Construction of Synthetic Models for Nitrogenase-Relevant NifB Biogenesis Intermediates and Iron-Carbide-Sulfide Clusters



Abstract
:1. Introduction
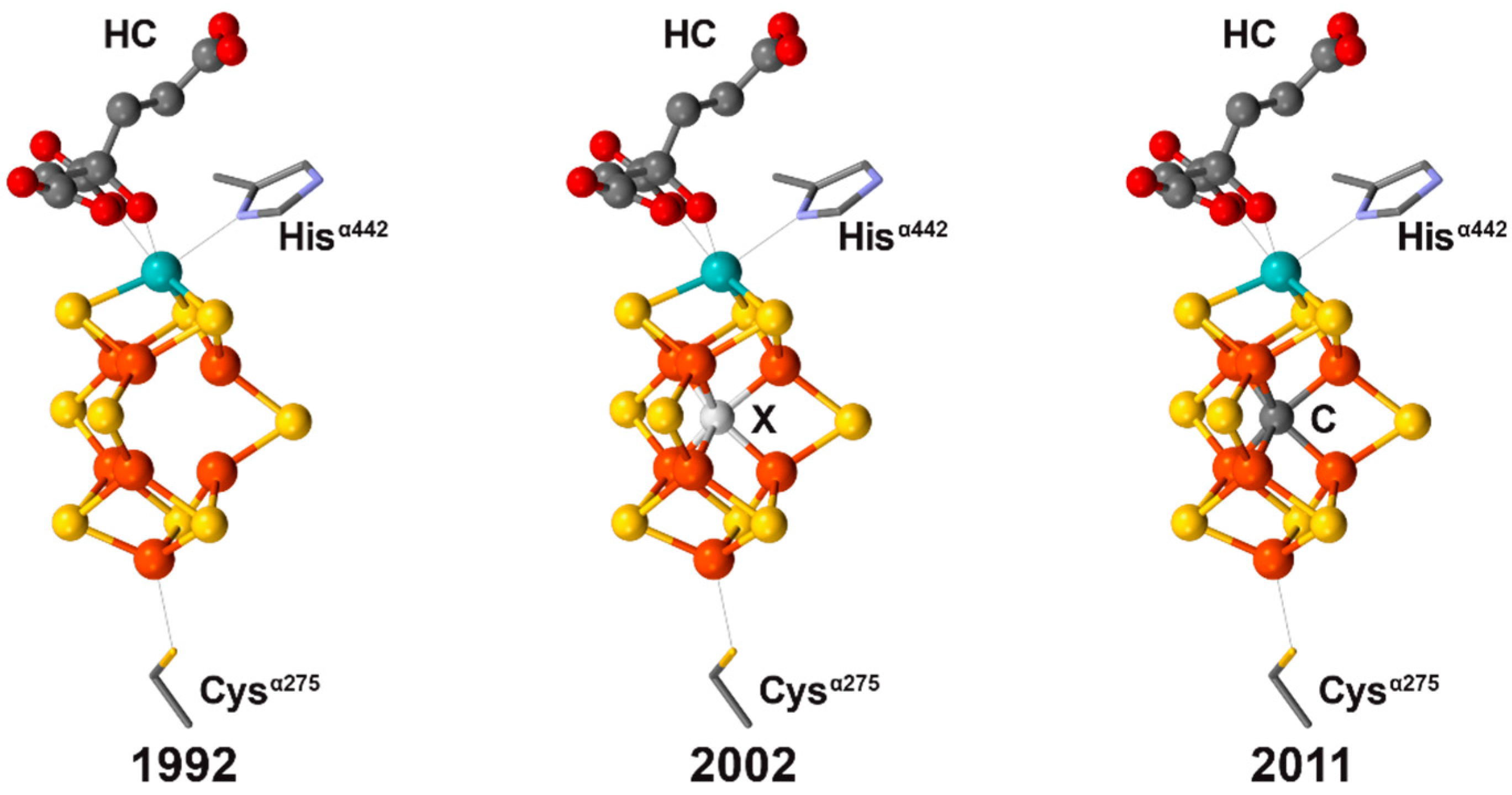
1.1. Structural Determination of the Resting Nitrogenase Active Site
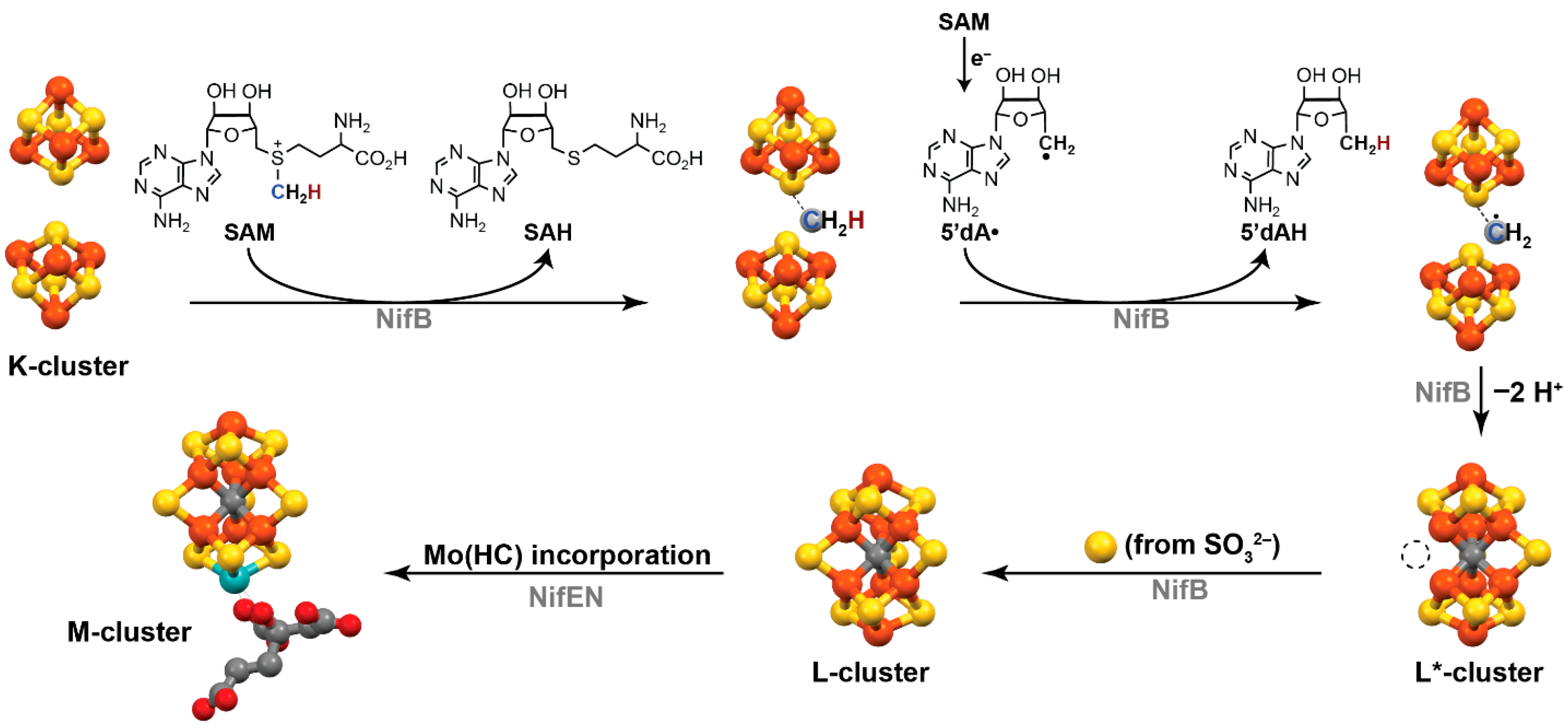
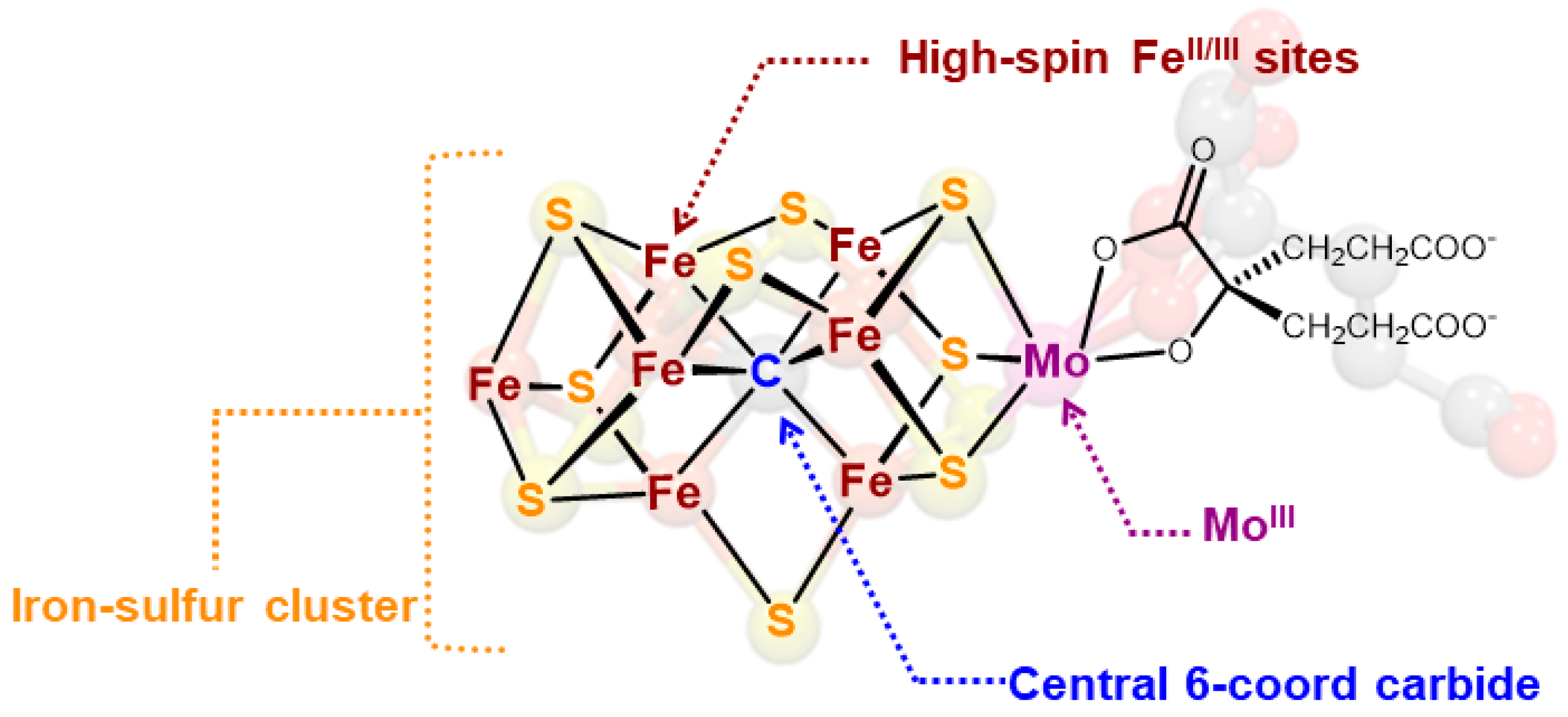
1.2. Biological Synthesis of the FeMoco Cluster
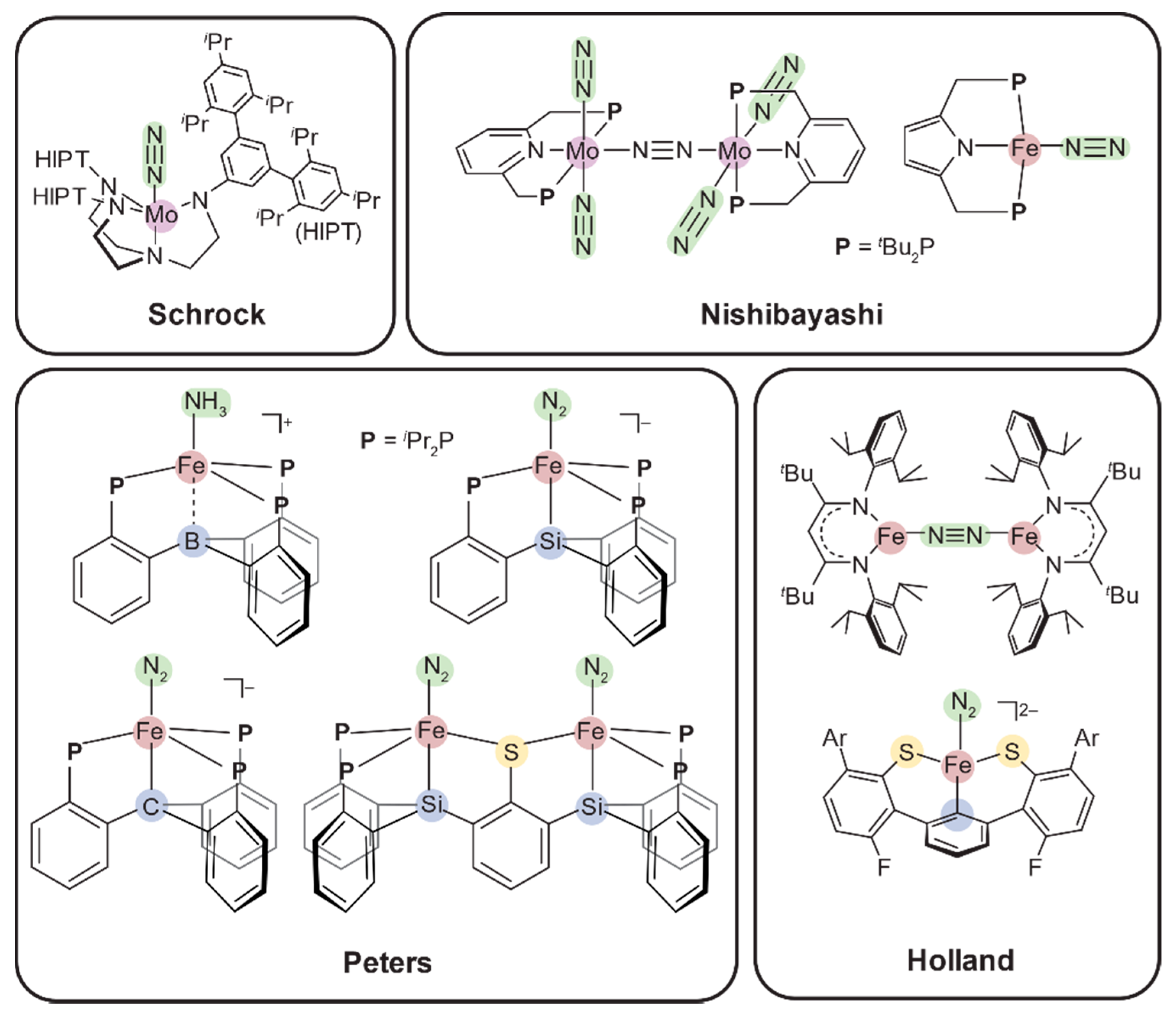
1.3. Multimetallic Co-Operativity toward Dinitrogen Activation
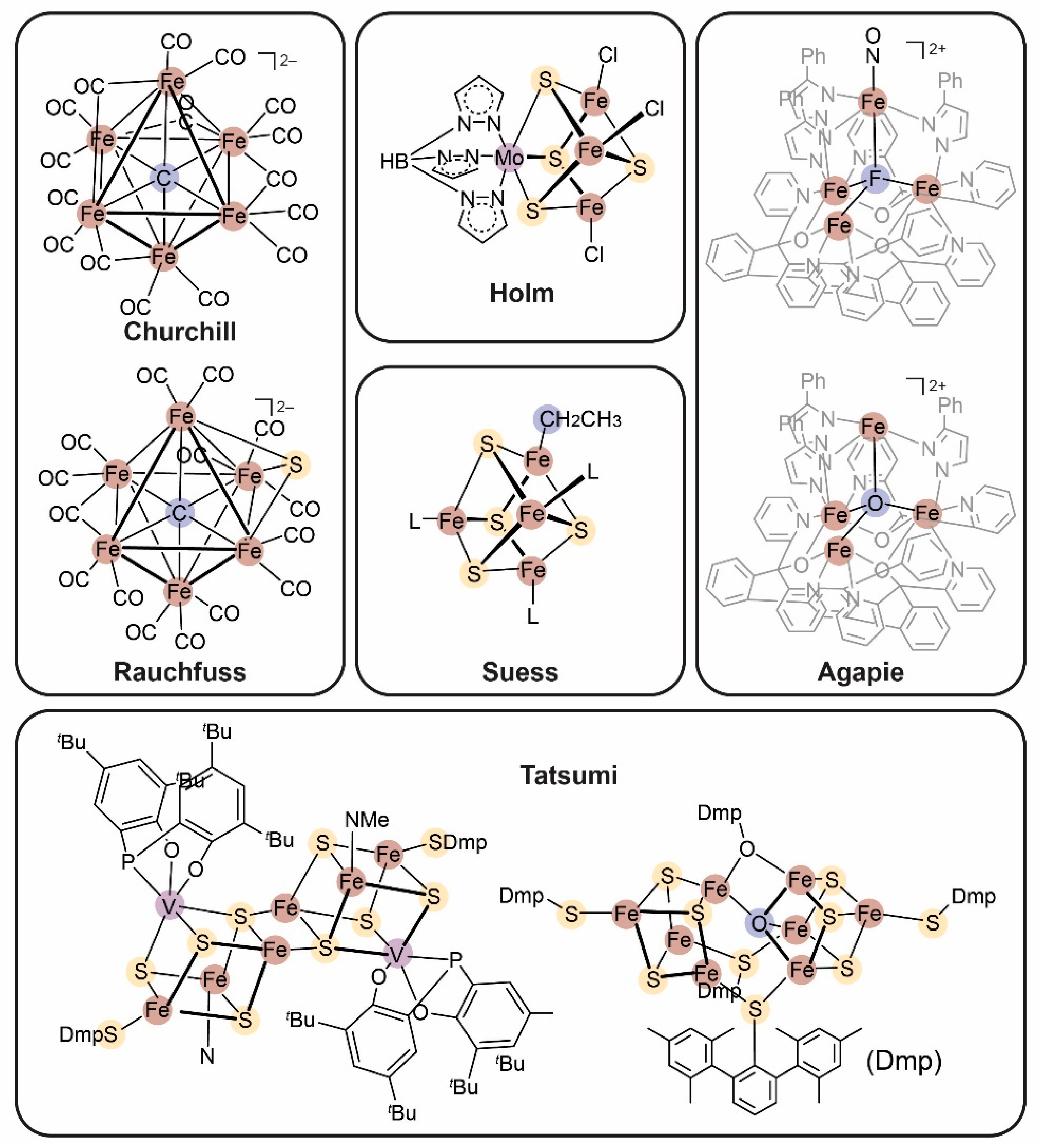
1.4. Synthetic Model Compounds in FeMoco Biomimetics
1.5. A Two-Fold Strategy toward Achieving FeMoco Construction
2. Synthetic Manipulations of CO-Supported FeS and FeC Clusters
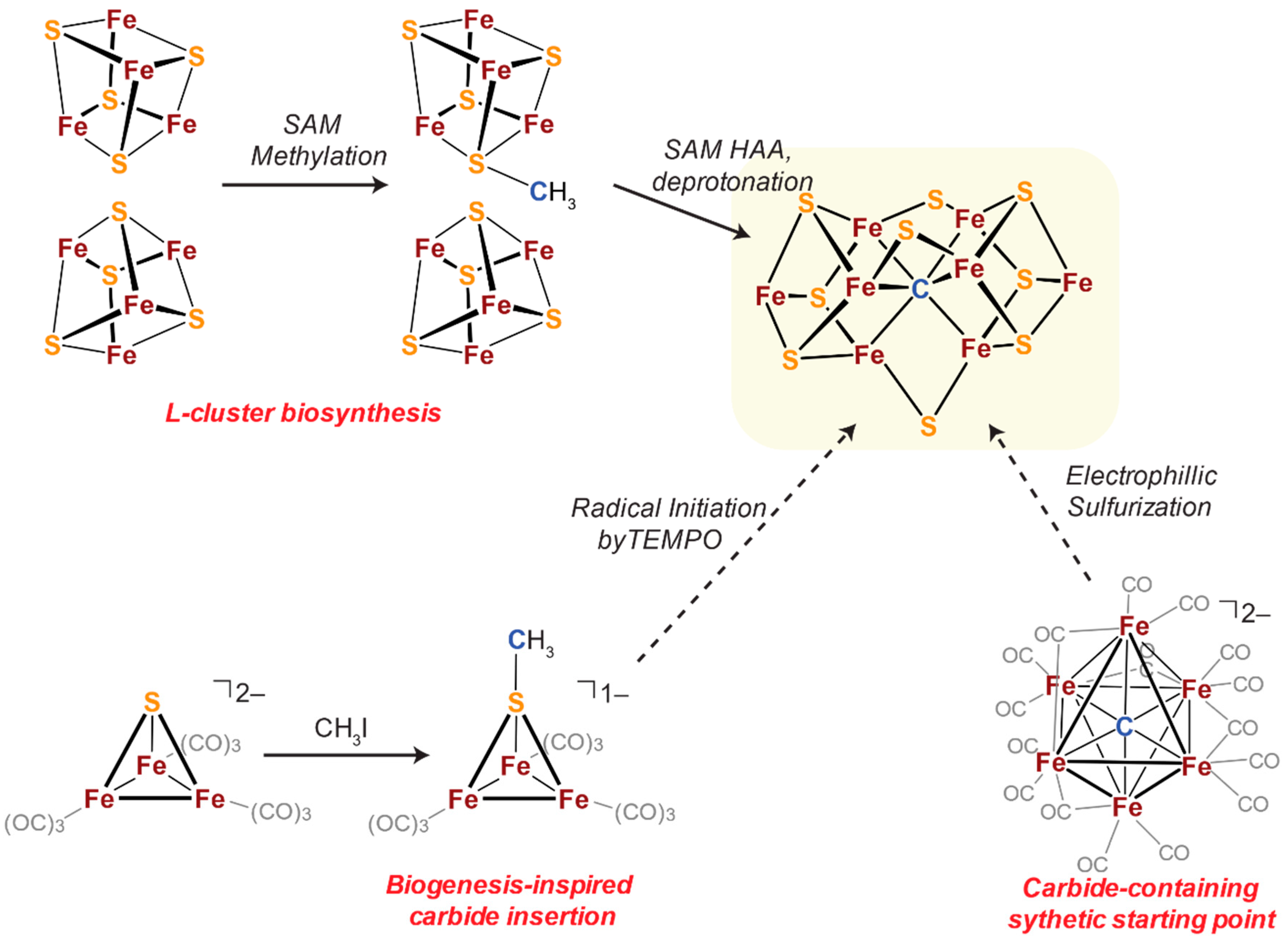
2.1. Biogenesis-Inspired Approach to FeSC Cluster Construction
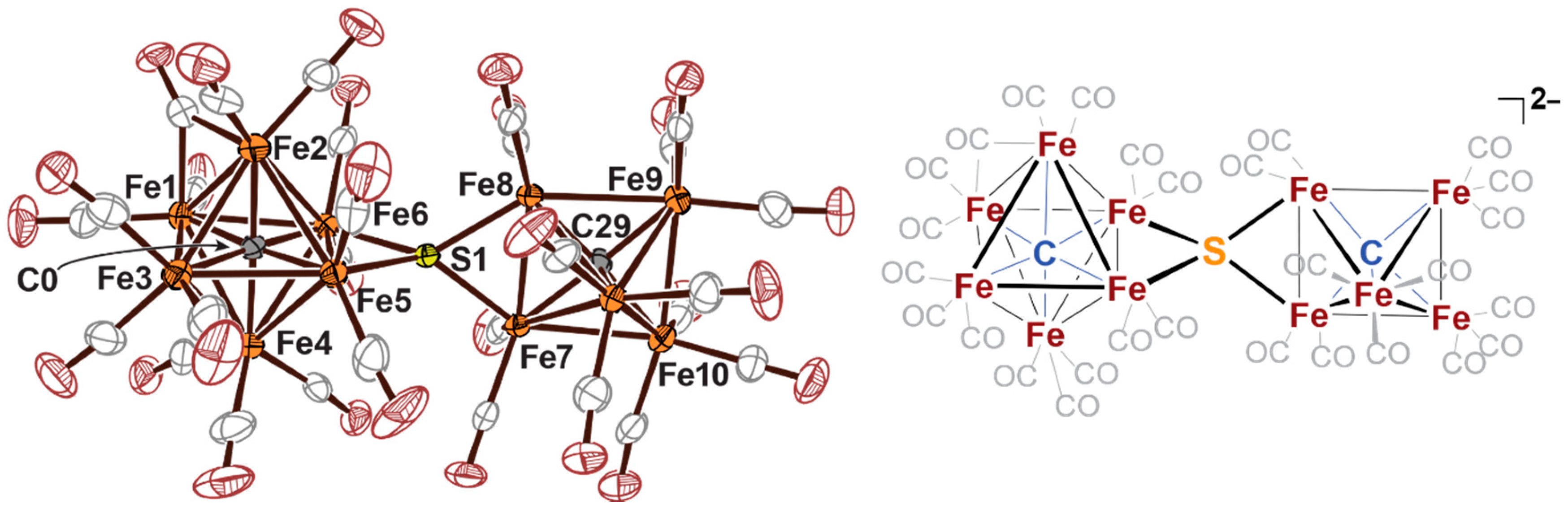
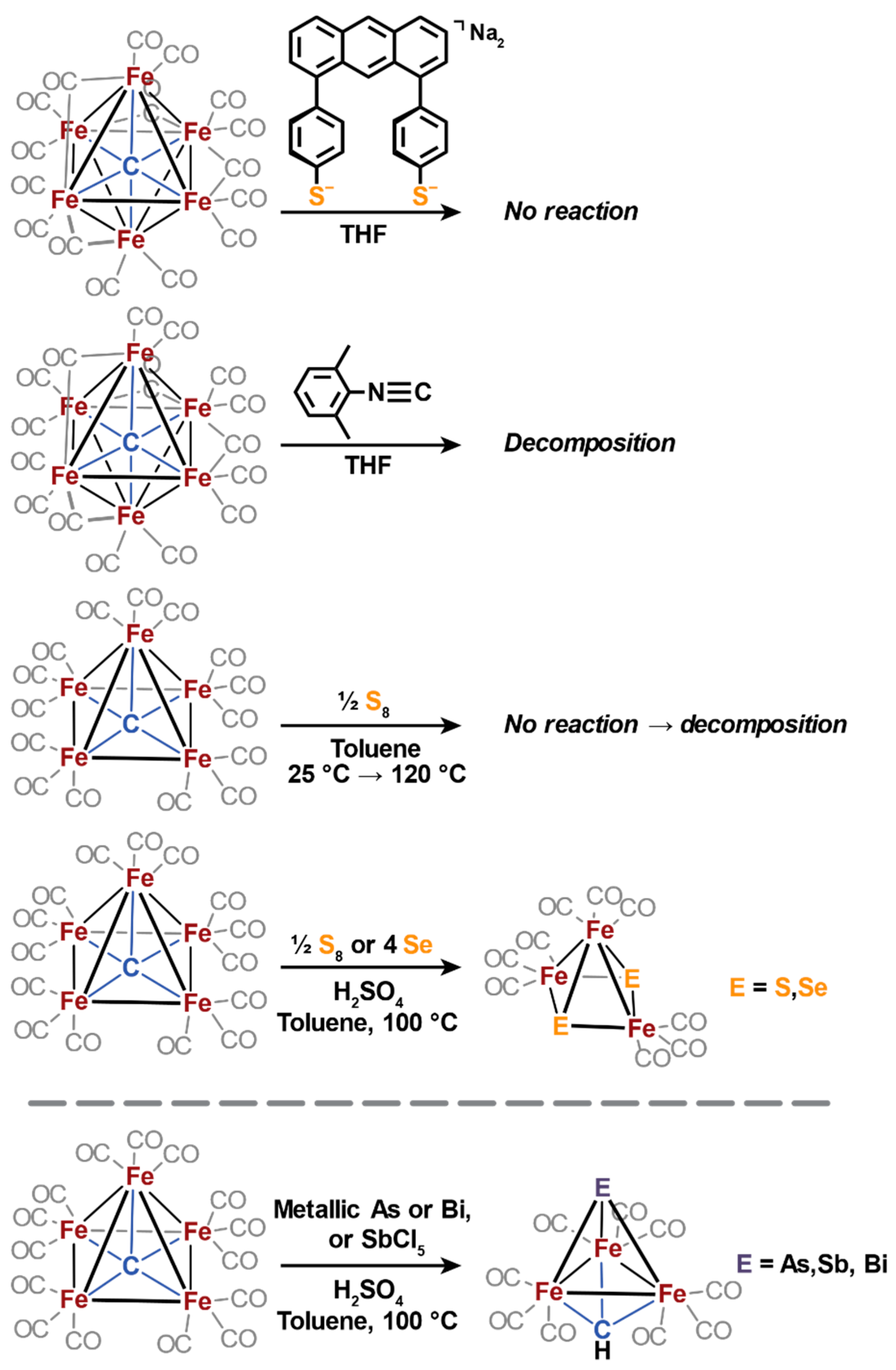
2.2. Sulfide Incorporation into Iron-Carbide Clusters: Initial Attempts
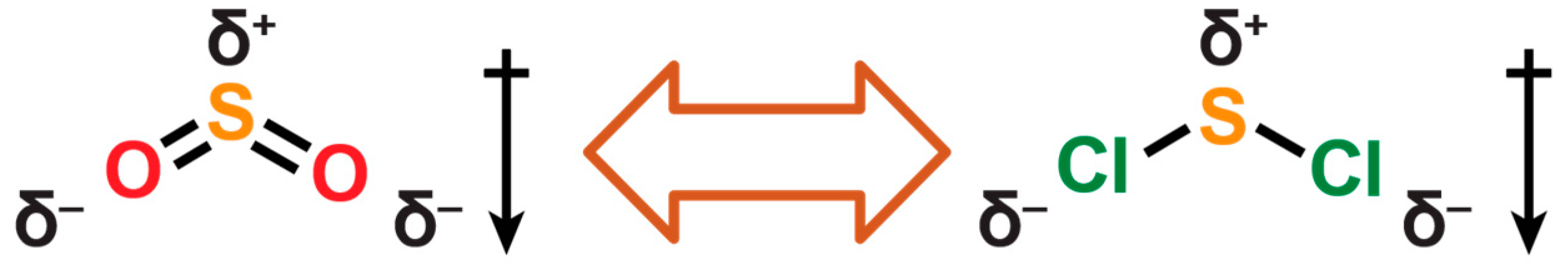
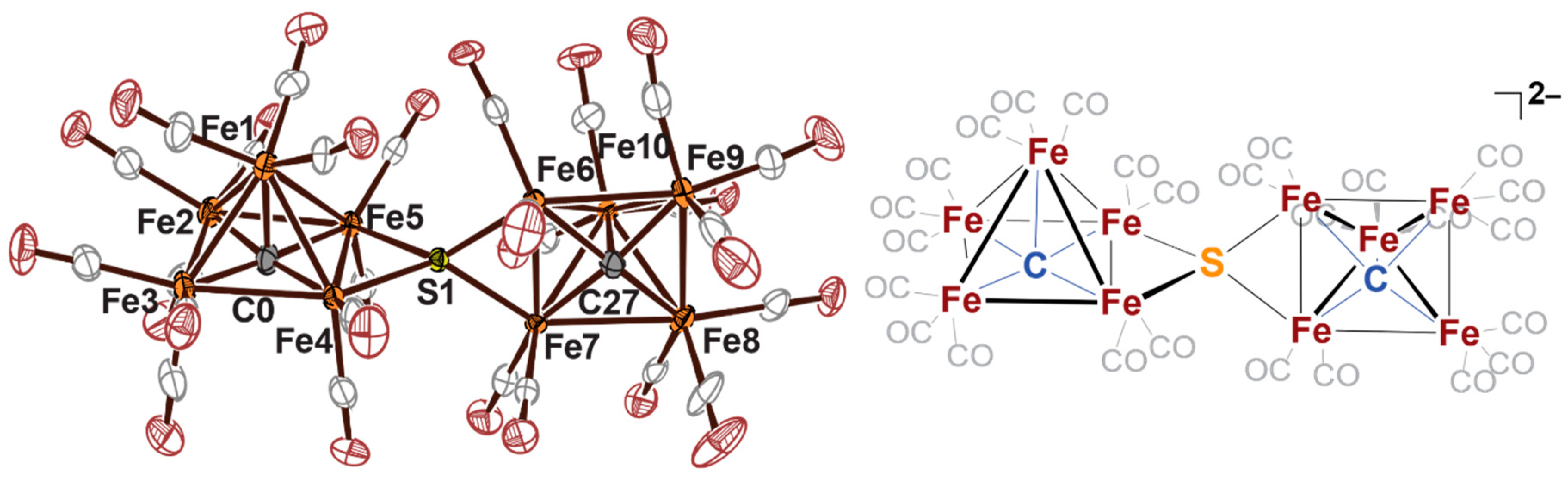
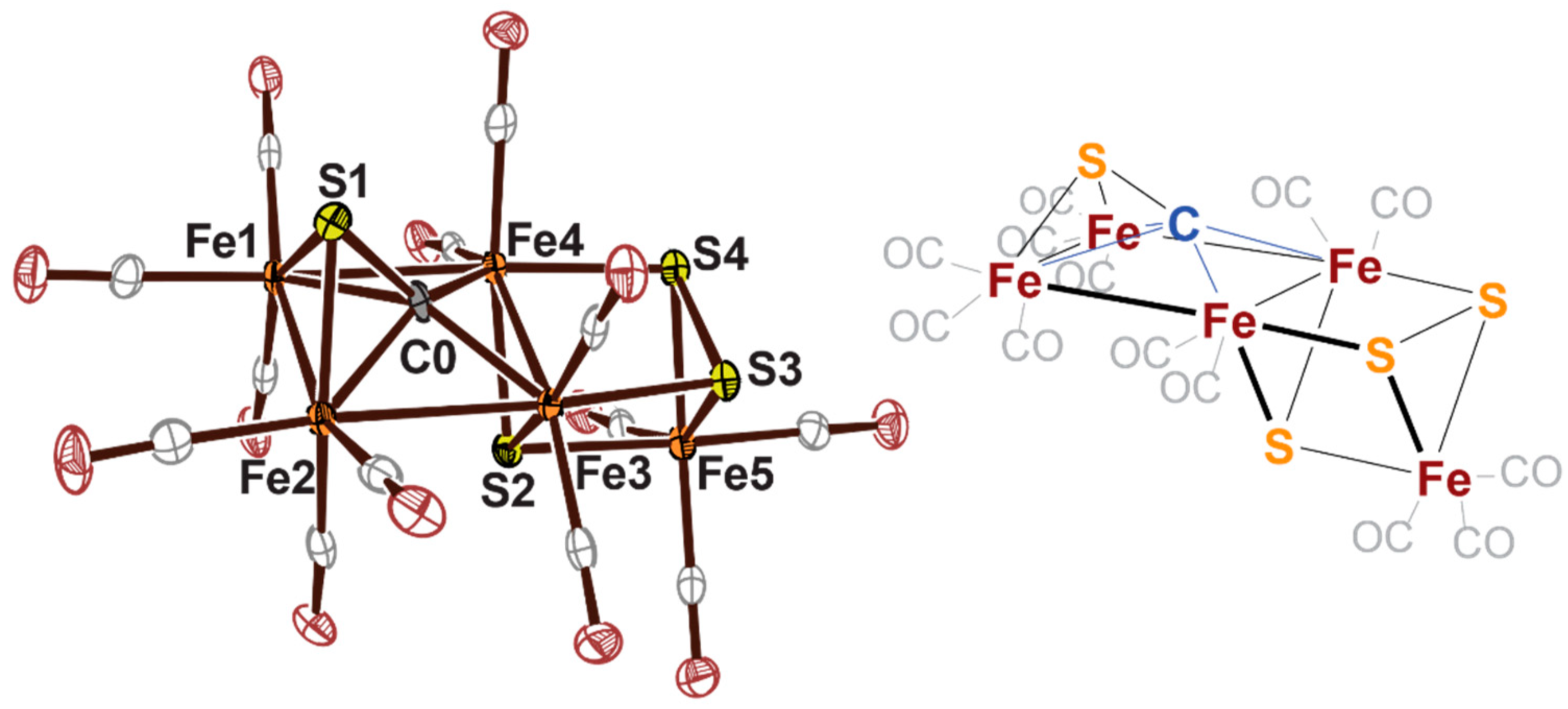
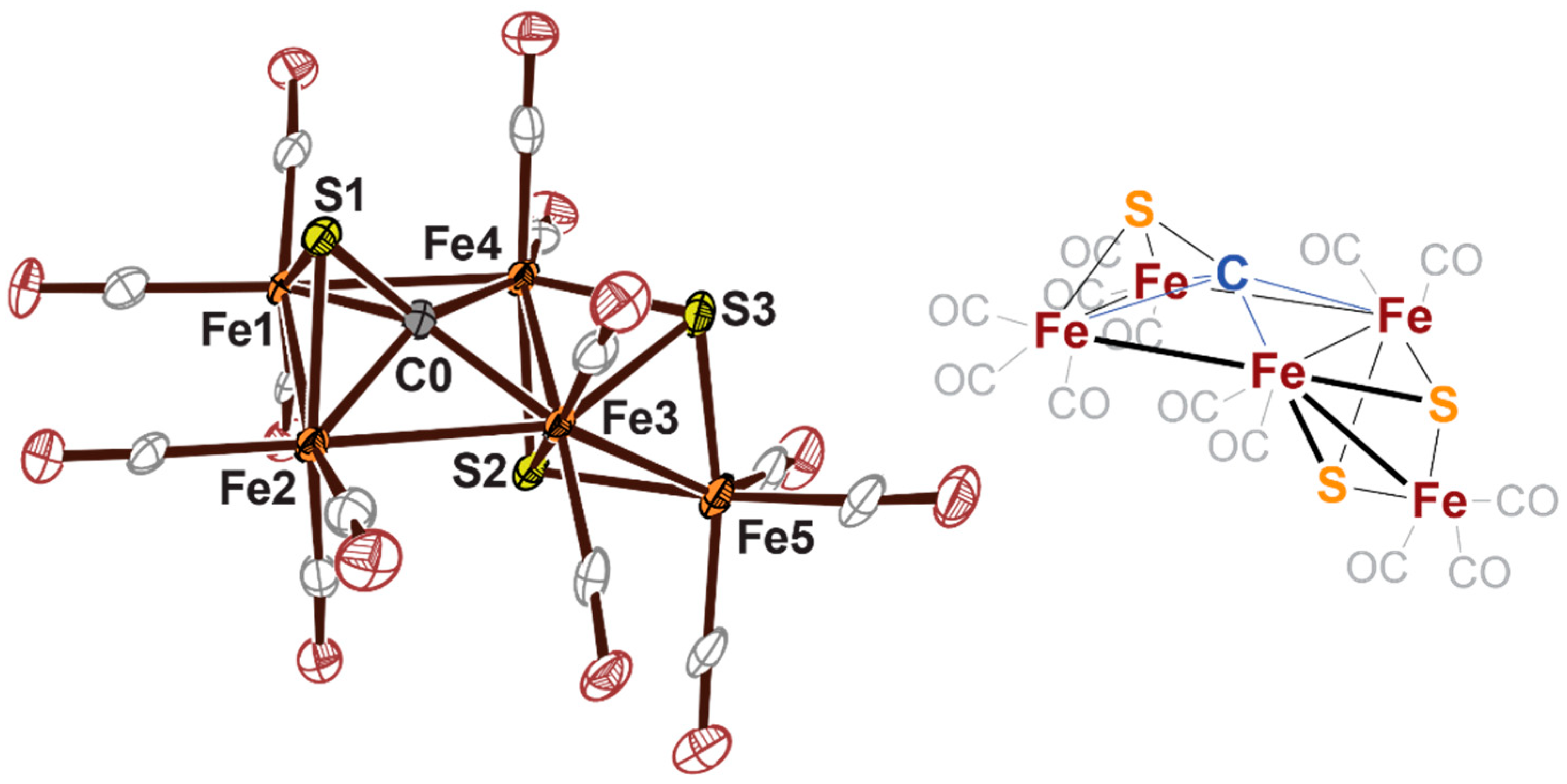
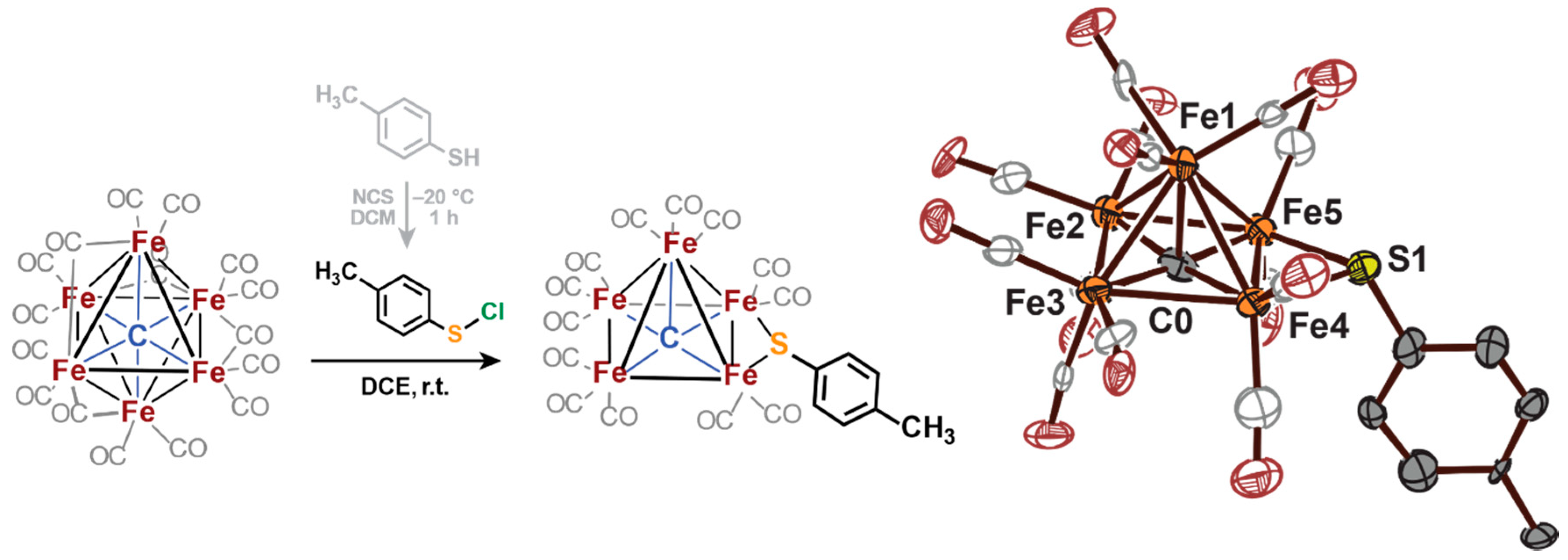
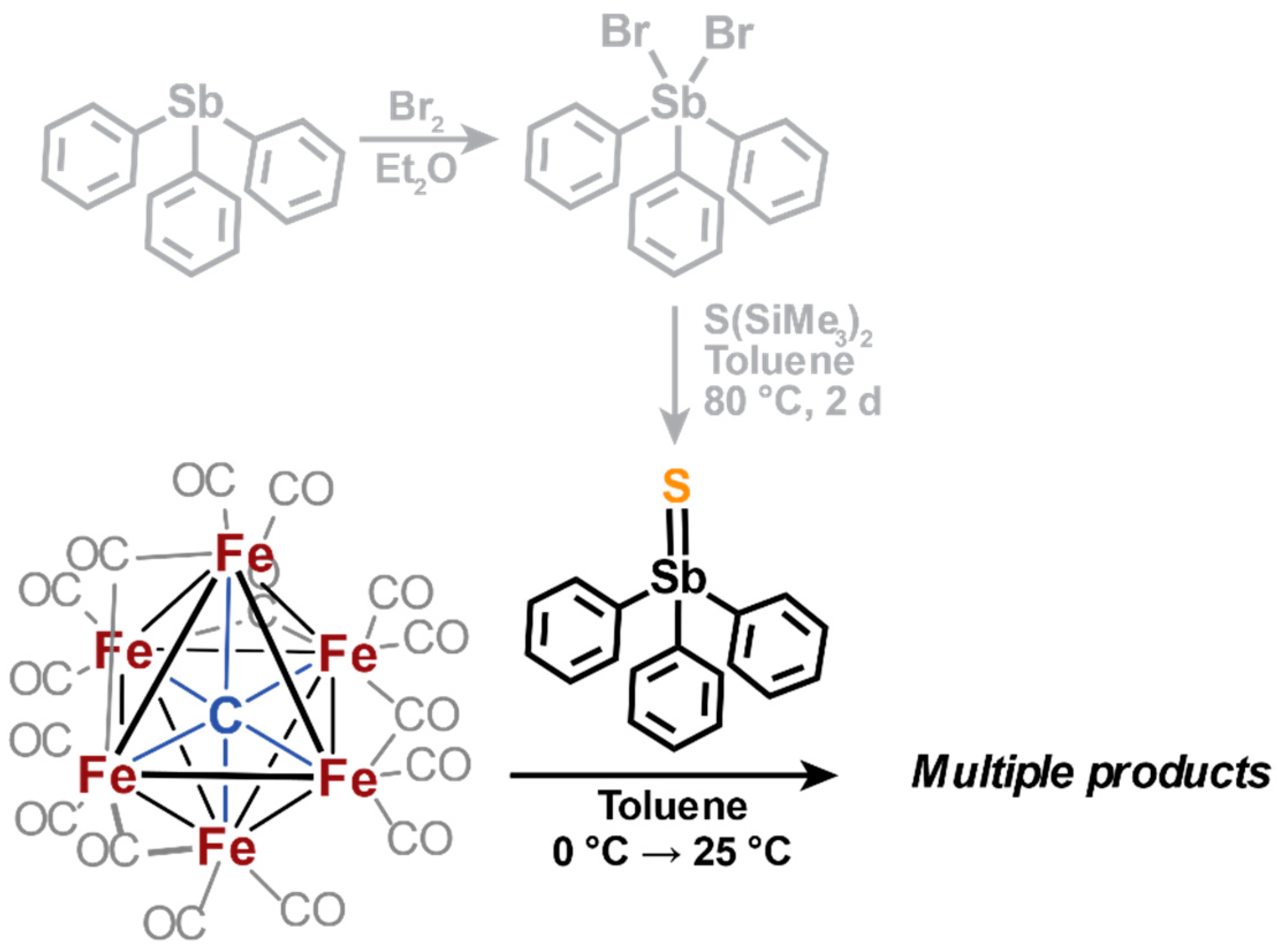
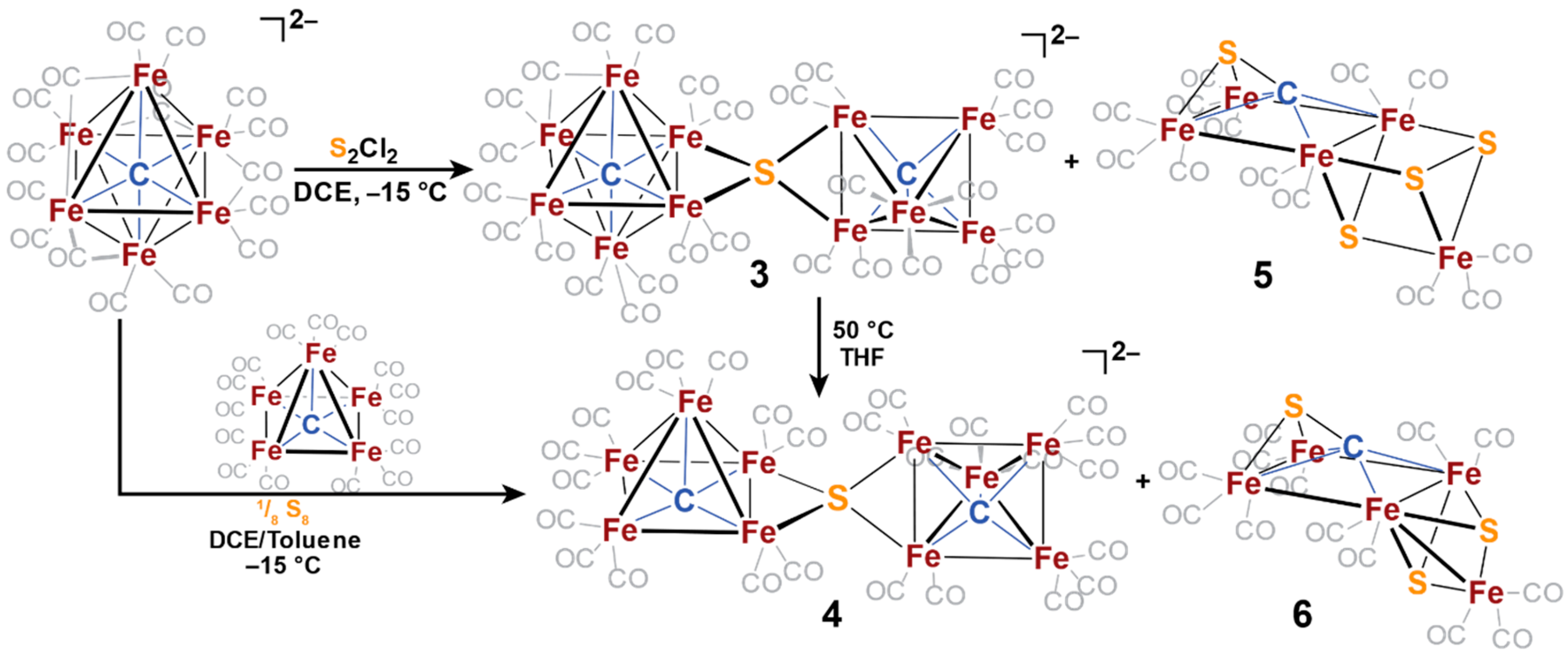
2.3. Electrophilic Sulfur Sources: Isolation of Sulfide-Containing Carbidocabonyl Iron Clusters
3. Extension of Electrophilic Sulfur Ligation toward Fine-Tuning of Cluster Thiolates
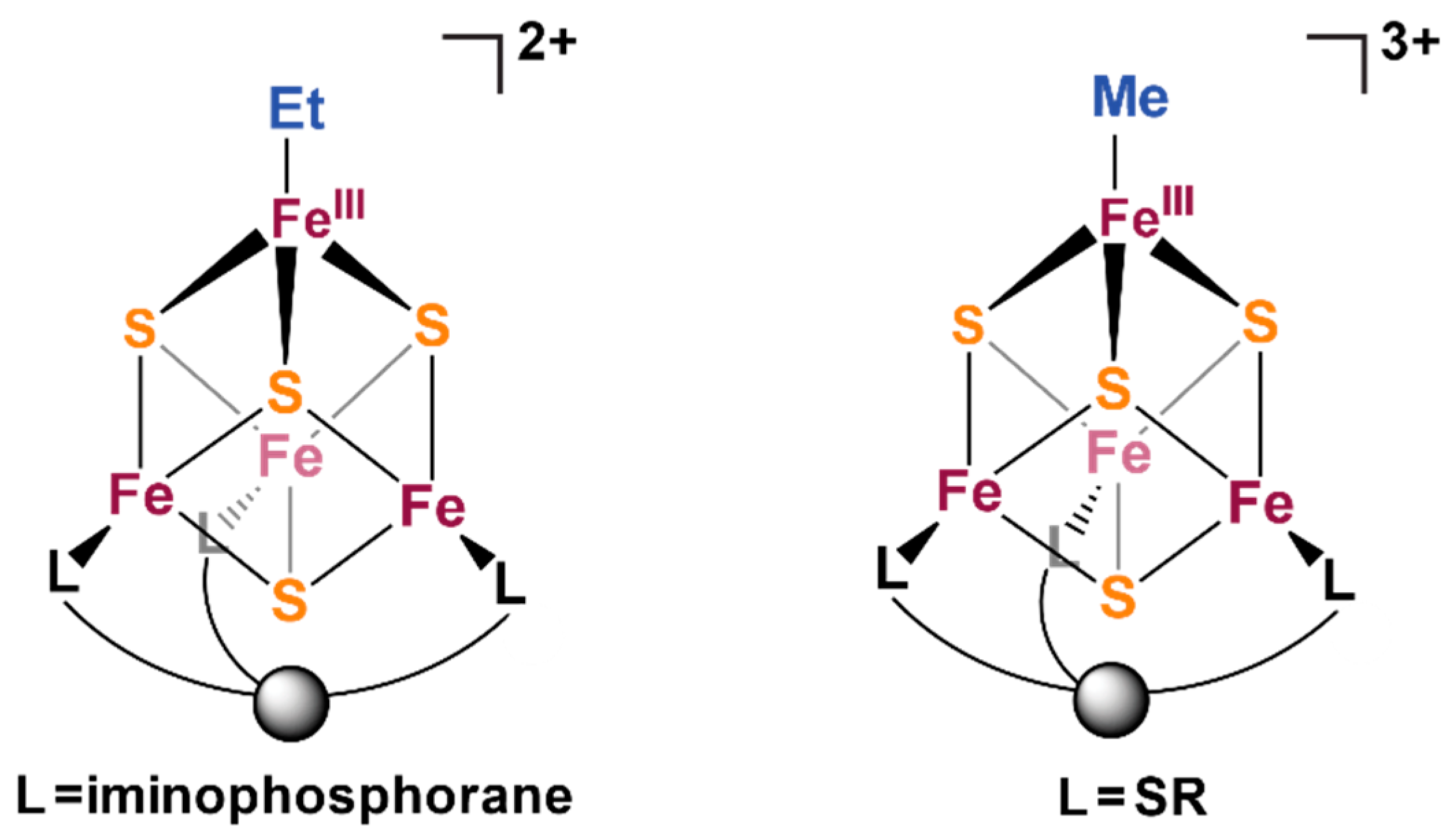
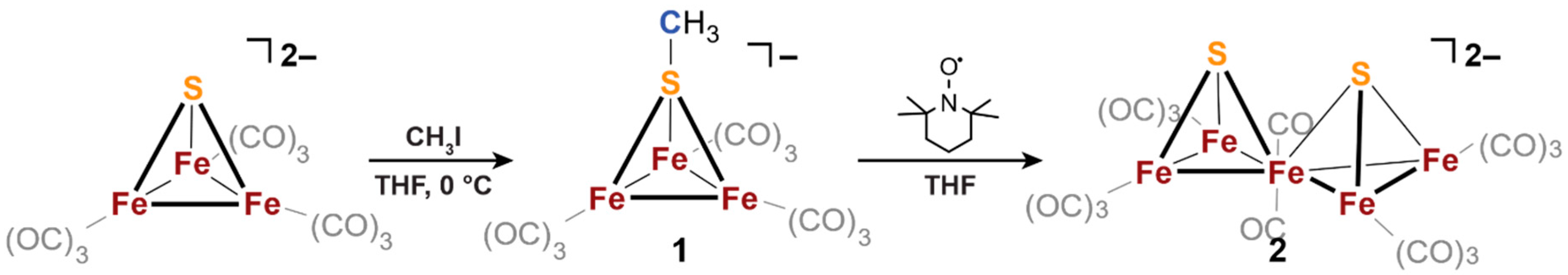
3.1. Electronic Effects of Fe-Alkyl Bonding in Iron-Sulfur Cubane Clusters
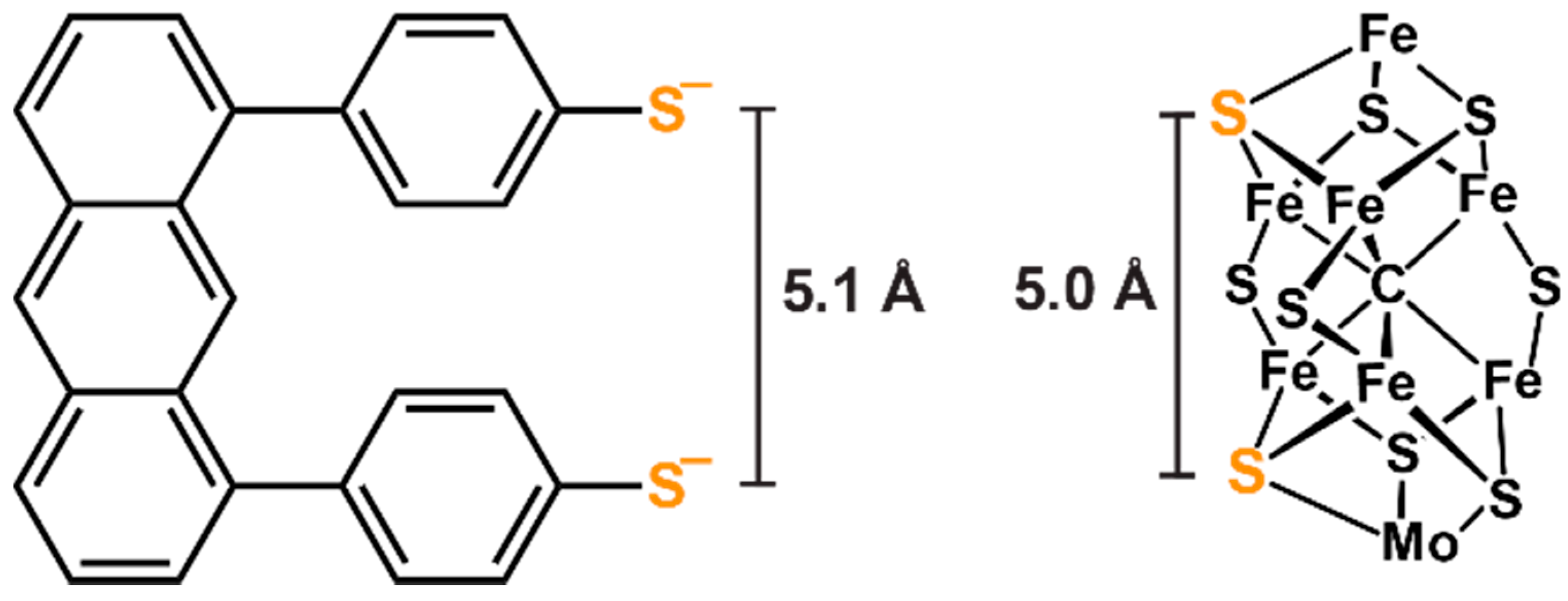
3.2. New Routes toward Carbide-Containing Iron-Thiolato Clusters
4. Closing Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Hoffman, B.M.; Lukoyanov, D.; Yang, Z.-Y.; Dean, D.R.; Seefeldt, L.C. Mechanism of nitrogen fixation by nitrogenase: The next stage. Chem. Rev. 2014, 114, 4041. [Google Scholar] [CrossRef] [PubMed]
- Eady, R.R. Structure−function relationships of alternative nitrogenases. Chem. Rev. 1996, 96, 3013. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, M.M.; Komiya, H.; Chakrabarti, P.; Woo, D.; Kornuc, J.J.; Rees, D.C. Crystallographic structure of the nitrogenase iron protein from Azotobacter vinelandii. Science 1992, 257, 1653. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Rees, D.C. Structural models for the metal centers in the nitrogenase molybdenum-iron protein. Science 1992, 257, 1677. [Google Scholar] [CrossRef]
