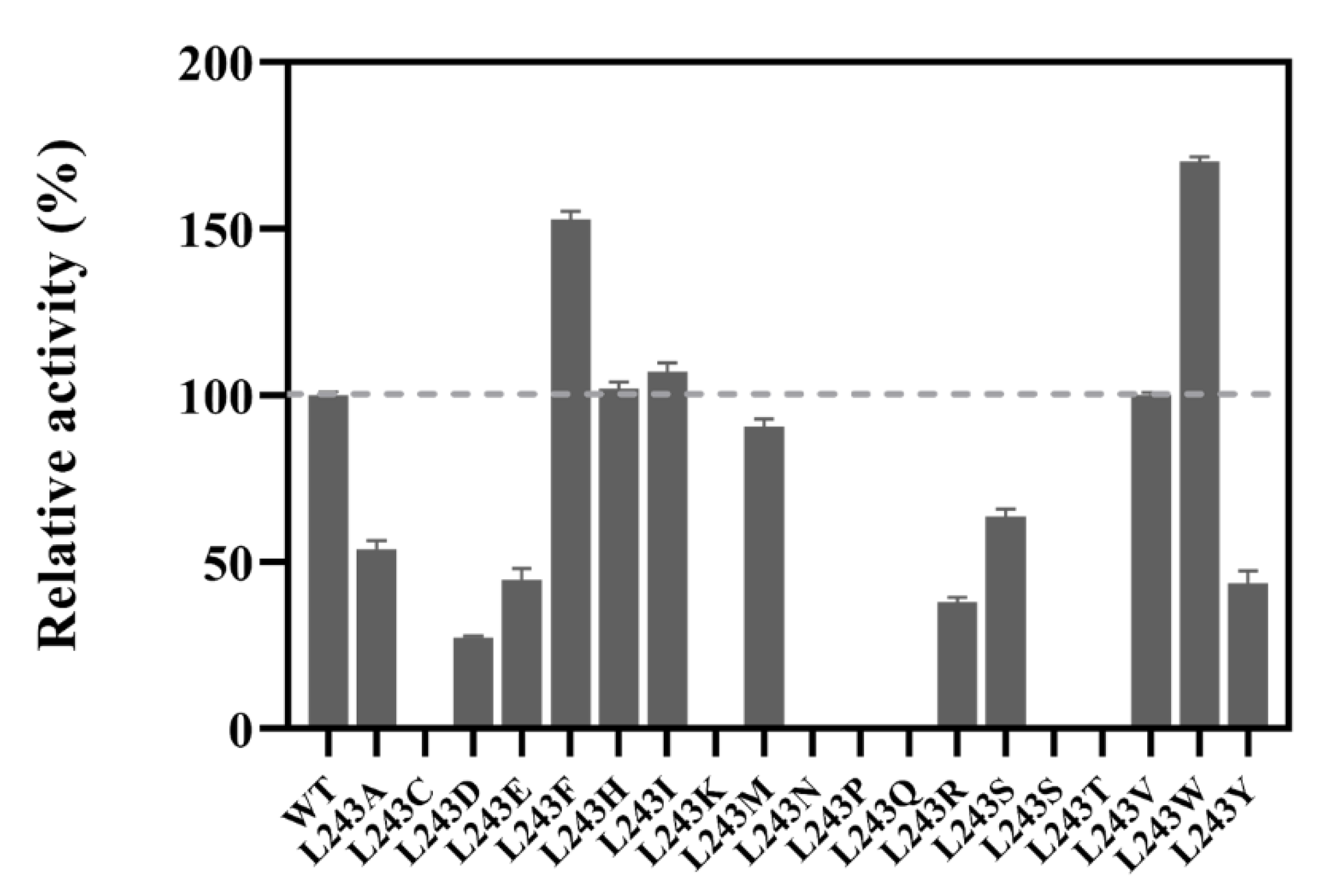
2.3. Site-Saturation Mutagenesis at Residue L243
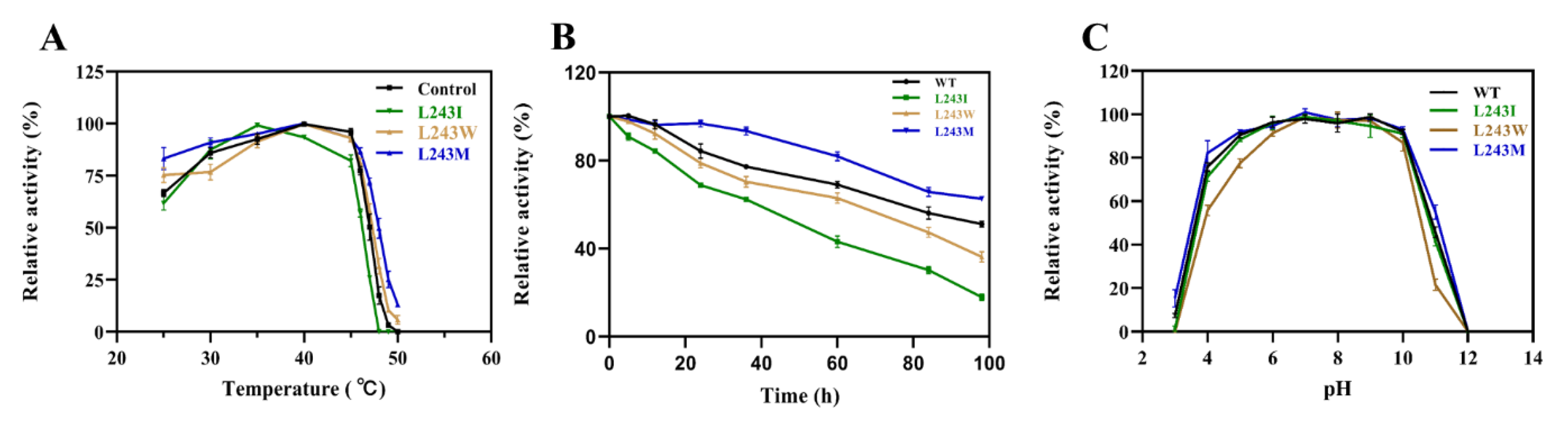
2.4. Enzyme Characterization and Kinetic Analysis of LbMDH and Variants
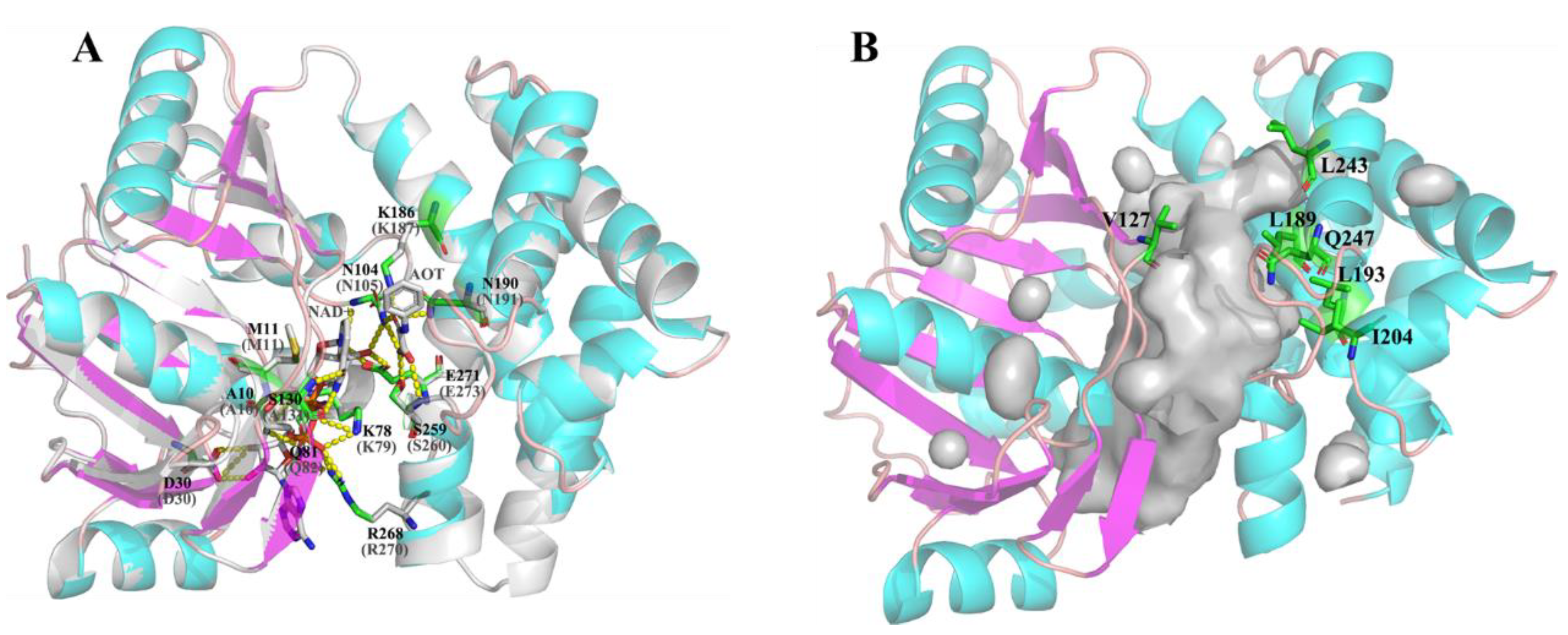
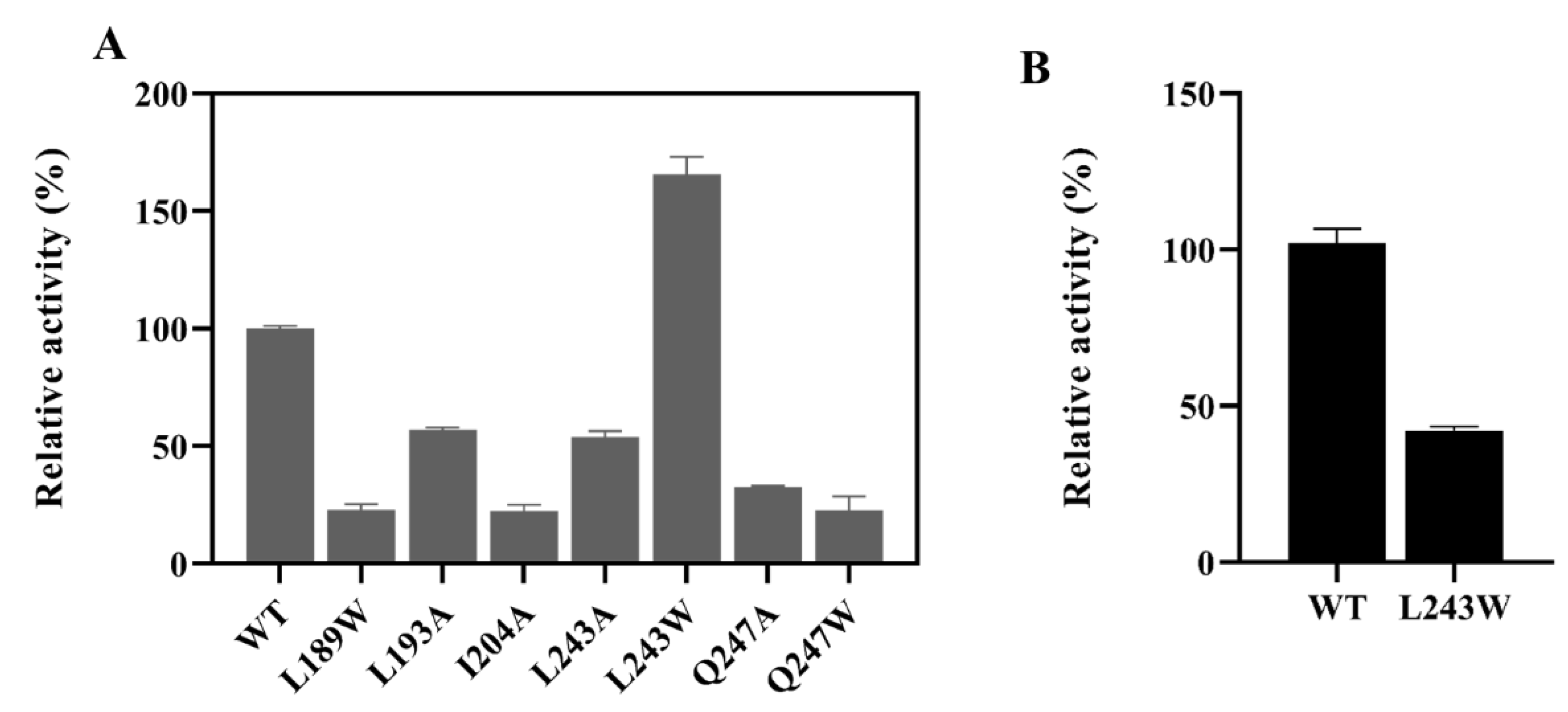
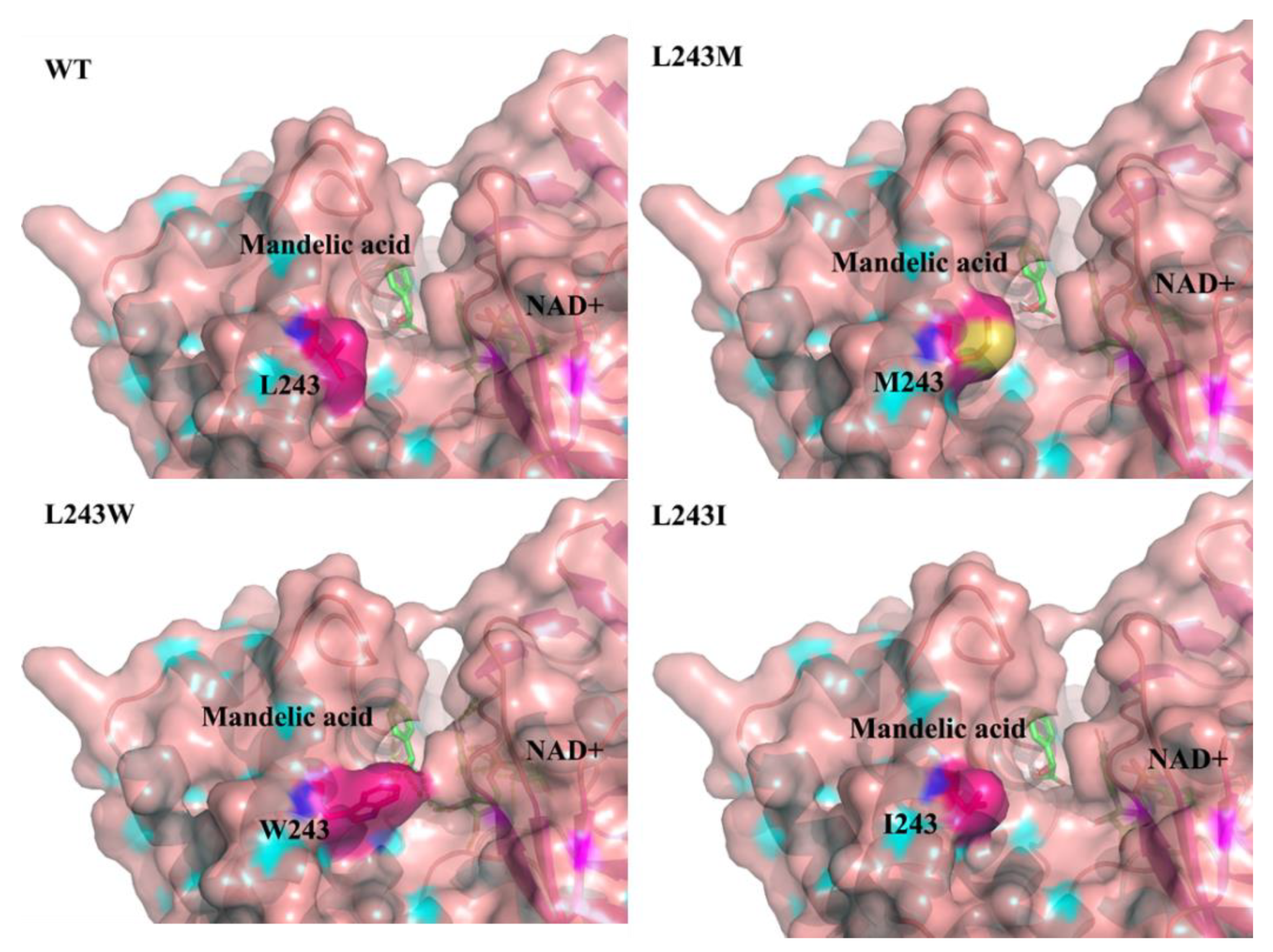
2.5. Structure Analysis of LbMDH and Variants
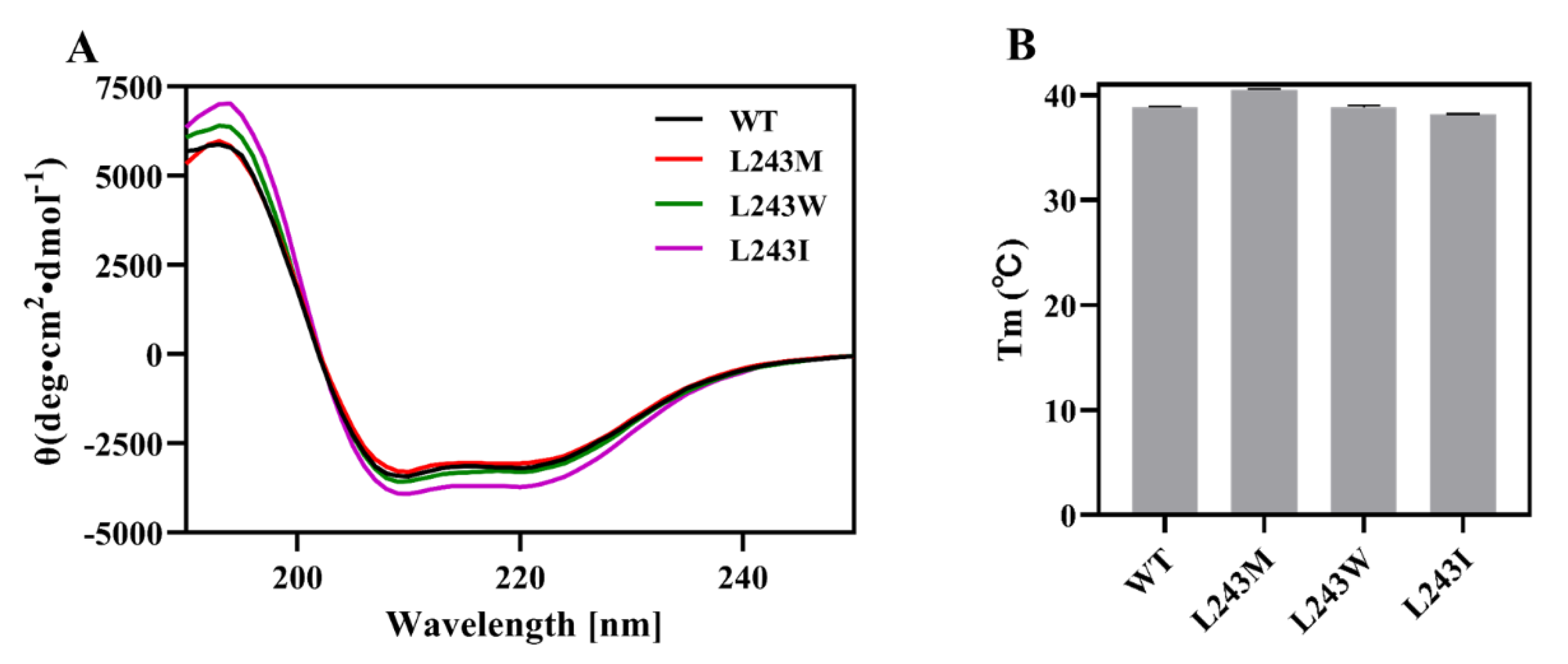
2.6. Secondary Structure and Thermostability Analysis by Circular Dichroism (CD)
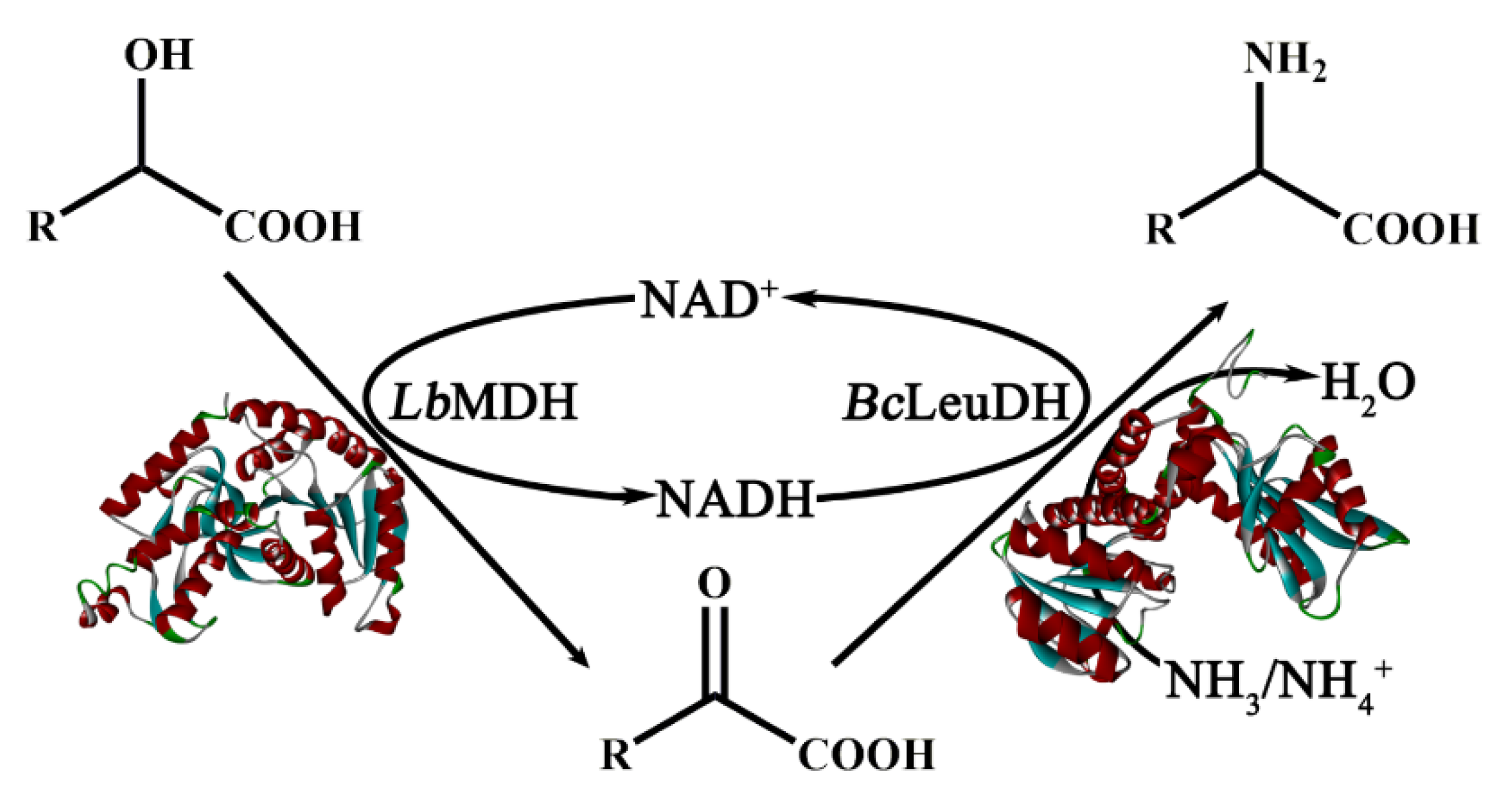
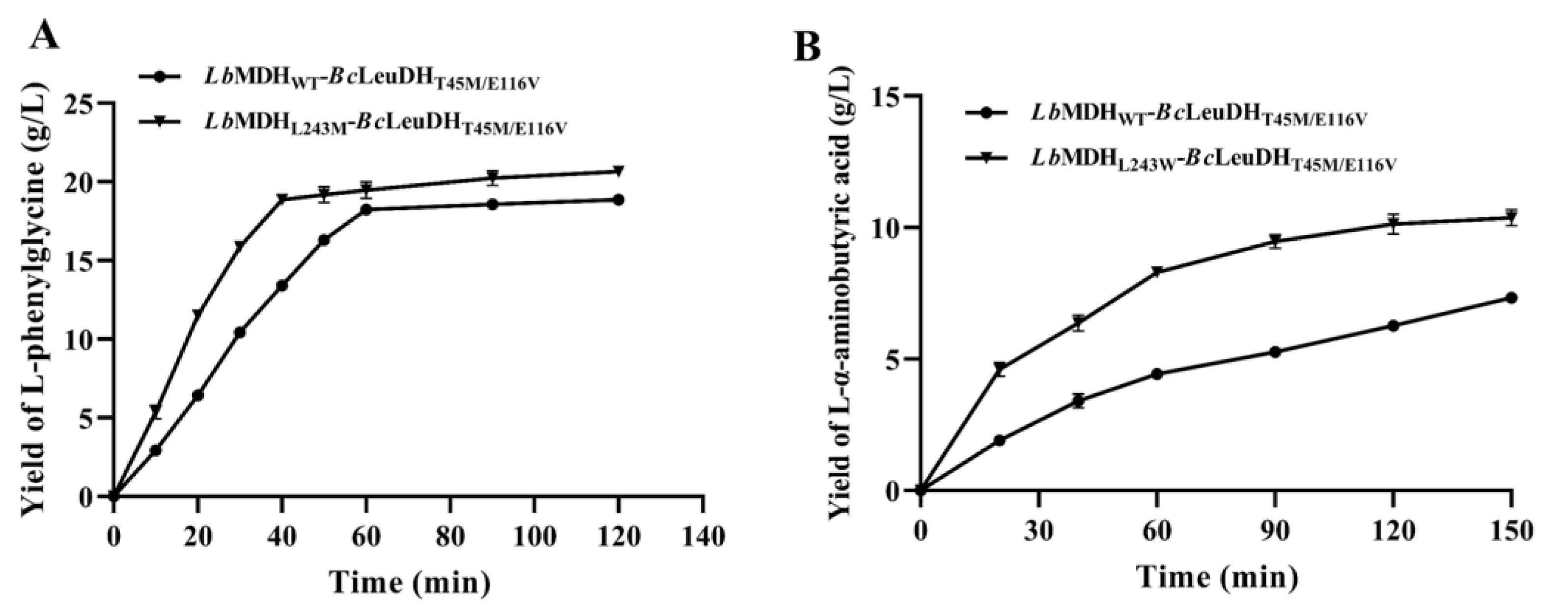
2.7. One-Pot Biocatalytic Preparation of UAAs with a Double-Enzyme System
3. Materials and Methods
3.1. Strains, Plasmids, and Materials
3.2. Site-Directed and Structure Analysis of LbMDH
3.3. Expression and Purification of LbMDH and Its Variants
3.4. Enzyme Activity Assays
3.5. Determination of Kinetic Characteristics
3.6. Circular Dichroism for Structure Analysis and Thermal Melts
3.7. Biocatalytic Preparation of UAAs with a Dual-Enzyme Hydrogen-Borrowing Cascade
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Narancic, T.; Almahboub, S.A.; O’Connor, K.E. Unnatural amino acids: Production and biotechnological potential. World J. Microbiol. Biotechnol. 2019, 35, 67. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, Y.A.; Wittmann, C.; Park, J.B. Production of non-proteinogenic amino acids from α-keto acid precursors with recombinant Corynebacterium glutamicum. Biotechnol. Bioeng. 2013, 110, 2846–2855. [Google Scholar] [CrossRef]
- Cui, Z.; Mureev, S.; Polinkovsky, M.E.; Tnimov, Z.; Guo, Z.; Durek, T.; Jones, A.; Alexandrov, K. Combining Sense and Nonsense Codon Reassignment for Site-Selective Protein Modification with Unnatural Amino Acids. ACS Synth. Biol. 2017, 6, 535–544. [Google Scholar] [CrossRef]
