
Targeting Myeloid-Derived Suppressor Cell Trafficking as a Novel Immunotherapeutic Approach in Microsatellite Stable Colorectal Cancer
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
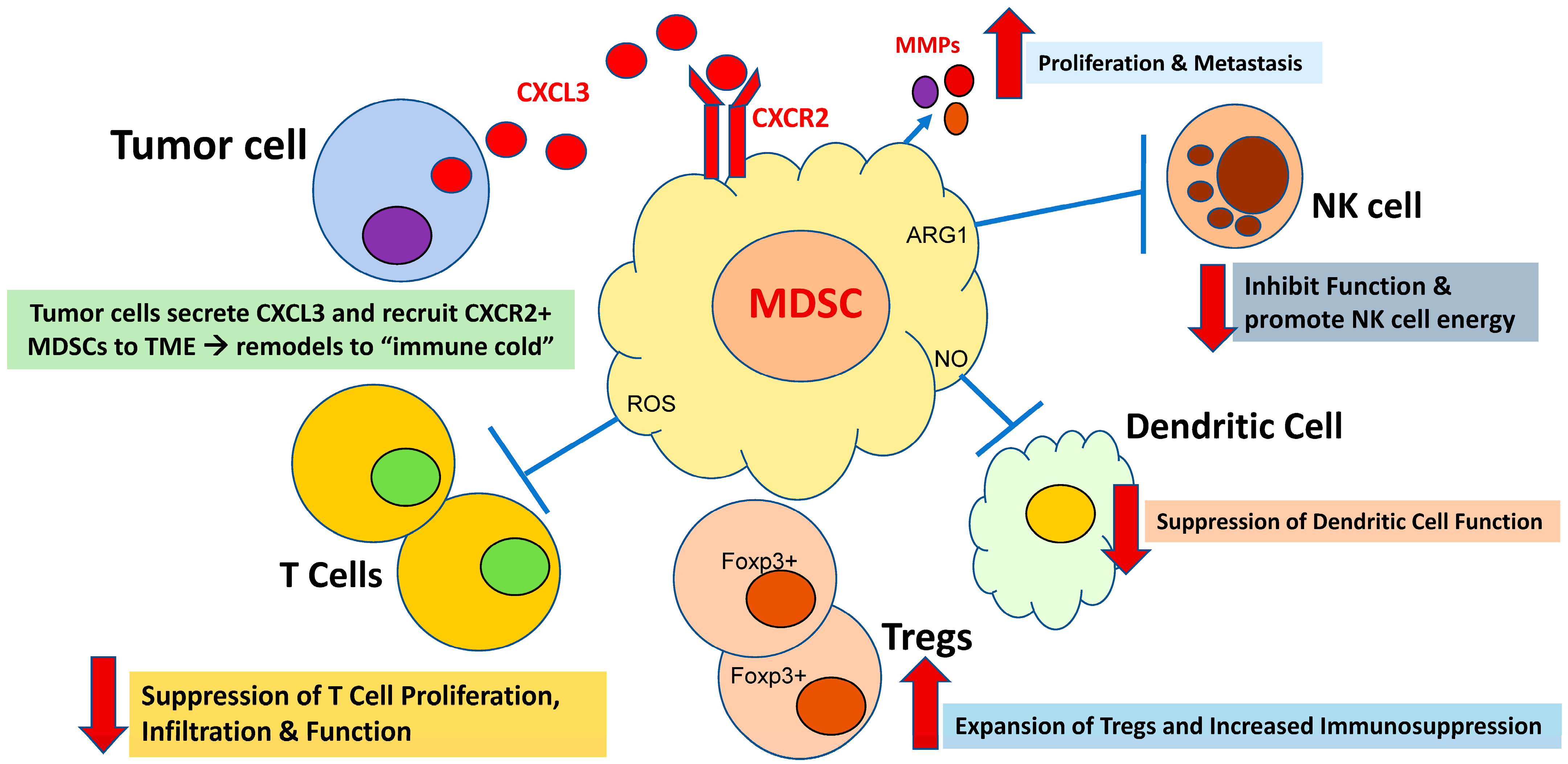
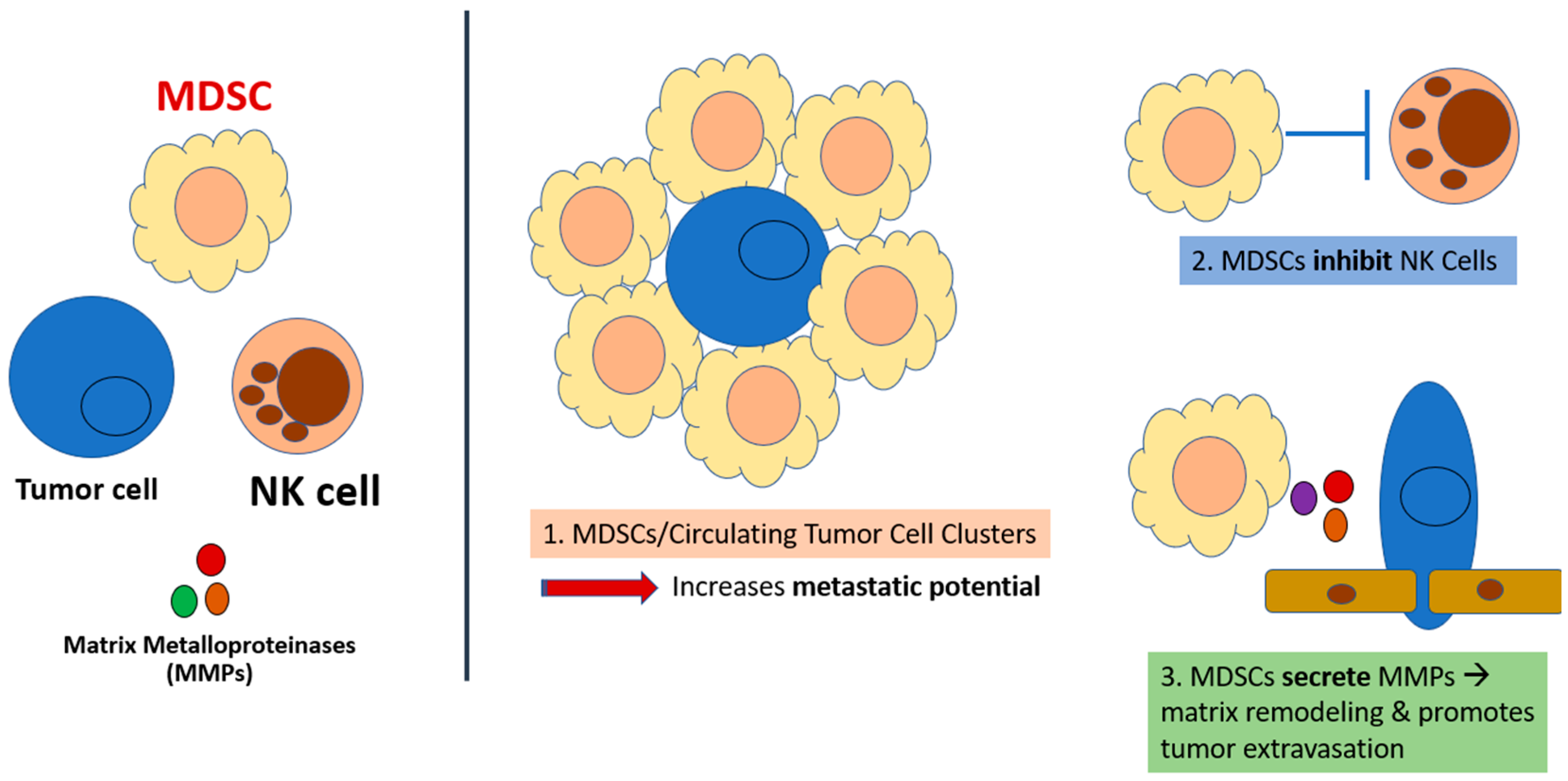
2. MDSC Immunosuppression: A Critical Mechanism of Immune Evasion in Cancer
3. CXCR1/2 Are Critical in the Tumor-Driven Recruitment of MDSCs to the Tumor
4. Core RAS Mutations in mCRC (KRAS or NRAS): Drivers of CXCR2 Chemokine Secretion by Tumor Cells
5. Mechanism of the KRAS-Promoted Immunosuppressive Tumor Microenvironment
6. Minimal Residual Disease (MRD) in Colorectal Cancer Represents an Innovative Space for Early Therapeutic Intervention: Can We Target MDSCs?