- Einsle, O.; Tezcan, F.A.; Andrade, S.L.A.; Schmid, B.; Yoshida, M.; Howard, J.B.; Rees, D.C. Nitrogenase MoFe-protein at 1.16 Å resolution: A central ligand in the FeMo-cofactor. Science 2002, 297, 1696. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.E. Nitrogenase reveals its inner secrets. Science 2002, 297, 1654. [Google Scholar] [CrossRef]
- Spatzal, T.; Aksoyoglu, M.; Zhang, L.; Andrade, S.L.A.; Schleicher, E.; Weber, S.; Rees, D.C.; Einsle, O. Evidence for interstitial carbon in nitrogenase FeMo cofactor. Science 2011, 334, 940. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, K.M.; Roemelt, M.; Ettenhuber, P.; Hu, Y.; Ribbe, M.W.; Neese, F.; Bergmann, U.; DeBeer, S. X-ray emission spectroscopy evidences a central carbon in the nitrogenase iron-molybdenum cofactor. Science 2011, 334, 974. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Jaime, M.U.; Dible, B.R.; Chiang, K.P.; Brennessel, W.W.; Bergmann, U.; Holland, P.L.; DeBeer, S. Identification of a single light atom within a multinuclear metal cluster using valence-to-core X-ray emission spectroscopy. Inorg. Chem. 2011, 50, 10709. [Google Scholar] [CrossRef] [Green Version]
- Frey, P.A.; Hegeman, A.D.; Ruzicka, F.J. The radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 63. [Google Scholar] [CrossRef]
- Wiig, J.A.; Hu, Y.; Lee, C.C.; Ribbe, M.W. Radical SAM-dependent carbon insertion into the nitrogenase M-cluster. Science 2012, 337, 1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiig, J.A.; Hu, Y.; Ribbe, M.W. Refining the pathway of carbide insertion into the nitrogenase M-cluster. Nat. Commun. 2015, 6, 8034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rettberg, L.A.; Wilcoxen, J.; Lee, C.C.; Stiebritz, M.T.; Tanifuji, K.; Britt, R.D.; Hu, Y. Probing the Coordination and Function of Fe4S4 Modules in Nitrogenase Assembly Protein NifB. Nat. Commun. 2018, 9, 2824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierre, S.; Guillot, A.; Benjdia, A.; Sandström, C.; Langella, P.; Berteau, O. Thiostrepton Tryptophan Methyltransferase Expands the Chemistry of Radical SAM Enzymes. Nat. Chem. Biol. 2012, 8, 957. [Google Scholar] [CrossRef]
- Marous, D.R.; Lloyd, E.P.; Buller, A.R.; Moshos, K.A.; Grove, T.L.; Blaszczyk, A.J.; Booker, S.J.; Townsend, C.A. Consecutive Radical S -Adenosylmethionine Methylations Form the Ethyl Side Chain in Thienamycin Biosynthesis. Proc. Natl. Acad. Sci. USA 2015, 112, 10354. [Google Scholar] [CrossRef] [Green Version]