- Blaskovich, M.A. Unusual Amino Acids in Medicinal Chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; He, X. Molecular design and genetic optimization of antimicrobial peptides containing unnatural amino acids against antibiotic-resistant bacterial infections. Biopolymers 2016, 106, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Maluch, I.; Czarna, J.; Drag, M. Applications of Unnatural Amino Acids in Protease Probes. Chem. Asian J. 2019, 14, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- Zerfas, B.L.; Coleman, R.A.; Salazar-Chaparro, A.F.; Macatangay, N.J.; Trader, D.J. Fluorescent Probes with Unnatural Amino Acids to Monitor Proteasome Activity in Real-Time. ACS Chem. Biol. 2020, 15, 2588–2596. [Google Scholar] [CrossRef]
- Soth, M.; Hermann, J.C.; Yee, C.; Alam, M.; Barnett, J.W.; Berry, P.; Browner, M.F.; Frank, K.; Frauchiger, S.; Harris, S.; et al. 3-Amido pyrrolopyrazine JAK kinase inhibitors: Development of a JAK3 vs JAK1 selective inhibitor and evaluation in cellular and in vivo models. J. Med. Chem. 2013, 56, 345–356. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, Y.; Chen, J.; Xu, M.; Yang, T.; Zheng, J.; Zhang, X.; Rao, Z. Rational Engineering of Bacillus cereus Leucine Dehydrogenase Towards α-keto Acid Reduction for Improving Unnatural Amino Acid Production. Biotechnol. J. 2019, 14, e1800253. [Google Scholar] [CrossRef]
- Al Toma, R.S.; Brieke, C.; Cryle, M.J.; Süssmuth, R.D. Structural aspects of phenylglycines, their biosynthesis and occurrence in peptide natural products. Nat. Prod. Rep. 2015, 32, 1207–1235. [Google Scholar] [CrossRef] [Green Version]