7. Conclusions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Jakubowski, C.D.; Fedewa, S.A.; Davis, A.; Azad, N.S. Colorectal Cancer in the Young: Epidemiology, Prevention, Management. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, e75–e88. [Google Scholar] [CrossRef] [PubMed]
- Mauri, G.; Sartore-Bianchi, A.; Russo, A.G.; Marsoni, S.; Bardelli, A.; Siena, S. Early-onset colorectal cancer in young individuals. Mol. Oncol. 2019, 13, 109–131. [Google Scholar] [CrossRef] [PubMed]
- Jácome, A.A.; Vreeland, T.J.; Johnson, B.; Kawaguchi, Y.; Wei, S.H.; You, Y.N.; Vilar, E.; Vauthey, J.N.; Eng, C. The prognostic impact of RAS on overall survival following liver resection in early versus late-onset colorectal cancer patients. Br. J. Cancer 2021, 124, 797–804. [Google Scholar] [CrossRef]
- Goswami, S.; Anandhan, S.; Raychaudhuri, D.; Sharma, P. Myeloid cell-targeted therapies for solid tumours. Nat. Rev. Immunol. 2023, 23, 106–120. [Google Scholar] [CrossRef]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e6, Erratum in Immunity 2017, 47, 597. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Denardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef]
- Dontu, G.; Al-Hajj, M.; Abdallah, W.M.; Clarke, M.F.; Wicha, M.S. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003, 36, 59–72. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Kim, S.; Takahashi, H.; Lin, W.W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.L.; Karin, M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009, 457, 102–106. [Google Scholar] [CrossRef]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572.e7. [Google Scholar] [CrossRef] [PubMed]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Lan, P.; Fu, L. The role of myeloid-derived suppressor cells in gastrointestinal cancer. Cancer Commun. 2021, 41, 442–471. [Google Scholar] [CrossRef] [PubMed]
- Väyrynen, S.A.; Zhang, J.; Yuan, C.; Väyrynen, J.P.; Dias Costa, A.; Williams, H.; Morales-Oyarvide, V.; Lau, M.C.; Rubinson, D.A.; Dunne, R.F.; et al. Composition, Spatial Characteristics, and Prognostic Significance of Myeloid Cell Infiltration in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 1069–1081. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Ramachandran, I.; Youn, J.I.; Gabrilovich, D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef]
- Rot, A.; von Andrian, U.H. Chemokines in innate and adaptive host defense: Basic chemokinese grammar for immune cells. Annu. Rev. Immunol. 2004, 22, 891–928. [Google Scholar] [CrossRef]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef]
- Di Mitri, D.; Toso, A.; Chen, J.J.; Sarti, M.; Pinton, S.; Jost, T.R.; D’Antuono, R.; Montani, E.; Garcia-Escudero, R.; Guccini, I.; et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature 2014, 515, 134–137. [Google Scholar] [CrossRef]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra67. [Google Scholar] [CrossRef] [PubMed]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Bierie, B.; Tada, M.; Mohri, D.; Miyabayashi, K.; Asaoka, Y.; Maeda, S.; et al. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J. Clin. Investig. 2011, 121, 4106–4117. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, T.; Clarke, M.; Steele, C.W.; Samuel, M.S.; Neumann, J.; Jung, A.; Huels, D.; Olson, M.F.; Das, S.; Nibbs, R.J.; et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Investig. 2012, 122, 3127–3144. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Wang, D.; Daikoku, T.; Sun, H.; Dey, S.K.; Dubois, R.N. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 2013, 24, 631–644. [Google Scholar] [CrossRef]
- Toh, B.; Wang, X.; Keeble, J.; Sim, W.J.; Khoo, K.; Wong, W.C.; Kato, M.; Prevost-Blondel, A.; Thiery, J.P.; Abastado, J.P. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011, 9, e1001162. [Google Scholar] [CrossRef]
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, P.C.; et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008, 13, 23–35. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Yaeger, R.; Weiss, J.; Pelster, M.S.; Spira, A.I.; Barve, M.; Ou, S.I.; Leal, T.A.; Bekaii-Saab, T.S.; Paweletz, C.P.; Heavey, G.A.; et al. Adagrasib with or without Cetuximab in Colorectal Cancer with Mutated KRAS G12C. N. Engl. J. Med. 2023, 388, 44–54. [Google Scholar] [CrossRef]
- Henry, J.T.; Coker, O.; Chowdhury, S.; Shen, J.P.; Morris, V.K.; Dasari, A.; Raghav, K.; Nusrat, M.; Kee, B.; Parseghian, C.; et al. Comprehensive Clinical and Molecular Characterization of KRASG12C-Mutant Colorectal Cancer. JCO Precis. Oncol. 2021, 5, PO.20.00256. [Google Scholar] [CrossRef]
- Maeda, D.Y.; Peck, A.M.; Schuler, A.D.; Quinn, M.T.; Kirpotina, L.N.; Wicomb, W.N.; Auten, R.L.; Gundla, R.; Zebala, J.A. Boronic acid-containing CXCR1/2 antagonists: Optimization of metabolic stability, in vivo evaluation, and a proposed receptor binding model. Bioorganic Med. Chem. Lett. 2015, 25, 2280–2284. [Google Scholar] [CrossRef]
- Maeda, D.Y.; Peck, A.M.; Schuler, A.D.; Quinn, M.T.; Kirpotina, L.N.; Wicomb, W.N.; Fan, G.H.; Zebala, J.A. Discovery of 2-[5-(4-Fluorophenylcarbamoyl)pyridin-2-ylsulfanylmethyl]phenylboronic Acid (SX-517): Noncompetitive Boronic Acid Antagonist of CXCR1 and CXCR2. J. Med. Chem. 2014, 57, 8378–8397. [Google Scholar] [CrossRef] [PubMed]
- Maeda, D.Y.; Quinn, M.T.; Schepetkin, I.A.; Kirpotina, L.N.; Zebala, J.A. Nicotinamide glycolates antagonize CXCR2 activity through an intracellular mechanism. J. Pharmacol. Exp. Ther. 2010, 332, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Schuler, A.D.; Engles, C.A.; Maeda, D.Y.; Quinn, M.T.; Kirpotina, L.N.; Wicomb, W.N.; Mason, S.N.; Auten, R.L.; Zebala, J.A. Boronic acid-containing aminopyridine- and aminopyrimidinecarboxamide CXCR1/2 antagonists: Optimization of aqueous solubility and oral bioavailability. Bioorganic Med. Chem. Lett. 2015, 25, 3793–3797. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Liu, X.; Hogg, G.D.; DeNardo, D.G. Rethinking immune checkpoint blockade: ‘beyond the T cell’. J. Immunother. Cancer 2021, 9, e001460. [Google Scholar] [CrossRef]
- Weissleder, R.; Pittet, M.J. The expanding landscape of inflammatory cells affecting cancer therapy. Nat. Biomed. Eng. 2020, 4, 489–498. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Li, T.; Liu, T.; Zhu, W.; Xie, S.; Zhao, Z.; Feng, B.; Guo, H.; Yang, R. Targeting MDSC for Immune-Checkpoint Blockade in Cancer Immunotherapy: Current Progress and New Prospects. Clin. Med. Insights Oncol. 2021, 15, 11795549211035540. [Google Scholar] [CrossRef]
- Gulhati, P.; Schalck, A.; Jiang, S.; Shang, X.; Wu, C.J.; Hou, P.; Ruiz, S.H.; Soto, L.S.; Parra, E.; Ying, H.; et al. Targeting T cell checkpoints 41BB and LAG3 and myeloid cell CXCR1/CXCR2 results in antitumor immunity and durable response in pancreatic cancer. Nat. Cancer 2023, 4, 62–80. [Google Scholar] [CrossRef]
- Faget, J.; Groeneveld, S.; Boivin, G.; Sankar, M.; Zangger, N.; Garcia, M.; Guex, N.; Zlobec, I.; Steiner, L.; Piersigilli, A.; et al. Neutrophils and Snail Orchestrate the Establishment of a Pro-tumor Microenvironment in Lung Cancer. Cell Rep. 2017, 21, 3190–3204. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Cassetta, L.; Kitamura, T. Targeting tumor-associated macrophages as a potential strategy to enhance the response to immune checkpoint inhibitors. Front. Cell Dev. Biol. 2018, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668.e5. [Google Scholar] [CrossRef] [PubMed]
- Gyori, D.; Lim, E.L.; Grant, F.M.; Spensberger, D.; Roychoudhuri, R.; Shuttleworth, S.J.; Okkenhaug, K.; Stephens, L.R.; Hawkins, P.T. Compensation between CSF1R+ macrophages and Foxp3+ Treg cells drives resistance to tumor immunotherapy. JCI Insight 2018, 3, e120631. [Google Scholar] [CrossRef] [PubMed]
- Yuen, K.C.; Liu, L.F.; Gupta, V.; Madireddi, S.; Keerthivasan, S.; Li, C.; Rishipathak, D.; Williams, P.; Kadel, E.E., 3rd; Koeppen, H.; et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat. Med. 2020, 26, 693–698, Erratum in Nat. Med. 2021, 27, 560. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Carleton, M.; Zhou, M.; Chen, T.; Feng, Y.; Huang, S.P.; Walsh, A.M.; Baxi, V.; Pandya, D.; Baradet, T.; et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat. Med. 2020, 26, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Linde, I.L.; Prestwood, T.R.; Qiu, J.; Pilarowski, G.; Linde, M.H.; Zhang, X.; Shen, L.; Reticker-Flynn, N.E.; Chiu, D.K.; Sheu, L.Y.; et al. Neutrophil-activating therapy for the treatment of cancer. Cancer Cell 2023, 41, 356–372.e10. [Google Scholar] [CrossRef]
- Johnson, B.; Kopetz, S.; Hwang, H.; Yuan, Y.; DePinho, R.A.; Zebala, J.; Overman, M.J. STOPTRAFFIC-1: A phase I/II trial of SX-682 in combination with nivolumab for refractory RAS-mutated microsatellite stable (MSS) metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2022, 40, TPS3638. [Google Scholar] [CrossRef]
- Johnson, B.; Haymaker, C.; Morris, V.K.; Dasari, A.; Higbie, V.S.; Shen, J.P.; Parseghian, C.; Morelli, M.P.; Huey, R.; Lee, M.S.; et al. A phase I/II trial of a CXCR1/2 inhibitor in combination with anti-PD-1 for circulating tumor DNA (ctDNA) positive & refractory RAS-mutated microsatellite stable (MSS) colorectal cancer [abstract]. In Proceedings of the American Association for Cancer Research Annual Meeting 2023, Orlando, FL, USA, 14–19 April 2023; Part 2 (Clinical Trials and Late-Breaking Research). AACR: Philadelphia, PA, USA, 2023. [Google Scholar]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients with Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131, Erratum in JAMA Oncol. 2019, 5, 1232. [Google Scholar] [CrossRef]
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, Y.; Guo, N.; Wang, S. MDSCs: Key Criminals of Tumor Pre-metastatic Niche Formation. Front. Immunol. 2019, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Heeke, S.; Mograbi, B.; Alix-Panabières, C.; Hofman, P. Never Travel Alone: The Crosstalk of Circulating Tumor Cells and the Blood Microenvironment. Cells 2019, 8, 714. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [PubMed]
- Sprouse, M.L.; Welte, T.; Boral, D.; Liu, H.N.; Yin, W.; Vishnoi, M.; Goswami-Sewell, D.; Li, L.; Pei, G.; Jia, P.; et al. PMN-MDSCs Enhance CTC Metastatic Properties through Reciprocal Interactions via ROS/Notch/Nodal Signaling. Int. J. Mol. Sci. 2019, 20, 1916. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Tanikawa, T.; Li, W.; Zhao, L.; Vatan, L.; Szeliga, W.; Wan, S.; Wei, S.; Wang, Y.; Liu, Y.; et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016, 76, 3156–3165. [Google Scholar] [CrossRef]
- Jackstadt, R.; van Hooff, S.R.; Leach, J.D.; Cortes-Lavaud, X.; Lohuis, J.O.; Ridgway, R.A.; Wouters, V.M.; Roper, J.; Kendall, T.J.; Roxburgh, C.S.; et al. Epithelial NOTCH Signaling Rewires the Tumor Microenvironment of Colorectal Cancer to Drive Poor-Prognosis Subtypes and Metastasis. Cancer Cell 2019, 36, 319–336.e7. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Taniguchi, H.; Nakamura, Y.; Kotani, D.; Yukami, H.; Mishima, S.; Sawada, K.; Shirasu, H.; Ebi, H.; Yamanaka, T.; Aleshin, A.; et al. CIRCULATE-Japan: Circulating tumor DNA-guided adaptive platform trials to refine adjuvant therapy for colorectal cancer. Cancer Sci. 2021, 112, 2915–2920. [Google Scholar] [CrossRef]
- Kotani, D.; Oki, E.; Nakamura, Y.; Yukami, H.; Mishima, S.; Bando, H.; Shirasu, H.; Yamazaki, K.; Watanabe, J.; Kotaka, M.; et al. Molecular residual disease and efficacy of adjuvant chemotherapy in patients with colorectal cancer. Nat. Med. 2023, 29, 127–134. [Google Scholar] [CrossRef]
- Jácome, A.A.; Johnson, B. Minimal Residual Disease in Colorectal Cancer: Are We Finding the Needle in a Haystack? Cells 2023, 12, 1068. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, B. Targeting Myeloid-Derived Suppressor Cell Trafficking as a Novel Immunotherapeutic Approach in Microsatellite Stable Colorectal Cancer. Cancers 2023, 15, 5484. https://doi.org/10.3390/cancers15225484
Johnson B. Targeting Myeloid-Derived Suppressor Cell Trafficking as a Novel Immunotherapeutic Approach in Microsatellite Stable Colorectal Cancer. Cancers. 2023; 15(22):5484. https://doi.org/10.3390/cancers15225484
Chicago/Turabian StyleJohnson, Benny. 2023. "Targeting Myeloid-Derived Suppressor Cell Trafficking as a Novel Immunotherapeutic Approach in Microsatellite Stable Colorectal Cancer" Cancers 15, no. 22: 5484. https://doi.org/10.3390/cancers15225484