- Sickerman, N.S.; Ribbe, M.W.; Hu, Y. Nitrogenase cofactor assembly: An elemental inventory. Acc. Chem. Res. 2017, 50, 2834. [Google Scholar] [CrossRef]
- Jasniewski, A.J.; Wilcoxen, J.; Tanifuji, K.; Hedman, B.; Hodgson, K.O.; Britt, R.D.; Hu, Y.; Ribbe, M.W. Spectroscopic characterization of an eight-iron nitrogenase cofactor precursor that lacks the “9th sulfur”. Angew. Chem. Int. Ed. 2019, 58, 14703. [Google Scholar] [CrossRef]
- Tanifuji, K.; Lee, C.C.; Sickerman, N.S.S.; Tatsumi, K.; Ohki, Y.; Hu, Y.; Ribbe, M.W. Tracing the ‘ninth sulfur’ of the nitrogenase cofactor via a semi-synthetic approach. Nat. Chem. 2018, 10, 568. [Google Scholar] [CrossRef]
- Van Stappen, C.; Thorhallsson, A.T.; Decamps, L.; Bjornsson, R.; DeBeer, S. Resolving the structure of the E1 state of Mo nitrogenase through Mo and Fe K-edge EXAFS and QM/MM calculations. Chem. Sci. 2019, 10, 9807. [Google Scholar] [CrossRef] [Green Version]
- Van Stappen, C.; Davydov, R.; Yang, Z.-Y.; Fan, R.; Guo, Y.; Bill, E.; Seefeldt, L.C.; Hoffman, B.M.; DeBeer, S. Spectroscopic description of the E1 state of Mo nitrogenase based on Mo and Fe X-ray absorption and Mössbauer studies. Inorg. Chem. 2019, 58, 12365. [Google Scholar] [CrossRef] [Green Version]
- Yandulov, D.V.; Schrock, R.R. Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Science 2003, 301, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.S.; Rittle, J.; Peters, J.C. Catalytic conversion of nitrogen to ammonia by an iron model complex. Nature 2013, 501, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spatzal, T.; Perez, K.A.; Einsle, O.; Howard, J.B.; Rees, D.C. Ligand binding to the FeMo-cofactor: Structures of CO-bound and reactivated nitrogenase. Science 2014, 345, 1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spatzal, T.; Perez, K.A.; Howard, J.B.; Rees, D.C. Catalysis-dependent selenium incorporation and migration in the nitrogenase active site iron-molybdenum cofactor. eLife 2015, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Sippel, D.; Rohde, M.; Netzer, J.; Trncik, C.; Gies, J.; Grunau, K.; Djurdjevic, I.; Decamps, L.; Andrade, S.L.A.; Einsle, O. A bound reaction intermediate sheds light on the mechanism of nitrogenase. Science 2018, 359, 1484. [Google Scholar] [CrossRef] [Green Version]
- Siegbahn, P.E.M. A major structural change of the homocitrate ligand of probable importance for the nitrogenase mechanism. Inorg. Chem. 2018, 57, 1090. [Google Scholar] [CrossRef]
- Kang, W.; Lee, C.C.; Jasniewski, A.J.; Ribbe, M.W.; Hu, Y. Structural evidence for a dynamic metallocofactor during N2 reduction by Mo-nitrogenase. Science 2020, 368, 1381. [Google Scholar] [CrossRef]
- Arashiba, K.; Miyake, Y.; Nishibayashi, Y.A. Molybdenum complex bearing PNP-type pincer ligands leads to the catalytic reduction of dinitrogen into ammonia. Nat. Chem. 2011, 3, 120. [Google Scholar] [CrossRef]
- Nishibayashi, Y. Recent progress in transition-metal-catalyzed reduction of molecular dinitrogen under ambient reaction conditions. Inorg. Chem. 2015, 54, 9234. [Google Scholar] [CrossRef]
- Kuriyama, S.; Arashiba, K.; Nakajima, K.; Matsuo, Y.; Tanaka, H.; Ishii, K.; Yoshizawa, K.; Nishibayashi, Y. Catalytic transformation of dinitrogen into ammonia and hydrazine by iron-dinitrogen complexes bearing pincer ligand. Nat. Commun. 2016, 7, 12181. [Google Scholar] [CrossRef]
- Lee, Y.; Mankad, N.P.; Peters, J.C. Triggering N2 uptake via redox-induced expulsion of coordinated NH3 and N2 silylation at trigonal bipyramidal iron. Nat. Chem. 2010, 2, 558. [Google Scholar] [CrossRef] [PubMed]
- Creutz, S.E.; Peters, J.C. Catalytic reduction of N2 to NH3 by an Fe–N2 complex featuring a C-atom anchor. J. Am. Chem. Soc. 2014, 136, 1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rittle, J.; Peters, J.C. An Fe-N2 complex that generates hydrazine and ammonia via Fe=NNH2: Demonstrating a hybrid distal-to-alternating pathway for N2 reduction. J. Am. Chem. Soc. 2016, 138, 4243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creutz, S.E.; Peters, J.C. Diiron bridged-thiolate complexes that bind N2 at the FeIIFeII, FeIIFeI, and FeIFeI redox states. J. Am. Chem. Soc. 2015, 137, 7310. [Google Scholar] [CrossRef] [Green Version]
- Čorić, I.; Mercado, B.Q.; Bill, E.; Vinyard, D.J.; Holland, P.L. Binding of dinitrogen to an iron–sulfur–carbon site. Nature 2015, 526, 96. [Google Scholar] [CrossRef] [Green Version]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. 2016, B72, 171. [Google Scholar] [CrossRef]
- Kovacs, J.A.; Holm, R.H. Assembly of vanadium-iron-sulfur cubane clusters from mononuclear and linear trinuclear reactants. J. Am. Chem. Soc. 1986, 108, 340. [Google Scholar] [CrossRef]
- Fomitchev, D.V.; McLauchlan, C.C.; Holm, R.H. Heterometal cubane-type MFe3S4 clusters (M = Mo, V) trigonally symmetrized with Hydrotris(Pyrazolyl)Borate(1−) and Tris(Pyrazolyl)Methanesulfonate(1−) capping ligands. Inorg. Chem. 2002, 41, 958. [Google Scholar] [CrossRef]
- Rees, J.A.; Bjornsson, R.; Kowalska, J.K.; Lima, F.A.; Schlesier, J.; Sippel, D.; Weyhermüller, T.; Einsle, O.; Kovacs, J.A.; DeBeer, S. Comparative electronic structures of nitrogenase FeMoco and FeVco. Dalt. Trans. 2017, 46, 2445. [Google Scholar] [CrossRef] [Green Version]
- Reed, C.J.; Agapie, T. Tetranuclear Fe clusters with a varied interstitial ligand: Effects on the structure, redox properties, and nitric oxide activation. Inorg. Chem. 2017, 56, 13360. [Google Scholar] [CrossRef] [Green Version]
- Arnett, C.H.; Kaiser, J.T.; Agapie, T. Remote ligand modifications tune electronic distribution and reactivity in site-differentiated, high-spin iron clusters: Flipping scaling relationships. Inorg. Chem. 2019, 58, 15971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniyama, N.; Ohki, Y.; Tatsumi, K. Synthesis of V/Fe/S clusters using Vanadium(III) thiolate complexes bearing a phenoxide-based tridentate ligand. Inorg. Chem. 2014, 53, 5438. [Google Scholar] [CrossRef] [PubMed]
- Ohta, S.; Ohki, Y.; Hashimoto, T.; Cramer, R.E.; Tatsumi, K. A nitrogenase cluster model [Fe8S6O] with an oxygen unsymmetrically bridging two proto-Fe4S3 cubes: Relevancy to the substrate binding mode of the FeMo cofactor. Inorg. Chem. 2012, 51, 11217. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Rauchfuss, T.B.; Woods, T.J. Iron carbide–sulfide carbonyl clusters. Inorg. Chem. 2019, 58, 8271. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Woods, T.J.; Rauchfuss, T.B. Reactions of [Fe6C(CO)14(S)]2-: Cluster growth, redox, sulfiding. Eur. J. Inorg. Chem. 2020, 36, 3460. [Google Scholar]
- Churchill, M.R.; Wormald, J.; Knight, J.; Mays, M.J. Synthesis and crystallographic characterization of bis(tetramethylammonium) carbidohexadecacarbonylhexaferrate, a hexanuclear carbidocarbonyl derivative of iron. J. Am. Chem. Soc. 1971, 93, 3073. [Google Scholar] [CrossRef]
- Churchill, M.R.; Wormald, J. Crystal and molecular structure of tetramethylammonium carbidohexadecacarbonylhexaferrate(2–), [Me4N]2 [Fe6(CO)16C], a hexanuclear iron cluster complex with an encapsulated six-co-ordinate carbon atom. J. Chem. Soc. Dalt. Trans. 1974, 2410–2415. [Google Scholar] [CrossRef]
- Ye, M.; Thompson, N.B.; Brown, A.C.; Suess, D.L.M. A synthetic model of enzymatic [Fe4S4]-alkyl intermediates. J. Am. Chem. Soc. 2019, 141, 13330. [Google Scholar] [CrossRef]
- McSkimming, A.; Sridharan, A.; Thompson, N.B.; Müller, P.; Suess, D.L.M. An [Fe4S4]3+–alkyl cluster stabilized by an expanded scorpionate ligand. J. Am. Chem. Soc. 2020, 142, 14314. [Google Scholar] [CrossRef]