- Langen, L.M.V.; Rantwijk, F.V.; Švedas, V.K.; Sheldon, R.A.J.T.A. Penicillin acylase-catalyzed peptide synthesis: A chemo-enzymatic route to stereoisomers of 3,6-diphenylpiperazine-2,5-dione. Tetrahedron Asymmetry 2000, 11, 1077–1083. [Google Scholar] [CrossRef]
- Weber, N.; Hatsch, A.; Labagnere, L.; Heider, H. Production of (S)-2-aminobutyric acid and (S)-2-aminobutanol in Saccharomyces cerevisiae. Microb. Cell Factories 2017, 16, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Tao, R.; Wang, Y.; Jiang, Y.; Lin, X.; Yang, Y.; Zheng, H.; Jiang, W.; Yang, S. Removal of L-alanine from the production of L-2-aminobutyric acid by introduction of alanine racemase and D-amino acid oxidase. Appl. Microbiol. Biotechnol. 2011, 90, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.N.; Comotti, A.; Sozzani, P.; Bracco, S.; Bonge-Hansen, T.; Hennum, M.; Görbitz, C.H. Microporous Molecular Materials from Dipeptides Containing Non-proteinogenic Residues. Angew. Chem. (Int. Ed. Engl.) 2015, 54, 15684–15688. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.M.; Li, J.Q.; Zhang, B.; Liu, Z.Q.; Zheng, Y.G. Fermentative production of the unnatural amino acid L-2-aminobutyric acid based on metabolic engineering. Microb. Cell Factories 2019, 18, 43. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Wang, J.H.; Wu, J.; Liu, J.; Chen, X.L.; Liu, L.M. Asymmetric assembly of high-value α-functionalized organic acids using a biocatalytic chiral-group-resetting process. Nat. Commun. 2018, 9, 3818. [Google Scholar] [CrossRef] [Green Version]
- Schrittwieser, J.H.; Velikogne, S.; Hall, M.; Kroutil, W. Artificial Biocatalytic Linear Cascades for Preparation of Organic Molecules. Chem. Rev. 2018, 118, 270–348. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Yu, L.; Zhou, L.; Zhou, Z. One-Pot Biosynthesis of l-Aspartate from Maleate via an Engineered Strain Containing a Dual-Enzyme System. Appl. Environ. Microbiol. 