
Dpep Inhibits Cancer Cell Growth and Survival via Shared and Context-Dependent Transcriptome Perturbations
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Cell Lines
2.2. Dpep
2.3. PLATE-Seq
2.4. Data Analysis
2.5. Bioinformatic Analysis
3. Results
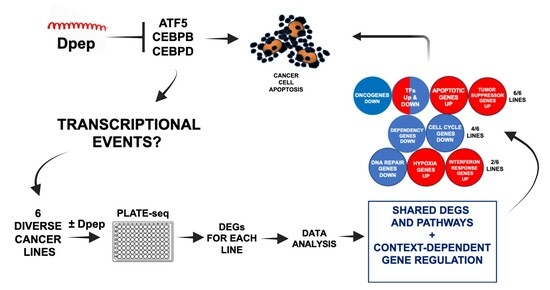
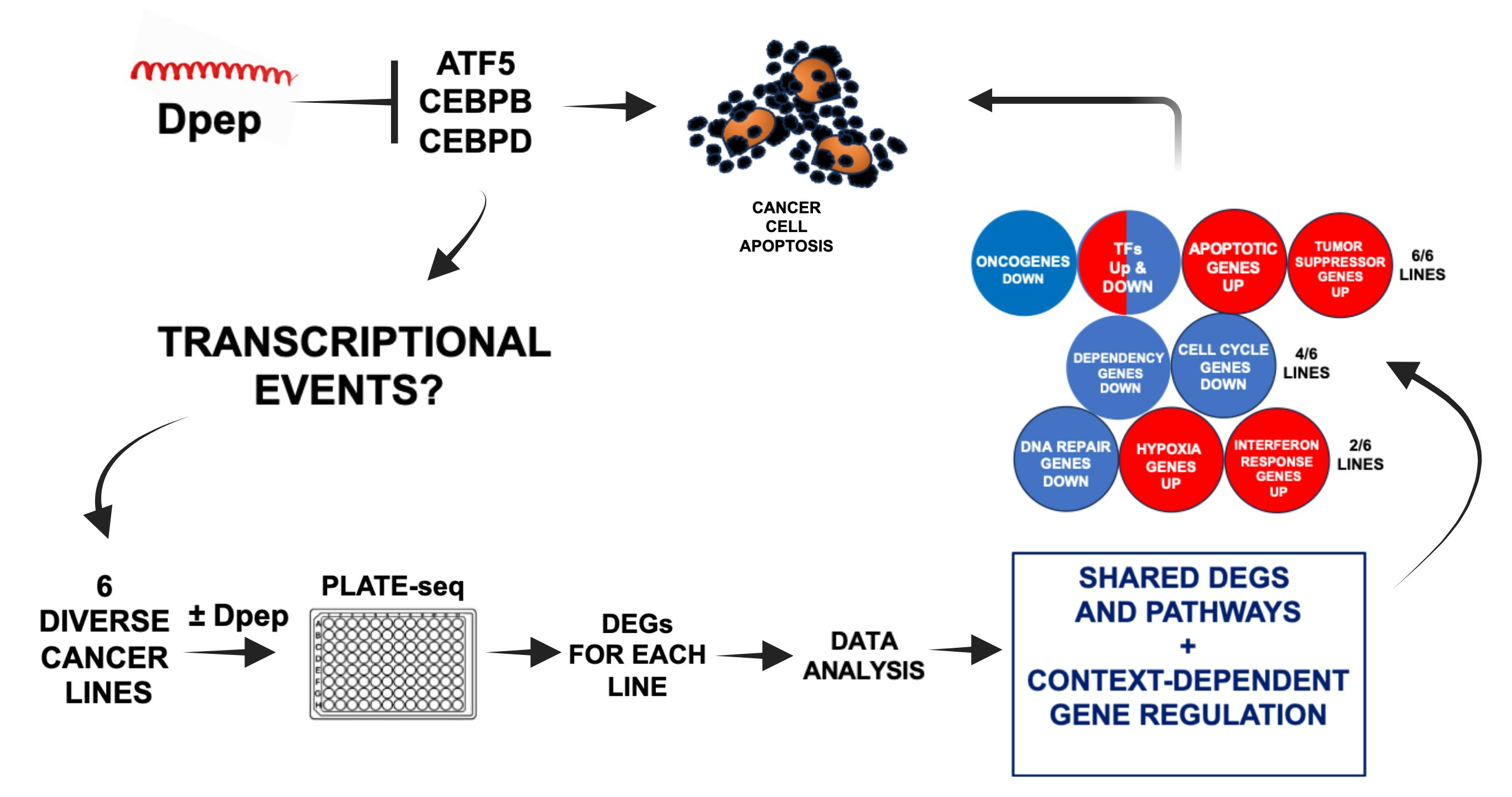
3.1. Experimental Scheme
3.2. ATF5, CEBPB, and CEBPD Are Differentially Expressed in Each Cell Line
3.3. Dpep Elicits Context-Dependent Changes in Gene Expression
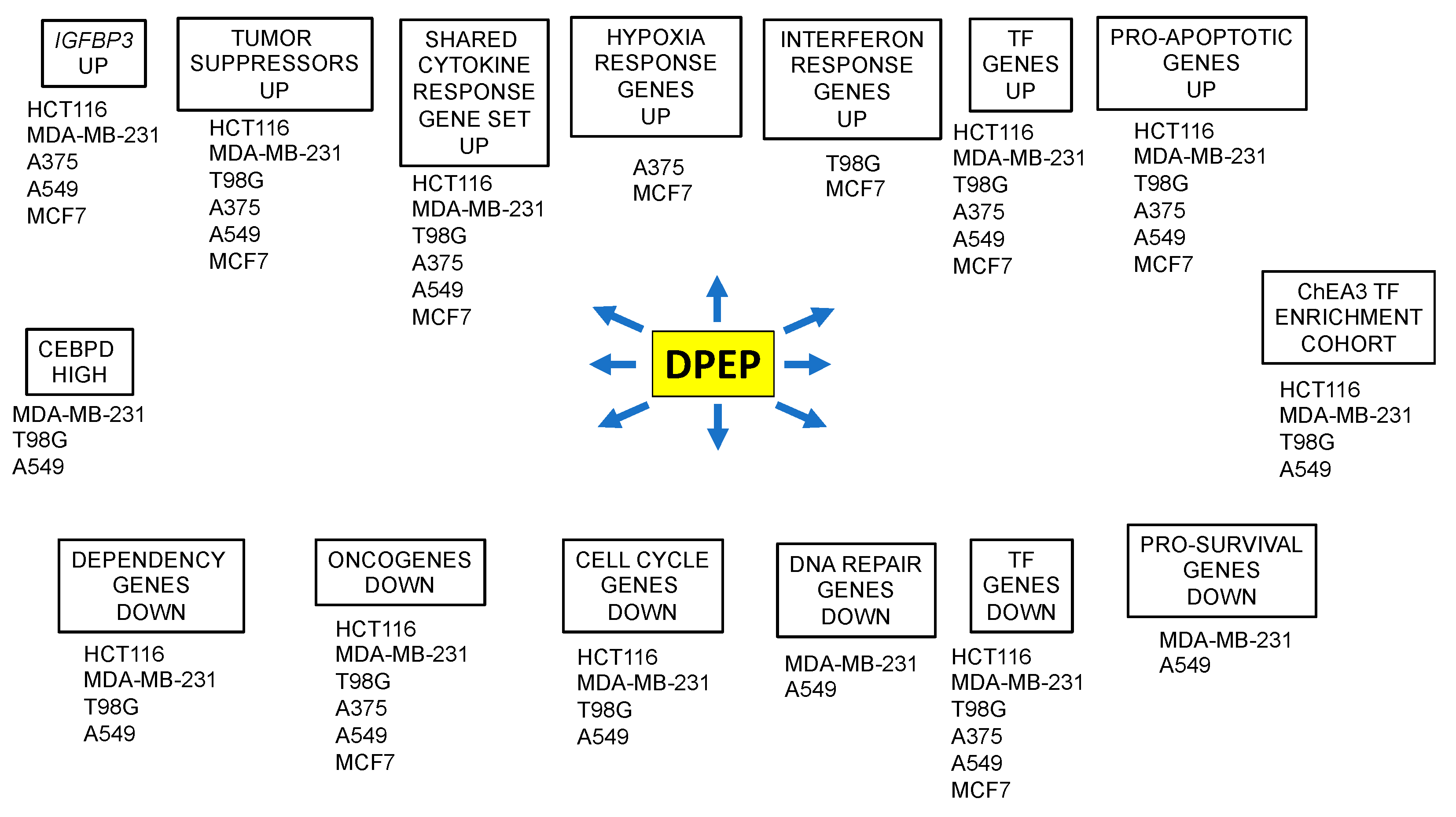
3.4. A Cohort of Genes Is Regulated across Multiple Cell Lines
3.5. Context-Dependent Regulation of Pro- and Anti-Apoptotic Genes
3.6. Dpep Downregulates Dependency Genes
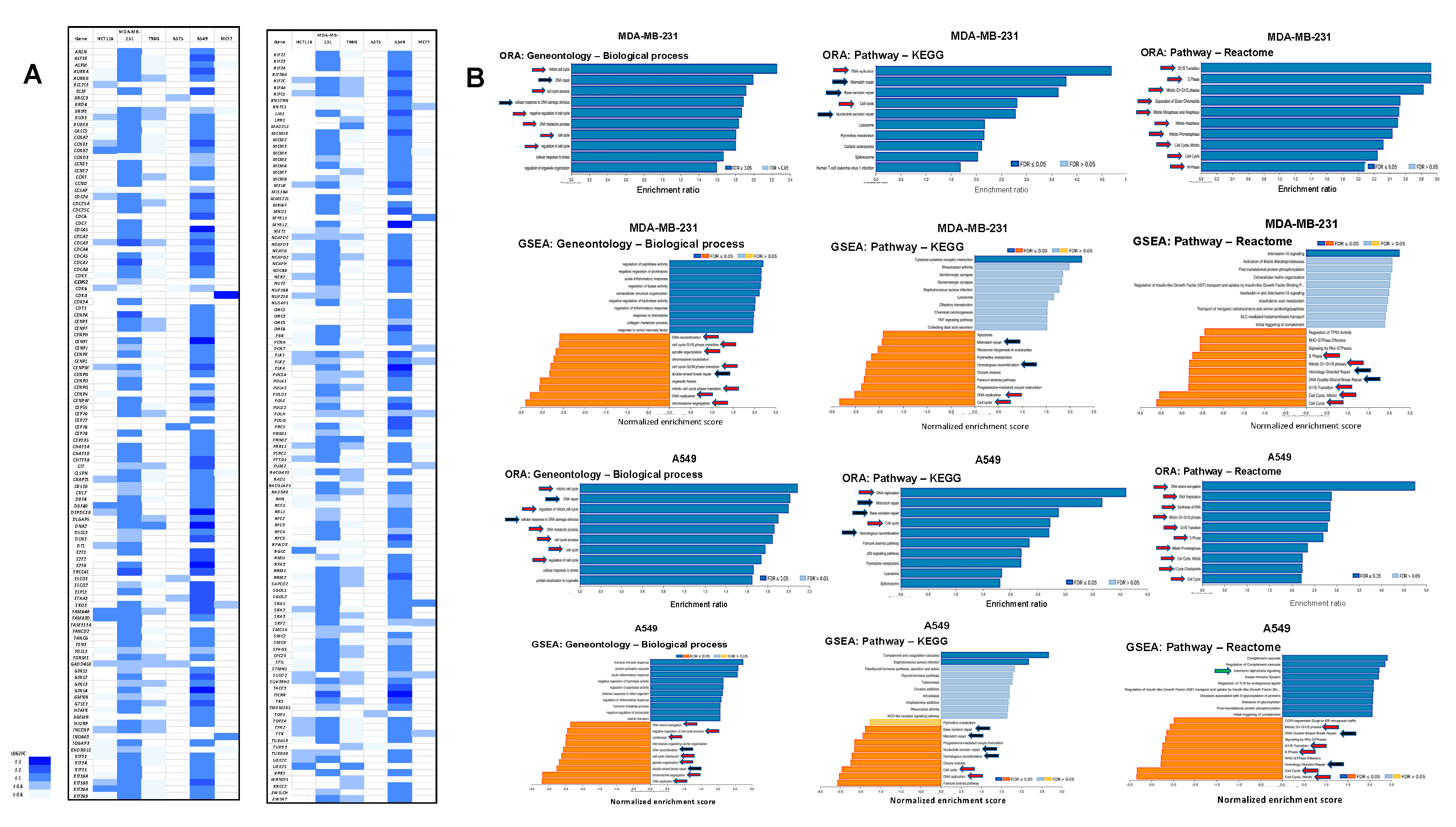
3.7. Dpep Downregulates Cell-Cycle-Related Genes in Multiple Lines
3.8. Downregulation of DEGs Associated with DNA Repair
3.9. Selective Induction of DEGs Associated with Interferon Responses
3.10. Dpep Upregulates Genes Indicative of a Response to Hypoxia
3.11. Downregulation of Oncogenes and Upregulation of Tumor Suppressors in Response to Dpep
3.12. Dpep Affects the Expression of Numerous Transcription Factors
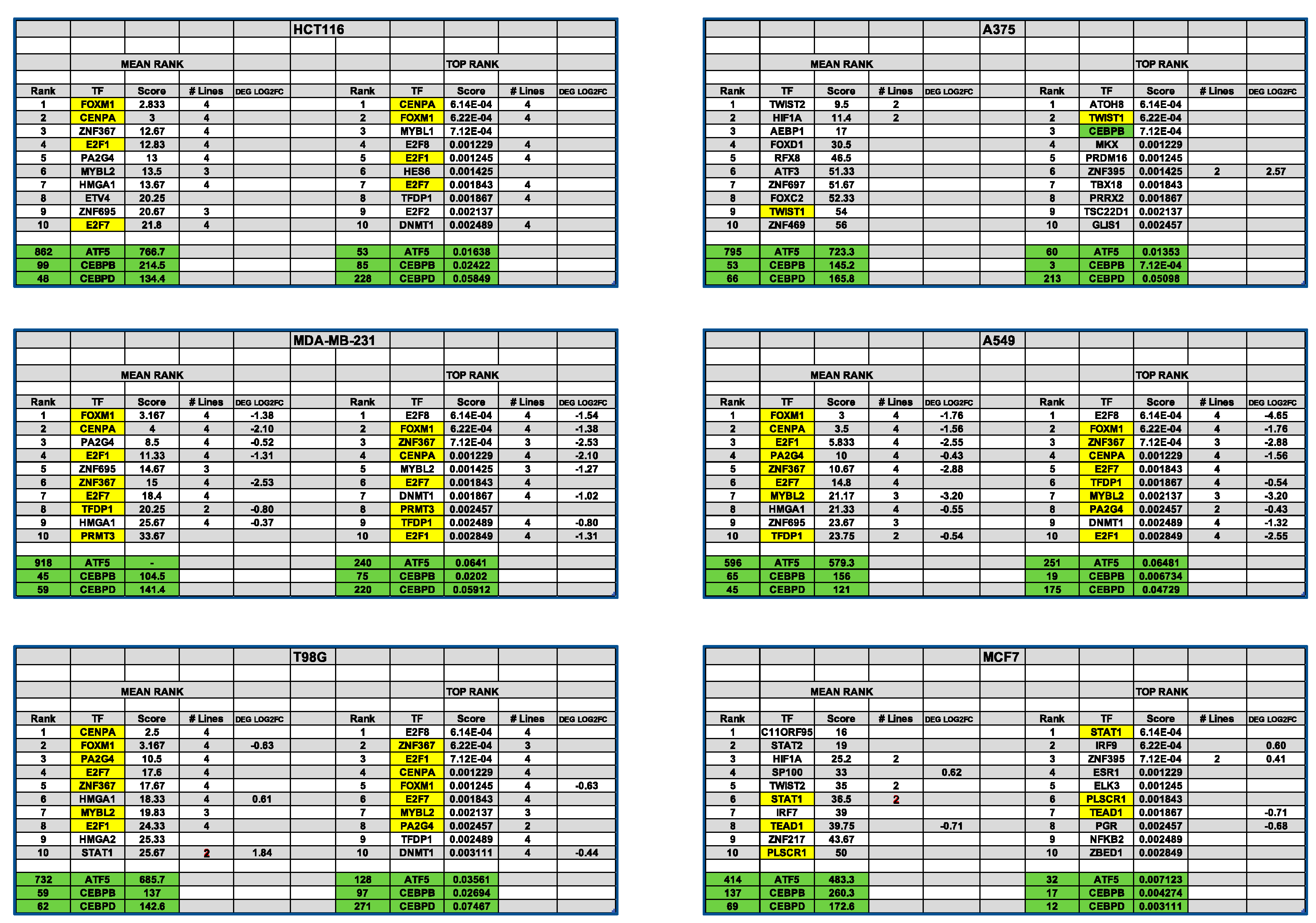
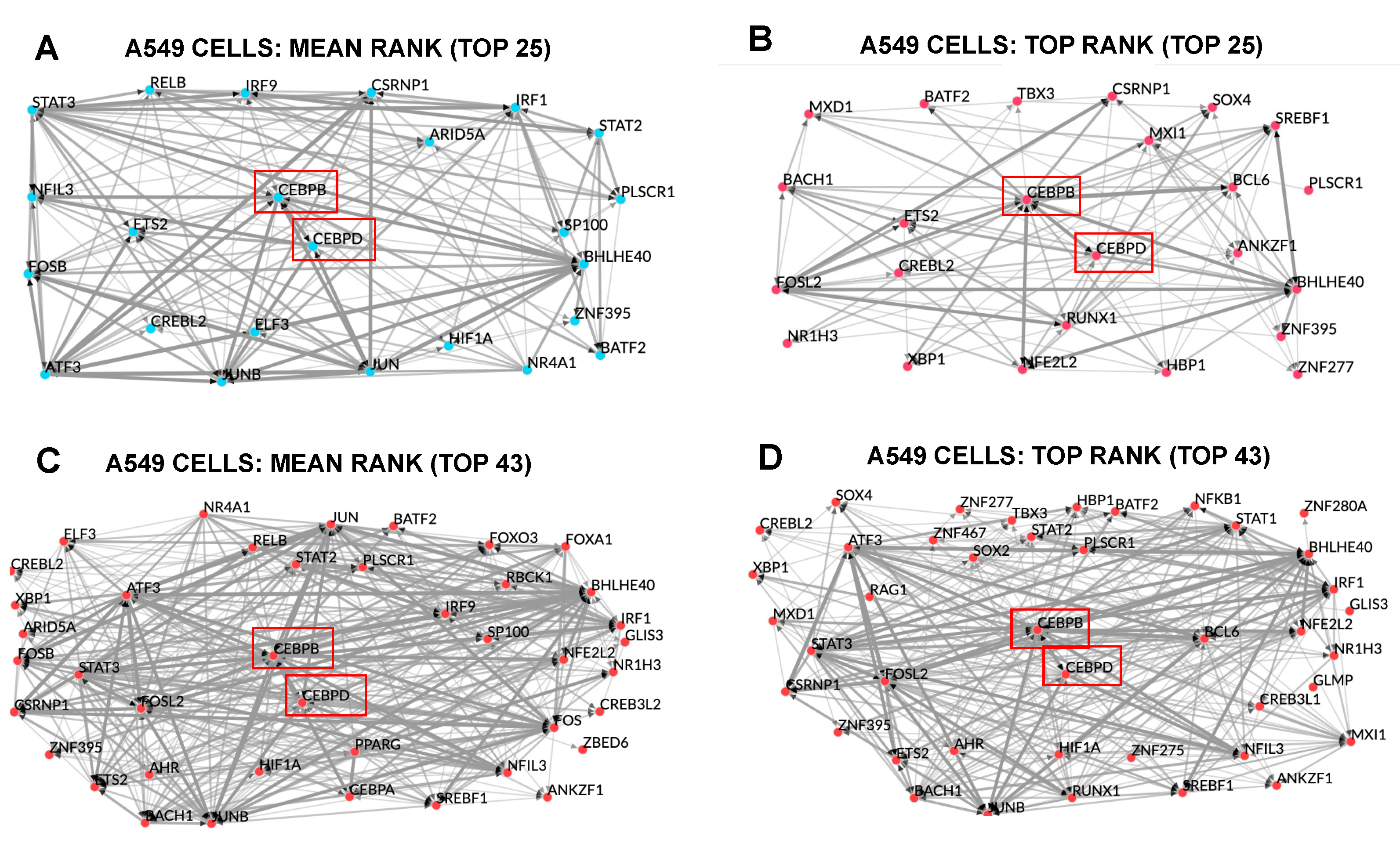
3.13. Transcription Factor Enrichment Analysis (TFEA) Indicates Shared and Cascading Transcriptional Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Greene, L.A.; Zhou, Q.; Siegelin, M.D.; Angelastro, J.M. Targeting Transcription Factors ATF5, CEBPB and CEBPD with Cell Penetrating Peptides to Treat Brain and Other Cancers. Cells 2023, 12, 581. [Google Scholar] [CrossRef] [PubMed]
- Reinke, A.W.; Baek, J.; Ashenberg, O.; Keating, A.E. Networks of bZIP protein-protein interactions diversified over a billion years of evolution. Science 2013, 340, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Vinson, C.R.; Hai, T.; Boyd, S.M. Dimerization specificity of the leucine zipper-containing bZIP motif on DNA binding: Prediction and rational design. Genes. Dev. 1993, 7, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Angelastro, J.M.; Ignatova, T.N.; Kukekov, V.G.; Steindler, D.A.; Stengren, G.B.; Mendelsohn, C.; Greene, L.A. Regulated expression of ATF5 is required for the progression of neural progenitor cells to neurons. J. Neurosci. 2003, 23, 4590–4600. [Google Scholar] [CrossRef]
- Zhou, Q.; Sun, X.; Pasquier, N.; Jefferson, P.; Nguyen, T.T.T.; Siegelin, M.D.; Angelastro, J.M.; Greene, L.A. Cell-Penetrating CEBPB and CEBPD Leucine Zipper Decoys as Broadly Acting Anti-Cancer Agents. Cancers 2021, 13, 2504. [Google Scholar] [CrossRef]
- Dupont, E.; Prochiantz, A.; Joliot, A. Penetratin Story: An Overview. Methods Mol. Biol. 2015, 1324, 29–37. [Google Scholar]
- Cates, C.C.; Arias, A.D.; Nakayama Wong, L.S.; Lame, M.W.; Sidorov, M.; Cayanan, G.; Rowland, D.J.; Fung, J.; Karpel-Massler, G.; Siegelin, M.D.; et al. Regression/eradication of gliomas in mice by a systemically-deliverable ATF5 dominant-negative peptide. Oncotarget 2016, 7, 12718–12730. [Google Scholar] [CrossRef]
- Sun, X.; Jefferson, P.; Zhou, Q.; Angelastro, J.M.; Greene, L.A. Dominant-Negative ATF5 Compromises Cancer Cell Survival by Targeting CEBPB and CEBPD. Mol. Cancer Res. 2020, 18, 216–228. [Google Scholar] [CrossRef]
- Karpel-Massler, G.; Horst, B.A.; Shu, C.; Chau, L.; Tsujiuchi, T.; Bruce, J.N.; Canoll, P.; Greene, L.A.; Angelastro, J.M.; Siegelin, M.D. A Synthetic Cell-Penetrating Dominant-Negative ATF5 Peptide Exerts Anticancer Activity against a Broad Spectrum of Treatment-Resistant Cancers. Clin. Cancer Res. 2016, 22, 4698–4711. [Google Scholar] [CrossRef]
- Sun, X.; Angelastro, J.M.; Merino, D.; Zhou, Q.; Siegelin, M.D.; Greene, L.A. Dominant-negative ATF5 rapidly depletes survivin in tumor cells. Cell Death Dis. 2019, 10, 709. [Google Scholar] [CrossRef]
- Dluzen, D.; Li, G.; Tacelosky, D.; Moreau, M.; Liu, D.X. BCL-2 is a downstream target of ATF5 that mediates the prosurvival function of ATF5 in a cell type-dependent manner. J. Biol. Chem. 2011, 286, 7705–7713. [Google Scholar] [CrossRef] [PubMed]
- Bush, E.C.; Ray, F.; Alvarez, M.J.; Realubit, R.; Li, H.; Karan, C.; Califano, A.; Sims, P.A. PLATE-Seq for genome-wide regulatory network analysis of high-throughput screens. Nat. Commun. 2017, 8, 105. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium; Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; et al. The Gene Ontology knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef]
- Keenan, A.B.; Torre, D.; Lachmann, A.; Leong, A.K.; Wojciechowicz, M.L.; Utti, V.; Jagodnik, K.M.; Kropiwnicki, E.; Wang, Z.; Ma’ayan, A. ChEA3: Transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019, 47, W212–W224. [Google Scholar] [CrossRef]
- Varma Shrivastav, S.; Bhardwaj, A.; Pathak, K.A.; Shrivastav, A. Insulin-Like Growth Factor Binding Protein-3 (IGFBP-3): Unraveling the Role in Mediating IGF-Independent Effects Within the Cell. Front. Cell Dev. Biol. 2020, 8, 286. [Google Scholar] [CrossRef]
- Cai, Q.; Dozmorov, M.; Oh, Y. IGFBP-3/IGFBP-3 Receptor System as an Anti-Tumor and Anti-Metastatic Signaling in Cancer. Cells 2020, 9, 1261. [Google Scholar] [CrossRef] [PubMed]
- Brosseau, C.; Pirianov, G.; Colston, K.W. Role of insulin-like growth factor binding protein-3 in 1, 25-dihydroxyvitamin-d 3 -induced breast cancer cell apoptosis. Int. J. Cell Biol. 2013, 2013, 960378. [Google Scholar] [CrossRef] [PubMed]
- Shahjee, H.M.; Bhattacharyya, N. Activation of various downstream signaling molecules by IGFBP-3. J. Cancer Ther. 2014, 5, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Gucev, Z.; Ng, L.; