Porosity and Structure of Hierarchically Porous Ni/Al2O3 Catalysts for CO2 Methanation

 , , , , , ,
, , , , , ,  and
and
Abstract
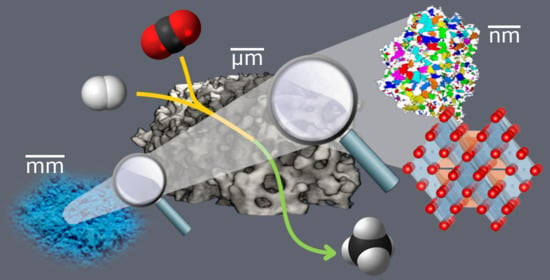
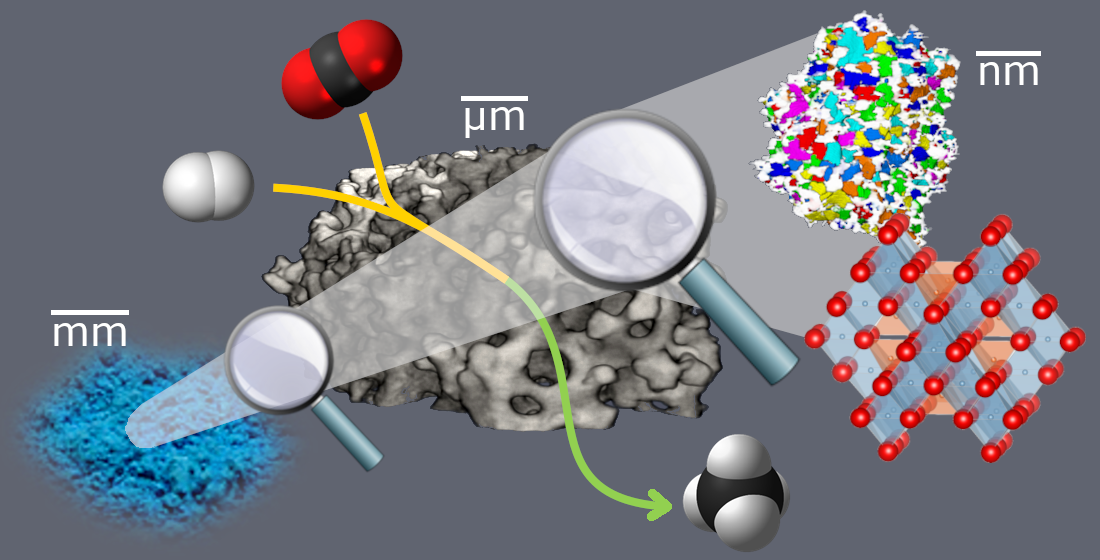
:
1. Introduction
2. Results and Discussion
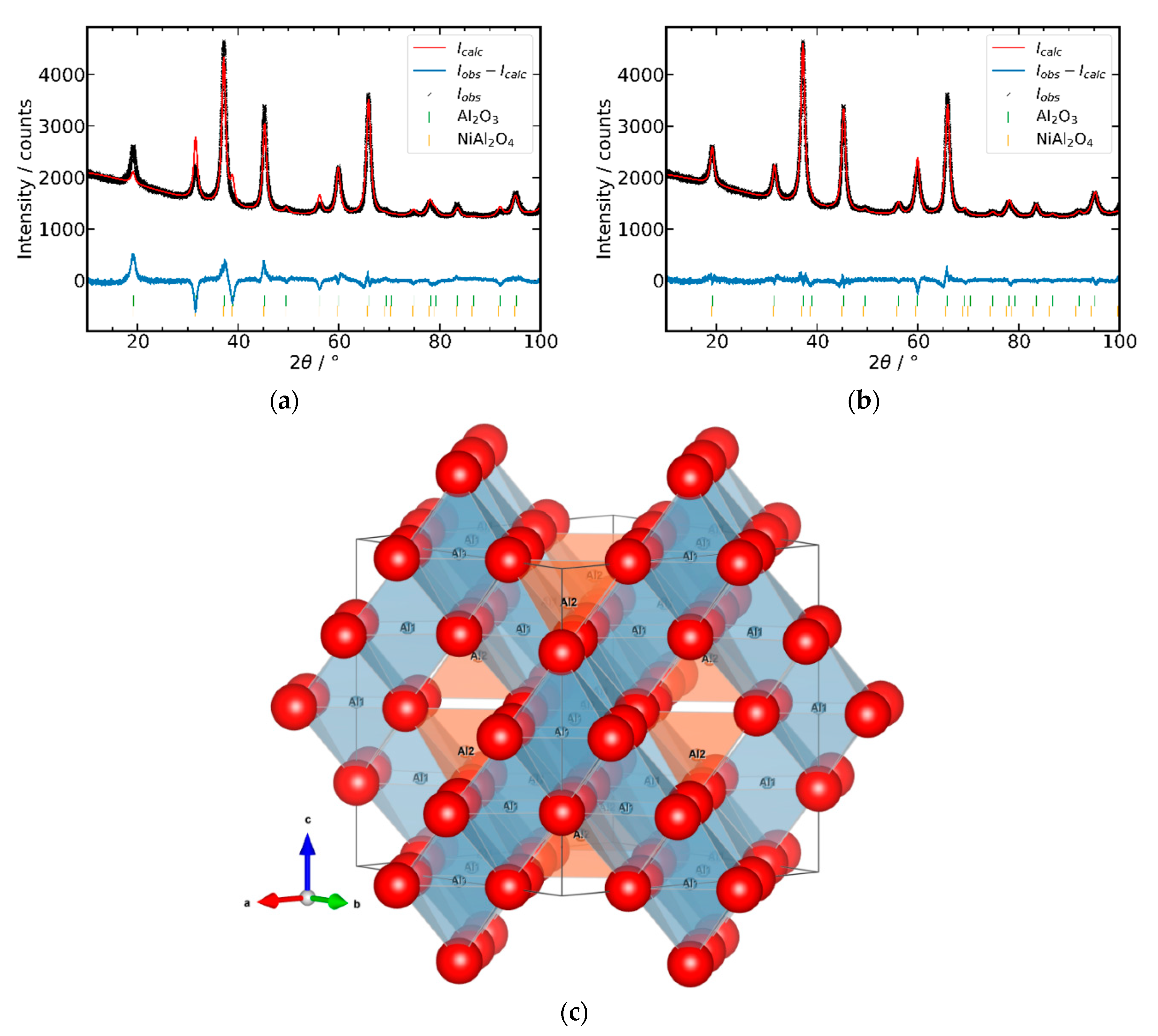
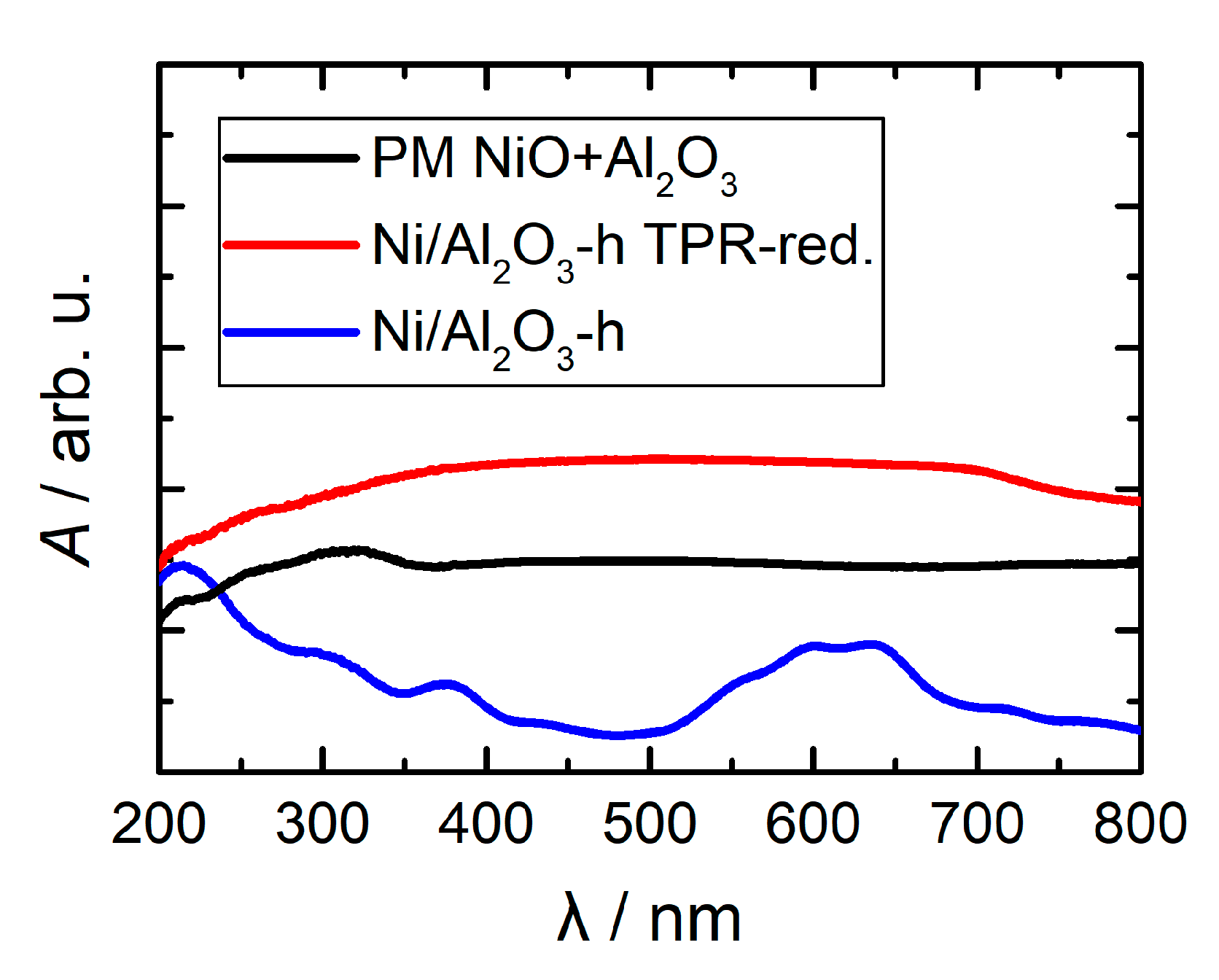
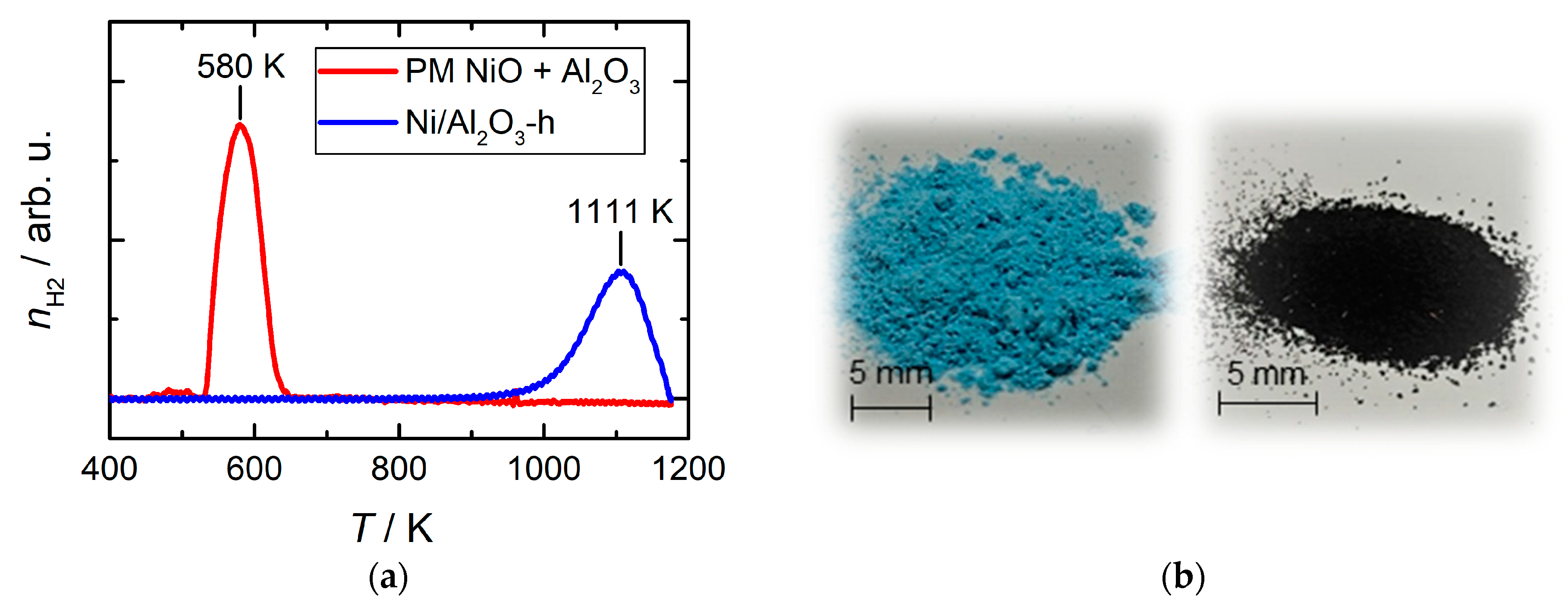
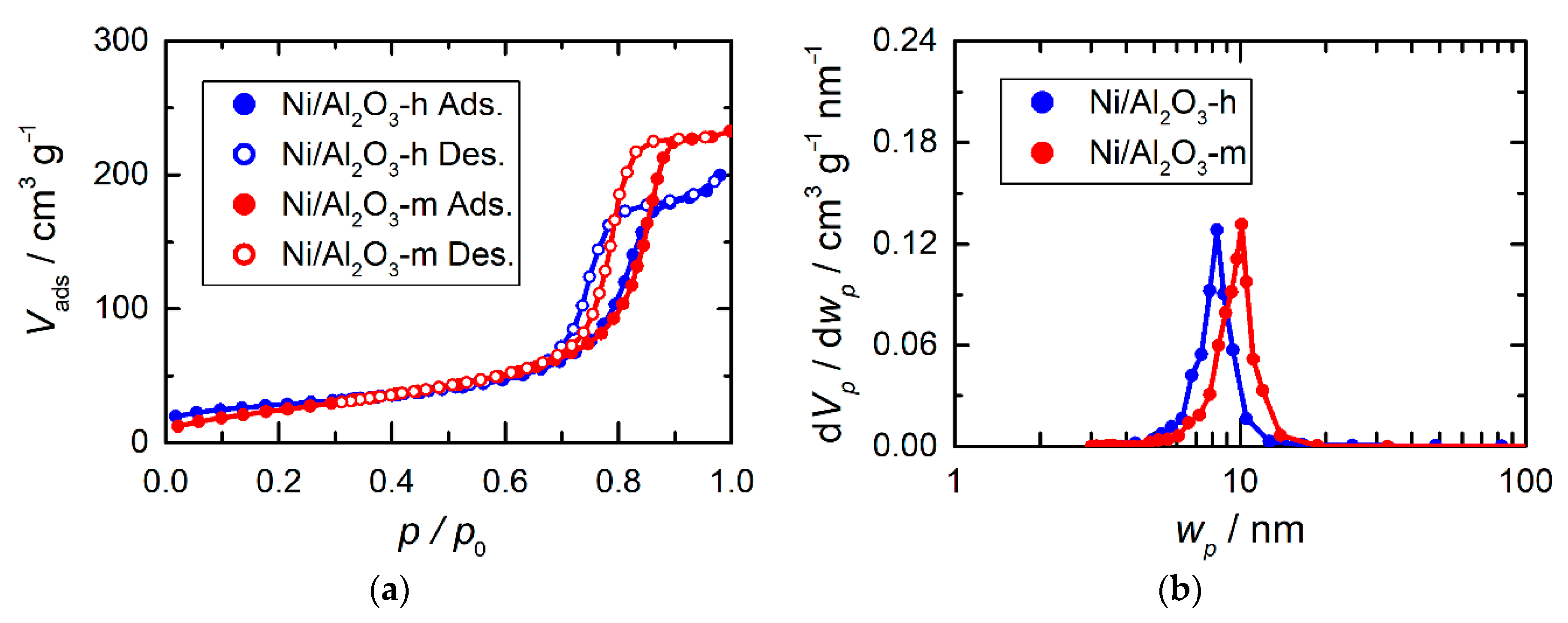
2.1. Synthesis and Structural Characterization
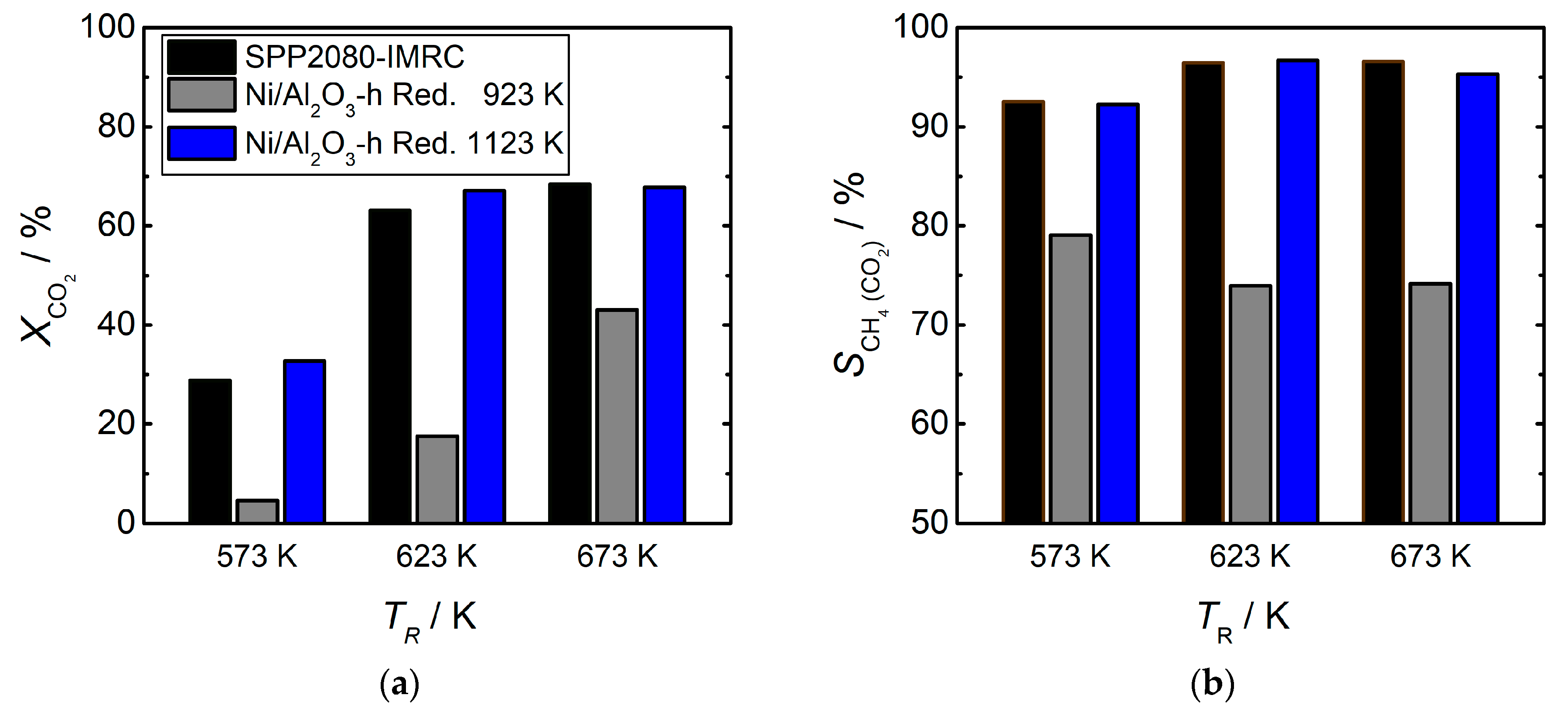
2.2. Catalytic Measurements
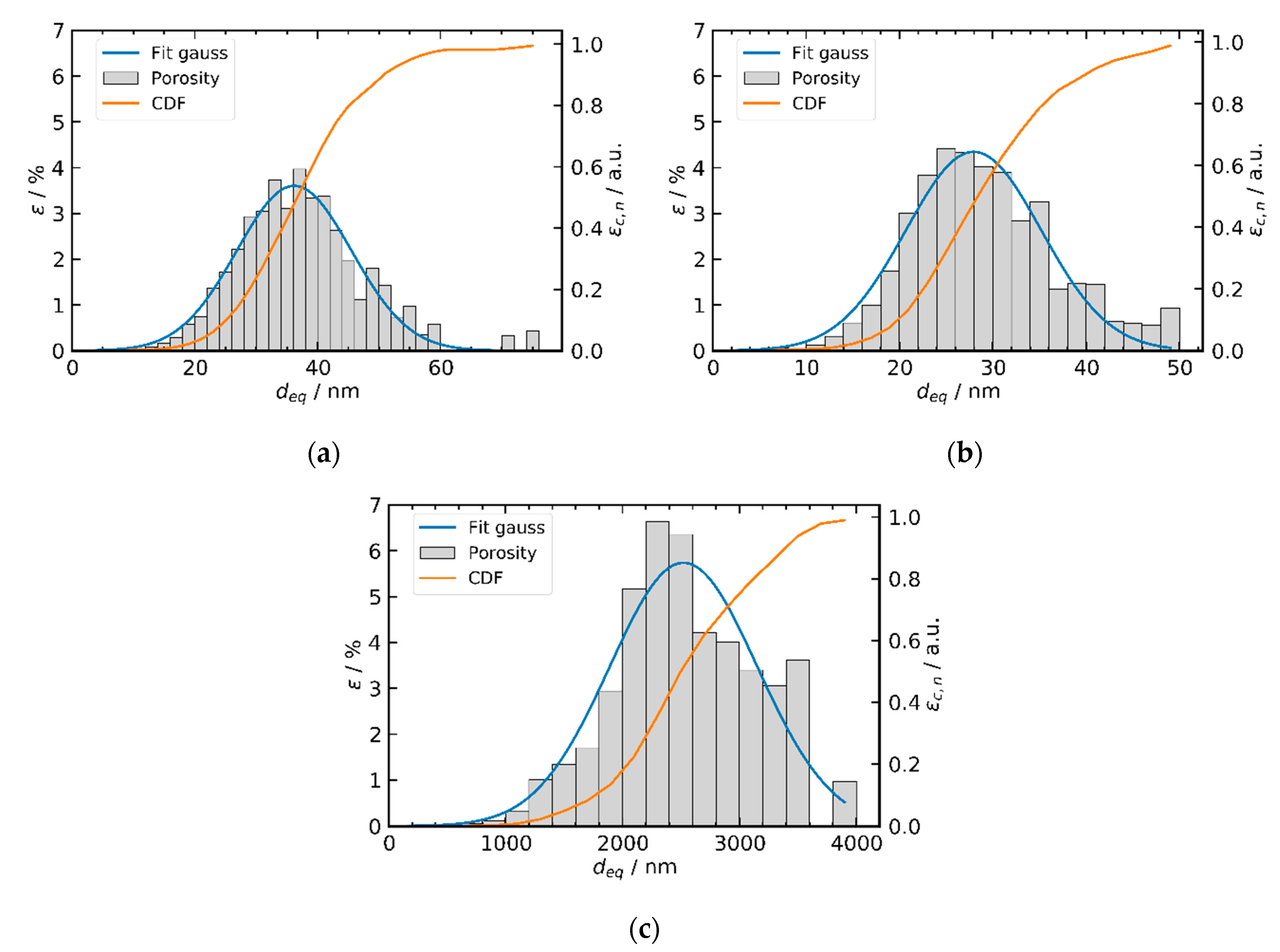
2.3. Tomography Studies
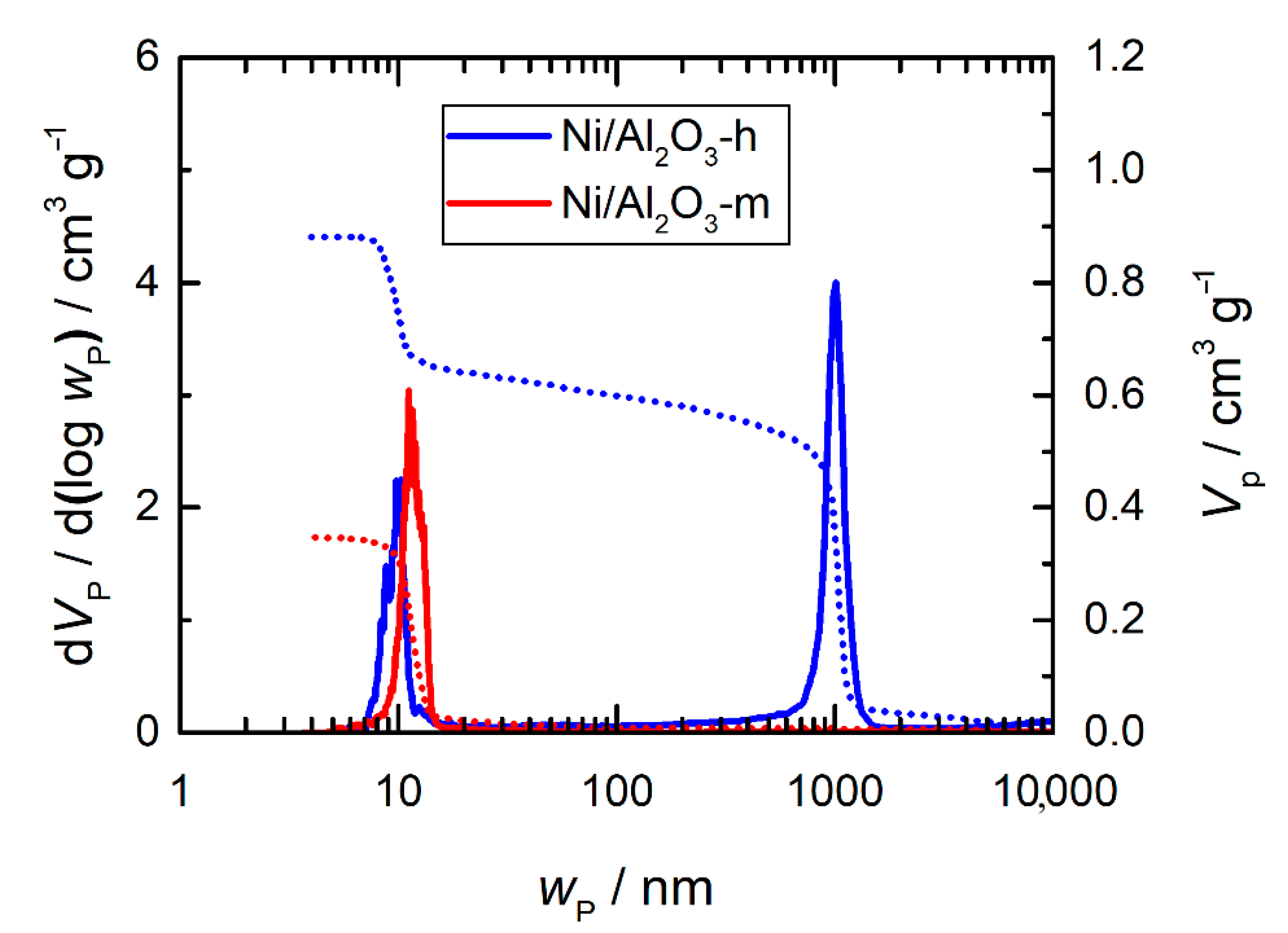
2.4. Porosity Analysis
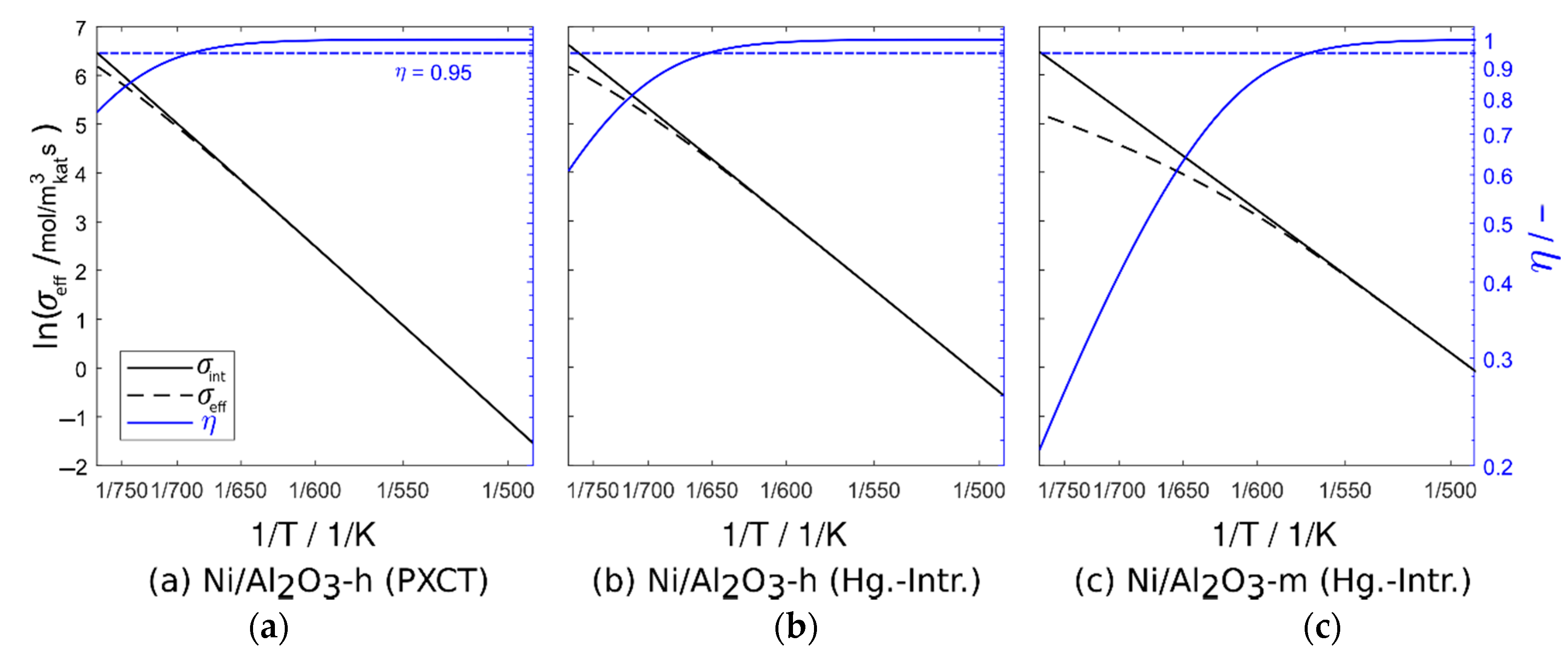
2.5. Macrokinetics
3. Materials and Methods
3.1. Materials and Synthesis
3.2. Characterization
3.3. Catalytic Measurements
3.4. Tomography Studies
3.5. Macrokinetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on methanation—From fundamentals to current projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P. Supported Catalysts for CO2 Methanation. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Gao, J.; Liu, Q.; Gu, F.; Liu, B.; Zhong, Z.; Su, F. Recent advances in methanation catalysts for the production of synthetic natural gas. RSC Adv. 2015, 5, 22759–22776. [Google Scholar] [CrossRef]
- Schlereth, D.; Hinrichsen, O. A fixed-bed reactor modeling study on the methanation of CO2. Chem. Eng. Res. Des. 2014, 92, 702–712. [Google Scholar] [CrossRef]
- Ghaib, K.; Ben-Fares, F.-Z. Power-to-Methane. Renew. Sustain. Energy Rev. 2018, 81, 433–446. [Google Scholar] [CrossRef]
- Zangouei, M.; Moghaddam, A.Z.; Arasteh, M. The influence of nickel loading on reducibility of NiO/Al2O3 catalysts synthesized by sol-gel method. Chem. Eng. Res. Bull. 2010, 14. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Li, Y.; Liu, X.; Guo, Y.; Pao, C.-W.; Chen, J.-L.; Hu, Y.; Wang, Y. NiAl2O4 Spinel Supported Pt Catalyst: High Performance and Origin in Aqueous-Phase Reforming of Methanol. ACS Catal. 2019, 9, 9671–9682. [Google Scholar] [CrossRef]
- Srisawad, N.; Chaitree, W.; Mekasuwandumrong, O.; Shotipruk, A.; Jongsomjit, B.; Panpranot, J. CO2 hydrogenation over Co/Al2O3 catalysts prepared via a solid-state reaction of fine gibbsite and cobalt precursors. React. Kinet. Mech. Cat. 2012, 107, 179–188. [Google Scholar] [CrossRef]
- Kim, Y.; Kwon, S.; Song, Y.; Na, K. Catalytic CO2 hydrogenation using mesoporous bimetallic spinel oxides as active heterogeneous base catalysts with long lifetime. J. CO2 Util. 2020, 36, 145–152. [Google Scholar] [CrossRef]
- Kiš, E.; Marinkovic-Neducin, R.; Lomic, G.; Boskovic, G.; Obadovic, D.Z.; Kiurski, J.; Putanov, P. Structural and textural properties of the NiO-AI2O3 catalyst. Polyhedron 1998, 17, 27–34. [Google Scholar]
- Sengupta, S.; Ray, K.; Deo, G. Effects of modifying Ni/Al2O3 catalyst with cobalt on the reforming of CH4 with CO2 and cracking of CH4 reactions. Int. J. Hydrogen Energy 2014, 39, 11462–11472. [Google Scholar] [CrossRef]
- Daroughegi, R.; Meshkani, F.; Rezaei, M. Enhanced activity of CO2 methanation over mesoporous nanocrystalline Ni–Al2O3 catalysts prepared by ultrasound-assisted co-precipitation method. Int. J. Hydrog. Energy 2017, 42, 15115–15125. [Google Scholar] [CrossRef]
- Moghaddam, S.V.; Rezaei, M.; Meshkani, F. Surfactant-Free Sol–Gel Synthesis Method for the Preparation of Mesoporous High Surface Area NiO–Al2O3 Nanopowder and Its Application in Catalytic CO2 Methanation. Energy Technol. 2020, 8, 1900778. [Google Scholar] [CrossRef]
- Salhi, N.; Boulahouache, A.; Petit, C.; Kiennemann, A.; Rabia, C. Steam reforming of methane to syngas over NiAl2O4 spinel catalysts. Int. J. Hydrog. Energy 2011, 36, 11433–11439. [Google Scholar] [CrossRef]
- Herwig, J.; Titus, J.; Kullmann, J.; Wilde, N.; Hahn, T.; Gläser, R.; Enke, D. Hierarchically Structured Porous Spinels via an Epoxide-Mediated Sol–Gel Process Accompanied by Polymerization-Induced Phase Separation. ACS Omega 2018, 3, 1201–1212. [Google Scholar] [CrossRef]
- Rahmani, S.; Rezaei, M.; Meshkani, F. Preparation of highly active nickel catalysts supported on mesoporous nanocrystalline γ-Al2O3 for CO2 methanation. J. Ind. Eng. Chem. 2014, 20, 1346–1352. [Google Scholar] [CrossRef]
- Lin, J.; Ma, C.; Luo, J.; Kong, X.; Xu, Y.; Ma, G.; Wang, J.; Zhang, C.; Li, Z.; Ding, M. Preparation of Ni based mesoporous Al2O3 catalyst with enhanced CO2 methanation performance. RSC Adv. 2019, 9, 8684–8694. [Google Scholar] [CrossRef] [Green Version]
- Tokudome, Y.; Fujita, K.; Nakanishi, K.; Miura, K.; Hirao, K. Synthesis of Monolithic Al2O3 with Well-Defined Macropores and Mesostructured Skeletons via the Sol−Gel Process Accompanied by Phase Separation. Chem. Mater. 2007, 19, 3393–3398. [Google Scholar] [CrossRef]
- Macedo, H.P.; Medeiros, R.L.; Ilsemann, J.; Melo, D.M.; Rezwan, K.; Wilhelm, M. Nickel-containing hybrid ceramics derived from polysiloxanes with hierarchical porosity for CO2 methanation. Microporous Mesoporous Mater. 2019, 278, 156–166. [Google Scholar] [CrossRef]
- Xiong, Q.; Baychev, T.G.; Jivkov, A.P. Review of pore network modelling of porous media: Experimental characterisations, network constructions and applications to reactive transport. J. Contam. Hydol. 2016, 192, 101–117. [Google Scholar] [CrossRef]
- Friedrich, H.; de Jongh, P.E.; Verkleij, A.J.; de Jong, K.P. Electron tomography for heterogeneous catalysts and related nanostructured materials. Chem. Rev. 2009, 109, 1613–1629. [Google Scholar] [CrossRef] [PubMed]
- Tariq, F.; Haswell, R.; Lee, P.D.; McComb, D.W. Characterization of hierarchical pore structures in ceramics using multiscale tomography. Acta Mater. 2011, 59, 2109–2120. [Google Scholar] [CrossRef]
- Andersson, L.; Jones, A.C.; Knackstedt, M.A.; Bergström, L. Three-dimensional structure analysis by X-ray micro-computed tomography of macroporous alumina templated with expandable microspheres. J. Eur. Ceram. 2010, 30, 2547–2554. [Google Scholar] [CrossRef]
- Shanti, N.O.; Chan, V.W.; Stock, S.R.; de Carlo, F.; Thornton, K.; Faber, K.T. X-ray micro-computed tomography and tortuosity calculations of percolating pore networks. Acta Mater. 2014, 71, 126–135. [Google Scholar] [CrossRef]
- Stoeckel, D.; Kübel, C.; Hormann, K.; Höltzel, A.; Smarsly, B.M.; Tallarek, U. Morphological analysis of disordered macroporous-mesoporous solids based on physical reconstruction by nanoscale tomography. Langmuir 2014, 30, 9022–9027. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.-J.; Svidrytski, A.; Höltzel, A.; Florek, J.; Kleitz, F.; Wang, W.; Kübel, C.; Hlushkou, D.; Tallarek, U. Hindered Diffusion in Ordered Mesoporous Silicas: Insights from Pore-Scale Simulations in Physical Reconstructions of SBA-15 and KIT-6 Silica. J. Phys. Chem. C 2018, 122, 12350–12361. [Google Scholar] [CrossRef]
- Reising, A.E.; Schlabach, S.; Baranau, V.; Stoeckel, D.; Tallarek, U. Analysis of packing microstructure and wall effects in a narrow-bore ultrahigh pressure liquid chromatography column using focused ion-beam scanning electron microscopy. J. Chromatogr. A 2017, 1513, 172–182. [Google Scholar] [CrossRef]
- Becher, J.; Sheppard, T.L.; Fam, Y.; Baier, S.; Wang, W.; Di Wang Kulkarni, S.; Keller, T.F.; Lyubomirskiy, M.; Brueckner, D.; Kahnt, M.; et al. Mapping the Pore Architecture of Structured Catalyst Monoliths from Nanometer to Centimeter Scale with Electron and X-ray Tomographies. J. Phys. Chem. C 2019, 123, 25197–25208. [Google Scholar] [CrossRef]
- Dierolf, M.; Menzel, A.; Thibault, P.; Schneider, P.; Kewish, C.M.; Wepf, R.; Bunk, O.; Pfeiffer, F. Ptychographic X-ray computed tomography at the nanoscale. Nature 2010, 467, 436–439. [Google Scholar] [CrossRef]
- Holler, M.; Diaz, A.; Guizar-Sicairos, M.; Karvinen, P.; Färm, E.; Härkönen, E.; Ritala, M.; Menzel, A.; Raabe, J.; Bunk, O. X-ray ptychographic computed tomography at 16 nm isotropic 3D resolution. Sci. Rep. 2014, 4, 3857. [Google Scholar] [CrossRef] [Green Version]
- Novák, V.; Dudák, M.; Kočí, P.; Marek, M. Understanding the gas transport in porous catalyst layers by using digital reconstruction techniques. Curr. Opin. Chem. Eng. 2015, 9, 16–26. [Google Scholar] [CrossRef]
- Wefers, K.M.; Misra, C. Oxides and Hydroxides of Aluminum, 19th ed.; Alcoa Laboratories: Pittsburgh, PA, USA, 1987. [Google Scholar]
- Zhou, R.S.; Snyder, R.L. Structures and transformation mechanisms of the η, γ and θ transition aluminas. Acta Crystallogr. B Struct. Sci. Cryst. 1991, 47, 617–630. [Google Scholar] [CrossRef]
- Smith, S.J.; Amin, S.; Woodfield, B.F.; Boerio-Goates, J.; Campbell, B.J. Phase progression of γ-Al2O3 nanoparticles synthesized in a solvent-deficient environment. Inorg. Chem. 2013, 52, 4411–4423. [Google Scholar] [CrossRef] [PubMed]
- Levin, I.; Brandon, D. Metastable Alumina Polymorphs: Crystal Structures and Transition Sequences. J. Am. Ceram. Soc. 1998, 81, 1995–2012. [Google Scholar] [CrossRef]
- O’Neill, H.S.C.; Dollase, W.A.; Ross, C.R., II. Temperature dependence of the cation distribution in nickel aluminate (NiAl2O4) spinel: A powder XRD study. Phys. Chem. Minerals 1991, 18, 302–319. [Google Scholar] [CrossRef]
- López-Fonseca, R.; Jiménez-González, C.; de Rivas, B.; Gutiérrez-Ortiz, J.I. Partial oxidation of methane to syngas on bulk NiAl2O4 catalyst. Comparison with alumina supported nickel, platinum and rhodium catalysts. Appl. Catal. A 2012, 437–438, 53–62. [Google Scholar] [CrossRef]
- Kim, P.; Kim, Y.; Kim, H.; Song, I.K.; Yi, J. Synthesis and characterization of mesoporous alumina with nickel incorporated for use in the partial oxidation of methane into synthesis gas. Appl. Catal. A 2004, 272, 157–166. [Google Scholar] [CrossRef]
- Scheffer, B.; Heijeinga, J.J.; Moulijn, J.A. An electron spectroscopy and x-ray diffraction study of nickel oxide/alumina and nickel-oxide-tungsten trioxide/alumina catalysts. J. Phys. Chem. 1987, 91, 4752–4759. [Google Scholar] [CrossRef]
- Li, C.; Chen, Y.-W. Temperature-programmed-reduction studies of nickel oxide/alumina catalysts: Effects of the preparation method. Thermochim. Acta 1995, 256, 457–465. [Google Scholar] [CrossRef]
- Xu, X.; Li, L.; Yu, F.; Peng, H.; Fang, X.; Wang, X. Mesoporous high surface area NiO synthesized with soft templates: Remarkable for catalytic CH4 deep oxidation. Mol. Catal. 2017, 441, 81–91. [Google Scholar] [CrossRef]
- Titus, J.; Kullmann, J.; Hill, J.; Enke, D.; Wilde, N.; Gläser, R. Preprints of the DGMK-Conference „Synthesis Gas Chemistry“; Deutsche Wissenschaftliche Gesellschaft für Erdöl, Erdgas und Kohle e.V.: Hamburg, Germany, 2015; p. 215. ISBN 978-3-941721-56-2. [Google Scholar]
- Miao, B.; Ma, S.S.K.; Wang, X.; Su, H.; Chan, S.H. Catalysis mechanisms of CO2 and CO methanation. Catal. Sci. Technol. 2016, 6, 4048–4058. [Google Scholar] [CrossRef]
- Winter, L.R.; Chen, R.; Chen, X.; Chang, K.; Liu, Z.; Senanayake, S.D.; Ebrahim, A.M.; Chen, J.G. Elucidating the roles of metallic Ni and oxygen vacancies in CO2 hydrogenation over Ni/CeO2 using isotope exchange and in situ measurements. Appl. Catal. B 2019, 245, 360–366. [Google Scholar] [CrossRef]
- Martínez, J.; Hernández, E.; Alfaro, S.; López Medina, R.; Valverde Aguilar, G.; Albiter, E.; Valenzuela, M. High Selectivity and Stability of Nickel Catalysts for CO2 Methanation. Catalysts 2019, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Odstrčil, M.; Holler, M.; Raabe, J.; Guizar-Sicairos, M. Alignment methods for nanotomography with deep subpixel accuracy. Opt. Express 2019, 27, 36637–36652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fam, Y.; Sheppard, T.L.; Diaz, A.; Scherer, T.; Holler, M.; Wang, W.; Di Wang Brenner, P.; Wittstock, A.; Grunwaldt, J.-D. Correlative Multiscale 3D Imaging of a Hierarchical Nanoporous Gold Catalyst by Electron, Ion and X-ray Nanotomography. ChemCatChem 2018, 10, 2858–2867. [Google Scholar] [CrossRef] [Green Version]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Liu, J.; Chu, M.; Yue, J.; Cui, Y.; Xu, G. Cooperation Between Active Metal and Basic Support in Ni-Based Catalyst for Low-Temperature CO2 Methanation. Catal. Lett. 2020, 150, 1418–1426. [Google Scholar] [CrossRef]
- Conner, W.C.; Christensen, S.; Topsøe, H.; Ferrero, M.; Pullen, A. The Estimation of Pore-network Dimensions and Structure: Analysis of Sorption and Comparison with Porosimetry. Stud. Surf. Sci. Catal. 1994, 87, 151–163. [Google Scholar] [CrossRef]
- Rijfkogel, L.S.; Ghanbarian, B.; Hu, Q.; Liu, H.-H. Clarifying pore diameter, pore width, and their relationship through pressure measurements: A critical study. Mar. Pet. Geol. 2019, 107, 142–148. [Google Scholar] [CrossRef]
- Diamond, S. Mercury porosimetry: An inappropriate method for the measurement of pore size distributions in cement-based materials. Cem. Concr. Res. 2000, 30, 1517–1525. [Google Scholar] [CrossRef]
- Alford, N.M.; Rahman, A.A. An assesment of porosity and pore sizes in hardened cement pastes. J. Mater. Sci. 1981, 16, 3105–3114. [Google Scholar] [CrossRef]
- Seong, H.; Choi, S.; Matusik, K.E.; Kastengren, A.L.; Powell, C.F. 3D pore analysis of gasoline particulate filters using X-ray tomography: Impact of coating and ash loading. J. Mater. Sci. 2019, 54, 6053–6065. [Google Scholar] [CrossRef]
- Washburn, E.W. The Dynamics of Capillary Flow. Phys. Rev. 1921, 17, 273–283. [Google Scholar] [CrossRef]
- Coelho, A.A.; Bruker, A.X.S. TOPAS and TOPAS-Academic an optimization program integrating computer algebra and crystallographic objects written in C++. J. Appl. Cryst. 2018, 51, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Rietveld, H.M. Line profiles of neutron powder-diffraction peaks for structure refinement. Acta Crystallogr. 1967, 22, 151–152. [Google Scholar] [CrossRef]
- Rietveld, H.M. A profile refinement method for nuclear and magnetic structures. J. Appl. Cryst. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption in Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of Pore Volume and Area Distributions in Porous Substances. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Kremer, J.R.; Mastronarde, D.N.; McIntosh, J.R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996, 116, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, P. Iterative Methods fo the Three-dimensional Reconstruction of an Object from Projections. J. Theor. Biol. 1972, 36, 105–107. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batenburg, K.J.; Bals, S.; Sijbers, J.; Kübel, C.; Midgley, P.A.; Hernandez, J.C.; Kaiser, U.; Encina, E.R.; Coronado, E.A.; van Tendeloo, G. 3D imaging of nanomaterials by discrete tomography. Ultramicroscopy 2009, 109, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Messaoudii, C.; Boudier, T.; Sanchez Sorzano, C.O.; Marco, S. TomoJ: Tomography software for three-dimensional reconstruction in transmission electron microscopy. BMC Bioinform. 2007, 8, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holler, M.; Raabe, J.; Wepf, R.; Shahmoradian, S.H.; Diaz, A.; Sarafimov, B.; Lachat, T.; Walther, H.; Vitins, M. OMNY PIN-A versatile sample holder for tomographic measurements at room and cryogenic temperatures. Rev. Sci. Instrum. 2017, 88, 113701. [Google Scholar] [CrossRef] [PubMed]
- Rau, C.; Wagner, U.; Pešić, Z. The Imaging and Coherence Beamline I13 at Diamond. AIP Conf. Proc. 2011, 132, 132–135. [Google Scholar] [CrossRef]
- Batey, D.; Cipiccia, S.; Shi, X.; Williams, S.; Wanelik, K.; Wilson, A.; Pérez-Tamarit, S.; Cimavilla, P.; Ródriguez-Pérez, M.A.; Rau, C. Coherence Branch at I13, DLS: The Multiscale, Multimodal, Ptycho-tomographic End Station. Microsc. Microanal. 2018, 24, 42–43. [Google Scholar] [CrossRef] [Green Version]
- Batey, D. Ptychographic Imaging of Mixed States. Ph.D. Thesis, University of Sheffield, Sheffield, UK, 2014. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tomogram | ET1 | ET2 | PXCT |
|---|---|---|---|
| Vs,tot/µm3 | 28,898.3 × 10−6 | 22,374.8 × 10−6 | 1952.39 |
| Vp,tot/µm3 | 25,402.1 × 10−6 | 16,795.3 × 10−6 | 1684.09 |
| εtomo/% | 46.8 | 42.9 | 46.3 |
| deq,w/nm | 37.4 ± 1.2 | 29.2 ± 1.3 | 2545 ± 23 |
| deq,f/nm | 36.2 | 27.9 | 2522 |
| Ni/Al2O3-h | Ni/Al2O3-m | |
|---|---|---|
| N2 sorption | ||
| ABET/m2 g−1 | 99 | 108 |
| VP/cm3 g−1 | 0.31 | 0.37 |
| wP/nm | 8.3 | 10.3 |
| Hg porosimetry | ||
| VP,meso/cm3 g−1 | 0.26 | 0.33 |
| VP,tot/cm3 g−1 | 0.88 | 0.35 |
| wP,meso/nm | 10.3 | 11.2 |
| wP,macro/nm | 1012 | - |
| εm/% | 23.0 | 55.8 |
| εM/% | 55.0 | 3.4 |
| He pycnometry | ||
| ρ/g cm−3 | 4.028 | 4.144 |
| εtot/% | 78.0 | 59.1 |
| Tomography | ||
| deq,meso/nm | 33.3 ± 4.1 | - |
| deq,macro/nm | 2545 ± 23 | - |
| εm/% | 24.1 | - |
| εM/% | 46.3 | - |
| εtot/% | 70.4 | - |
| Sample | Data Set | |||||
|---|---|---|---|---|---|---|
| Ni/Al2O3-h | Tomography | 46.3 | 2545 | 24.1 | 33.3 | 5.89∙10−6 |
| Ni/Al2O3-h | Hg porosimetry | 55.0 | 1012 | 23.0 | 10.30 | 6.67∙10−6 |
| Ni/Al2O3-m | Hg porosimetry | n.a. a | n.a. | 55.8 | 11.2 | 5.74∙10−7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weber, S.; Abel, K.L.; Zimmermann, R.T.; Huang, X.; Bremer, J.; Rihko-Struckmann, L.K.; Batey, D.; Cipiccia, S.; Titus, J.; Poppitz, D.; et al. Porosity and Structure of Hierarchically Porous Ni/Al2O3 Catalysts for CO2 Methanation. Catalysts 2020, 10, 1471. https://doi.org/10.3390/catal10121471
Weber S, Abel KL, Zimmermann RT, Huang X, Bremer J, Rihko-Struckmann LK, Batey D, Cipiccia S, Titus J, Poppitz D, et al. Porosity and Structure of Hierarchically Porous Ni/Al2O3 Catalysts for CO2 Methanation. Catalysts. 2020; 10(12):1471. https://doi.org/10.3390/catal10121471
Chicago/Turabian StyleWeber, Sebastian, Ken L. Abel, Ronny T. Zimmermann, Xiaohui Huang, Jens Bremer, Liisa K. Rihko-Struckmann, Darren Batey, Silvia Cipiccia, Juliane Titus, David Poppitz, and et al. 2020. "Porosity and Structure of Hierarchically Porous Ni/Al2O3 Catalysts for CO2 Methanation" Catalysts 10, no. 12: 1471. https://doi.org/10.3390/catal10121471