2019, 85. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Zhou, Y.; Wang, T.; Too, H.P.; Wang, D.I.; Li, Z. Highly regio- and enantioselective multiple oxy- and amino-functionalizations of alkenes by modular cascade biocatalysis. Nat. Commun. 2016, 7, 11917. [Google Scholar] [CrossRef] [Green Version]
- Resch, V.; Fabian, W.M.F.; Kroutil, W.J.A.S. Deracemisation of Mandelic Acid to Optically Pure Non-Natural L-Phenylglycine via a Redox-Neutral Biocatalytic Cascade. Adv. Synth. Catal. 2010, 352, 993–997. [Google Scholar] [CrossRef]
- Böhmer, W.; Knaus, T.; Mutti, F.G. Hydrogen-Borrowing Alcohol Bioamination with Coimmobilized Dehydrogenases. ChemCatChem 2018, 10, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.W.; Xu, G.C.; Ma, B.D.; Bai, Y.P.; Zhang, J.; Xu, J.H. A novel D-mandelate dehydrogenase used in three-enzyme cascade reaction for highly efficient synthesis of non-natural chiral amino acids. J. Biotechnol. 2015, 195, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.D.; Shi, H.L.; Jia, Y.Y.; Li, X.; Wang, L.F.; Xu, J.H.; Yao, L.G.; Kan, Y.C. High level and enantioselective production of L-phenylglycine from racemic mandelic acid by engineered Escherichia coli using response surface methodology. Enzym. Microb. Technol. 2020, 136, 109513. [Google Scholar] [CrossRef] [PubMed]
- Arnold, F.H. Directed Evolution: Bringing New Chemistry to Life. Angew. Chem. (Int. Ed. Engl.) 2018, 57, 4143–4148. [Google Scholar] [CrossRef] [Green Version]
- Almhjell, P.J.; Boville, C.E.; Arnold, F.H. Engineering enzymes for noncanonical amino acid synthesis. Chem. Soc. Rev. 2018, 47, 8980–8997. [Google Scholar] [CrossRef] [Green Version]
- Klaus, M.; Grininger, M. Engineering strategies for rational polyketide synthase design. Nat. Prod. Rep. 2018, 35, 1070–1081. [Google Scholar] [CrossRef] [Green Version]
- Kokkonen, P.; Bednar, D.; Pinto, G.; Prokop, Z.; Damborsky, J. Engineering enzyme access tunnels. Biotechnol. Adv. 2019, 37, 107386. [Google Scholar] [CrossRef]
- Bao, L.; Li, J.J.; Jia, C.; Li, M.; Lu, X. Structure-oriented substrate specificity engineering of aldehyde-deformylating oxygenase towards aldehydes carbon chain length. Biotechnol. Biofuels 2016, 9, 185. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhou, J.; Yang, T.; Zhang, X.; Xu, M.; Rao, Z. Efficient biosynthesis of L-phenylglycine by an engineered Escherichia coli with a tunable multi-enzyme-coordinate expression system. Appl. Microbiol. Biotechnol. 2018, 102, 2129–2141. [Google Scholar] [CrossRef]
- Miyanaga, A.; Fujisawa, S.; Furukawa, N.; Arai, K.; Nakajima, M.; Taguchi, H. The crystal structure of D-mandelate dehydrogenase reveals its distinct substrate and coenzyme recognition mechanisms from those of 2-ketopantoate reductase. Biochem. Biophys. Res. Commun. 2013, 439, 109–114. [Google Scholar] [CrossRef]
- Furukawa, N.; Miyanaga, A.; Nakajima, M.; Taguchi, H. The ternary complex structure of d-mandelate dehydrogenase with NADH and anilino(oxo)acetate. Biochem. Biophys. Res. Commun. 2017, 486, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Enugala, T.R.; Corbella, M.; Kamerlin, S.C.L.; Widersten, M.J.A.C. The Role of Substrate-Coenzyme Crosstalk in Determining Turnover Rates in Rhodococcus ruber Alcohol Dehydrogenase. ACS Catal. 2020, 10, 9115–9128. [Google Scholar] [CrossRef]
- Zhu, X.; Shin, W.H.; Kim, H.; Kihara, D. Combined Approach of Patch-Surfer and PL-PatchSurfer for Protein-Ligand Binding Prediction in CSAR 2013 and 2014. J. Chem. Inf. Modeling 2016, 56, 1088–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Jiang, T.; Liu, R.; Feng, X.; Li, C. Tuning the pH profile of β-glucuronidase by rational site-directed mutagenesis for efficient transformation of glycyrrhizin. Appl. Microbiol. Biotechnol. 2019, 103, 4813–4823. [Google Scholar] [CrossRef] [PubMed]
- Chmelova, K.; Sebestova, E.; Liskova, V.; Beier, A.; Bednar, D.; Prokop, Z.; Chaloupkova, R.; Damborsky, J. A Haloalkane Dehalogenase from Saccharomonospora viridis Strain DSM 43017, a Compost Bacterium with Unusual Catalytic Residues, Unique (S)-Enantiopreference, and High Thermostability. Appl. Environ. Microbiol. 2020, 86, e02820-19. [Google Scholar] [CrossRef] [PubMed]
- Sanchis, J.; Fernández, L.; Carballeira, J.D.; Drone, J.; Gumulya, Y.; Höbenreich, H.; Kahakeaw, D.; Kille, S.; Lohmer, R.; Peyralans, J.J.; et al. Improved PCR method for the creation of saturation mutagenesis libraries in directed evolution: Application to difficult-to-amplify templates. Appl. Microbiol. Biotechnol. 2008, 81, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Szilárd, P.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. arxiv 2014, arXiv:1506.00716. [Google Scholar]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Bartolomeo, M.P.; Maisano, F. Validation of a reversed-phase HPLC method for quantitative amino acid analysis. J. Biomol. Tech. JBT 2006, 17, 131–137. [Google Scholar]
- Zhang, T.; Zhang, R.; Xu, M.; Zhang, X.; Yang, T.; Liu, F.; Yang, S.; Rao, Z.J.P.B. Glu56Ser mutation improves the enzymatic activity and catalytic stability of Bacillus subtilis L-aspartate α-decarboxylase for an efficient β-alanine production. Process Biochem. 2018, 70, 117–123. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Enzyme | D-Mandelic Acid (U/mg) | D-α-Hydroxybutyric Acid (U/mg) | D-α-Hydroxycaproic Acid (U/mg) | Optimum pH | Cofactor |
|---|---|---|---|---|---|---|
| L. brevis | LbMDH | 243.5 ± 13.5 | 60.2 ± 4.7 | 118.9 ± 10.6 | 10.5 | NAD+ |
| E. faecalis | Ef2D2R | 12.4 ± 1.3 | 13.2 ± 3.9 | 10.5 ± 2.4 | 10.5 | NAD+ |
| S. aureus | SaDlacDH | 6.5 ± 0.9 | n/a | n/a | 11.0 | NAD+ |
| P. aeruginosa | PaDlacDH | 0.9 ± 0.4 | n/a | n/a | 11.5 | NAD+ |
| P. aeruginosa | Pa2D2R | n/a | 0.7 ± 0.2 | 0.8 ± 0.4 | 11 | NADP+ |
| Substrate | Relative Activity (%) | ||||||
|---|---|---|---|---|---|---|---|
| WT | L243M | L243W | L243V | L243I | L243F | L243H | |
| D-Mandelic acid | 100 | 231.2 ± 5.6 | 42.1 ± 0.8 | 169.5 ± 6.7 | 192.7 ± 2.5 | 50.8 ± 1.9 | 105.2 ± 2.1 |
| D-α-Hydroxybutanoic acid | 100 | 90.0 ± 1.3 | 171.0 ± 8.1 | 99.9 ± 4.2 | 107.5 ± 5.3 | 152.9 ± 9.3 | 98.3 ± 3.0 |
| D-α-hydroxycaproic acid | 100 | 121.6 ± 3.7 | 85.8 ± 6.0 | 130.8 ± 7.9 | 146.9 ± 7.8 | 93.7 ± 4.1 | 106.6 ± 3.5 |
| D-Phenyllactic acid | 100 | 78.5 ± 4.3 | 79.7 ± 3.2 | 67.0 ± 4.8 | 138.8 ± 9.2 | 81.4 ± 3.9 | 92.8 ± 6.2 |
| D-Lactic acid | 100 | 92.6 ± 8.9 | 339.1 ± 15.9 | 126.3 ± 6.3 | 75.9 ± 4.4 | 424.2 ± 29.7 | 130.5 ± 5.0 |
| Substrate | WT | L243M | ||||
| Km | kcat | kcat/Km | Km | kcat | kcat/Km | |
| (mM) | (s−1) | (s−1mM−1) | (mM) | (s−1) | (s−1mM−1) | |
| NAD+ | 0.25 ± 0.012 | 0.41 ± 0.004 | 1.661 | 0.45 ± 0.071 | 0.42 ± 0.027 | 0.933 |
| D-mandelic acid | 3.71 ± 0.031 | 0.44 ± 0.011 | 0.118 | 3.09 ± 0.196 | 0.52 ± 0.090 | 0.168 |
| D-α-hydroxybutanoic acid | 31.7 ± 0.580 | 0.22 ± 0.005 | 0.007 | 25.9 ± 0.207 | 0.05 ± 0.006 | 0.002 |
| D-α-hydroxycaproic acid | 20.9 ± 0.106 | 0.41 ± 0.037 | 0.02 | 18.7 ± 0.583 | 0.51 ± 0.032 | 0.027 |
| D-phenyllactic acid | 47.2 ± 0.237 | 0.16 ± 0.002 | 0.003 | 50.4 ± 1.070 | 0.13 ± 0.028 | 0.003 |
| D-lactic acid | 60.4 ± 2.934 | 0.05 ± 0.001 | 0.001 | 61.7 ± 4.535 | 0.08 ± 0.005 | 0.001 |
| Substrate | L243W | L243I | ||||
| Km | kcat | kcat/Km | Km | kcat | kcat/Km | |
| (mM) | (s−1) | (s−1mM−1) | (mM) | (s−1) | (s−1mM−1) | |
| NAD+ | 0.72 ± 0.008 | 0.15 ± 0.031 | 0.038 | 0.49 ± 0.014 | 0.61 ± 0.007 | 1.245 |
| D-mandelic acid | 5.88 ± 0.119 | 0.13 ± 0.008 | 0.022 | 4.01 ± 0.193 | 0.46 ± 0.005 | 0.114 |
| D-α-hydroxybutanoic acid | 9.2 ± 0.470 | 0.11 ± 0.003 | 0.012 | 49.8 ± 1.981 | 0.24 ± 0.073 | 0.005 |
| D-α-hydroxycaproic acid | 21.5 ± 0.134 | 0.12 ± 0.022 | 0.006 | 15.8 ± 0.335 | 0.49 ± 0.006 | 0.031 |
| D-phenyllactic acid | 49.8 ± 0.576 | 0.10 ± 0.019 | 0.002 | 33.7 ± 0.985 | 0.18 ± 0.038 | 0.005 |
| D-lactic acid | 48.2 ± 1.339 | 0.13 ± 0.009 | 0.003 | 65.3 ± 1.805 | 0.09 ± 0.002 | 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, F.; Zhou, J.; Xu, M.; Yang, T.; Shao, M.; Zhang, X.; Rao, Z. One-Pot Biocatalytic Preparation of Enantiopure Unusual α-Amino Acids from α-Hydroxy Acids via a Hydrogen-Borrowing Dual-Enzyme Cascade. Catalysts 2020, 10, 1470. https://doi.org/10.3390/catal10121470
Liu F, Zhou J, Xu M, Yang T, Shao M, Zhang X, Rao Z. One-Pot Biocatalytic Preparation of Enantiopure Unusual α-Amino Acids from α-Hydroxy Acids via a Hydrogen-Borrowing Dual-Enzyme Cascade. Catalysts. 2020; 10(12):1470. https://doi.org/10.3390/catal10121470
Chicago/Turabian StyleLiu, Fei, Junping Zhou, Meijuan Xu, Taowei Yang, Minglong Shao, Xian Zhang, and Zhiming Rao. 2020. "One-Pot Biocatalytic Preparation of Enantiopure Unusual α-Amino Acids from α-Hydroxy Acids via a Hydrogen-Borrowing Dual-Enzyme Cascade" Catalysts 10, no. 12: 1470. https://doi.org/10.3390/catal10121470