- Ni, C.; Power, P.P. Methyl-bridged transition metal complexes (M = Cr - Fe) supported by bulky terphenyl ligands. Organometallics 2009, 28, 6541. [Google Scholar] [CrossRef]
- Sun, C.L.; Krause, H.; Fürstner, A. A practical procedure for iron-catalyzed cross-coupling reactions of sterically hindered aryl-grignard reagents with primary alkyl halides. Adv. Synth. Catal. 2014, 356, 1281. [Google Scholar] [CrossRef] [Green Version]
- Klose, A.; Solari, E.; Floriani, C.; Chiesi-Villa, A.; Rizzoli, C.; Re, N. Magnetic Properties Diagnostic for the Existence of Iron(II)-Iron(II) Bonds in Dinuclear Complexes Which Derive from Stepwise Insertion Reactions on Unsupported Iron-Aryl Bonds. J. Am. Chem. Soc. 1994, 116, 9123. [Google Scholar] [CrossRef]
- Yogendra, S.; Weyhermüller, T.; Hahn, A.W.; DeBeer, S. From Ylides to Doubly Yldiide-Bridged Iron(II) High Spin Dimers via Self-Protolysis. Inorg. Chem. 2019, 58, 9358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagelski, A.L.; Fataftah, M.S.; Bollmeyer, M.M.; McWilliams, S.F.; MacMillan, S.N.; Mercado, B.Q.; Lancaster, K.M.; Holland, P.L. The influences of carbon donor ligands on biomimetic multi-iron complexes for N2 reduction. Chem. Sci. 2020, in press. [Google Scholar] [CrossRef]
- Booker, S.J.; Grove, T.L. Mechanistic and functional versatility of radical SAM enzymes. F1000 Biol. Rep. 2010, 2, 52. [Google Scholar] [CrossRef]
- Braye, E.H.; Dahl, L.F.; Hubel, W.; Wampler, D.L. The preparation, properties and structure of the iron carbonyl carbide Fe5(CO)15C. J. Am. Chem. Soc. 1962, 84, 4633. [Google Scholar] [CrossRef]
- Kuppuswamy, S.; Wofford, J.D.; Joseph, C.; Xie, Z.-L.; Ali, A.K.; Lynch, V.M.; Lindahl, P.A.; Rose, M.J. Structures, interconversions, and spectroscopy of iron carbonyl clusters with an interstitial carbide: Localized metal center reduction by overall cluster oxidation. Inorg. Chem. 2017, 56, 5998. [Google Scholar] [CrossRef]
- Joseph, C.; Kuppuswamy, S.; Lynch, V.M.; Rose, M.J. Fe5Mo cluster with iron-carbide and molybdenum-carbide bonding motifs: Structure and selective alkyne reductions. Inorg. Chem. 2018, 57, 20. [Google Scholar] [CrossRef]
- Rees, J.A.; Bjornsson, R.; Schlesier, J.; Sippel, D.; Einsle, O.; DeBeer, S. The Fe–V cofactor of vanadium nitrogenase contains an interstitial carbon atom. Angew. Chem. Int. Ed. 2015, 54, 13249. [Google Scholar] [CrossRef] [Green Version]
- Cherng, J.-J.; Tsai, Y.-C.; Ueng, C.-H.; Lee, G.-H.; Peng, S.-M.; Shieh, M. New synthesis of [SFe3(CO)9]2− and its reactivity toward electrophiles. Organometallics 2002, 17, 255. [Google Scholar] [CrossRef]
- Kagalwala, H.N.; Lalaoui, N.; Li, Q.L.; Liu, L.; Woods, T.; Rauchfuss, T.B. Redox and “antioxidant” properties of Fe2(μ-SH)2(CO)4(PPh3)3. Inorg. Chem. 2019, 58, 2761. [Google Scholar] [CrossRef] [PubMed]
- Wiig, J.A.; Hu, Y.; Ribbe, M.W. NifEN-B complex of Azotobacter vinelandii is fully functional in nitrogenase FeMo cofactor assembly. Proc. Natl. Acad. Sci. USA 2011, 108, 8623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokhtarzadeh, C.C.; Margulieux, G.W.; Carpenter, A.E.; Weidemann, N.; Moore, C.E.; Rheingold, A.L.; Figueroa, J.S. Synthesis and protonation of an encumbered iron tetraisocyanide dianion. Inorg. Chem. 2015, 54, 5579. [Google Scholar] [CrossRef] [PubMed]
- Caballero, C.; Nuber, B.; Ziegler, M.L. Darstellung und Charakterisierung von trigonal bipyramidalen methylidin-Clustern des Typs EFe3(CO)9CH (E = As, Sb, Bi) durch Umsetzung des Carbidoclusters Fe5C(CO)15 mit metallischem Arsen und Wismut Bzw. SbCl5. J. Organomet. Chem. 1990, 386, 209. [Google Scholar] [CrossRef]
- Capps, K.B.; Wixmerten, B.; Bauer, A.; Hoff, C.D. Thermochemistry of sulfur atom transfer. Enthalpies of reaction of phosphines with sulfur, selenium, and tellurium, and of desulfurization of triphenylarsenic sulfide, triphenylantimony sulfide, and benzyl trisulfide. Inorg. Chem. 1998, 37, 2861. [Google Scholar] [CrossRef]
- Bogdan, P.L.; Sabat, M.; Sunshine, S.A.; Woodcock, C.; Shriver, D.F. Anionic iron carbido carbonyl clusters with sulfur dioxide ligands. Inorg. Chem. 1988, 27, 1904. [Google Scholar] [CrossRef]
- Joseph, C.; Cobb, C.R.; Rose, M.J. Single-Step Insertion of Sulfides and Thiolate into Iron Carbide-Carbonyl Clusters: Unlocking the Synthetic Door to FeMoco Analogues. Angew. Chemie Int. Ed. 2020, in press. [Google Scholar] [CrossRef]
- Zhang, L.-M.; Morrison, C.N.; Kaiser, J.T.; Rees, D.C. Nitrogenase MoFe protein from Clostridium pasteurianum at 1.08 Å resolution: Comparison with the Azotobacter vinelandii MoFe protein. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 274. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.H.; Dahl, L.F. Crystal structure of a 1:1 mixture of two iron carbonyl sulfur complexes, S2Fe3(CO)9 and S2Fe2(CO)6. Inorg. Chem. 1965, 4, 493. [Google Scholar] [CrossRef]
- Bard, A.J.; Cowley, A.H.; Leland, J.K.; Thomas, G.J.N.; Norman, N.C.; Jutzi, P.; Morley, C.P.; Schlüter, E. Synthesis, structures, and reactivities of some pentamethylcyclopentadienyl–sulphur compounds. J. Chem. Soc. Dalt. Trans. 1985, 1303–1307. [Google Scholar] [CrossRef]
- Broadhurst, P.V.; Johnson, B.F.G.; Lewis, J.; Raithby, P.R. Reaction of carbon disulfide with metal cluster carbonyls of the iron triad: Synthesis and X-ray structure of [Fe5(CO)13S2(CS)] containing a six-electron-donor thiocarbonyl group. J. Am. Chem. Soc. 1981, 103, 3198. [Google Scholar] [CrossRef]
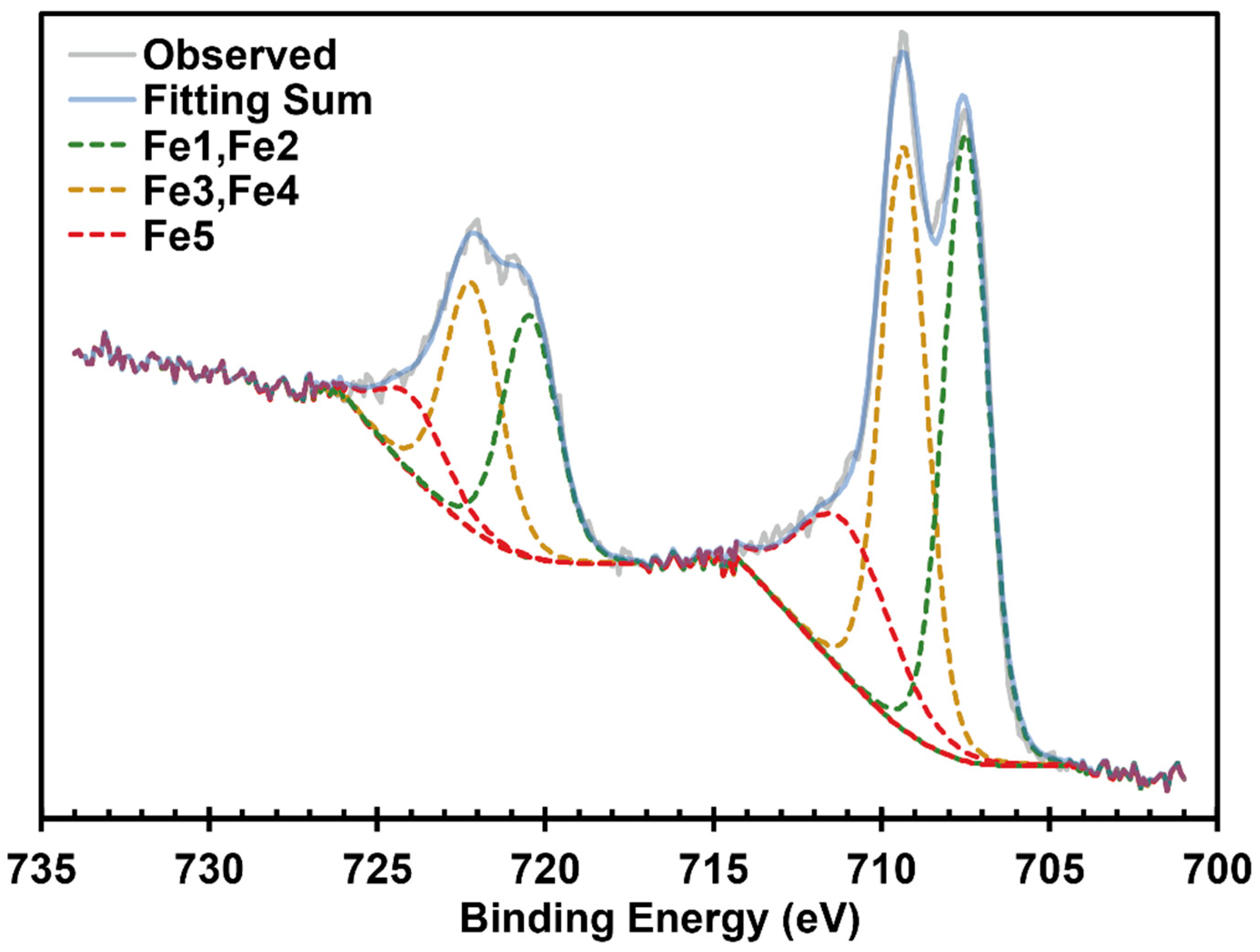
- Grosvenor, A.P.; Kobe, B.A.; Biesinger, M.C.; McIntyre, N.S. Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds. Surf. Interface Anal. 2004, 36, 1564. [Google Scholar] [CrossRef]
- Barber, M.; Connor, J.A.; Guest, M.F.; Hall, M.B.; Hillier, I.H.; Meredith, W.N.E. High energy photoelectron spectroscopy of transition metal complexes. Part 1.—Bonding in substituted and unsubstituted first row carbonyls. Faraday Discuss. Chem. Soc. 1972, 54, 219. [Google Scholar] [CrossRef]
- Brown, A.C.; Suess, D.L.M. Reversible formation of alkyl radicals at [Fe4S4] clusters and its implications for selectivity in radical SAM enzymes. J. Am. Chem. Soc. 2020, 142, 14240. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.C.; Suess, D.L.M. Controlling substrate binding to Fe4S4 clusters through remote steric effects. Inorg. Chem. 2019, 58, 5273. [Google Scholar] [CrossRef] [PubMed]
- Wawzonek, S.; Wearring, D. Polarographic studies in acetonitrile and dimethylformamide. IV. stability of anion-free radicals1,2. J. Am. Chem. Soc. 1959, 81, 2067. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3 | 4 | 5 | 6 | FeMoco | |
|---|---|---|---|---|---|
| Fe–Fe | 2.65 ± 0.06 | 2.64 ± 0.05 | 2.66 ± 0.09 | 2.7 ± 0.1 | 2.63 ± 0.04 |
| Fe–C | 1.88 ± 0.03 | 1.88 ± 0.04 | 1.97 ± 0.04 | 1.96 ± 0.02 | 2.00 ± 0.02 |
| Fe–S | 2.19 ± 0.01 | 2.175 ± 0.005 | 2.27 ± 0.03 | 2.25 ± 0.03 | 2.25 ± 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joseph, C.; Shupp, J.P.; Cobb, C.R.; Rose, M.J. Construction of Synthetic Models for Nitrogenase-Relevant NifB Biogenesis Intermediates and Iron-Carbide-Sulfide Clusters. Catalysts 2020, 10, 1317. https://doi.org/10.3390/catal10111317
Joseph C, Shupp JP, Cobb CR, Rose MJ. Construction of Synthetic Models for Nitrogenase-Relevant NifB Biogenesis Intermediates and Iron-Carbide-Sulfide Clusters. Catalysts. 2020; 10(11):1317. https://doi.org/10.3390/catal10111317
Chicago/Turabian StyleJoseph, Chris, John Patrick Shupp, Caitlyn R. Cobb, and Michael J. Rose. 2020. "Construction of Synthetic Models for Nitrogenase-Relevant NifB Biogenesis Intermediates and Iron-Carbide-Sulfide Clusters" Catalysts 10, no. 11: 1317. https://doi.org/10.3390/catal10111317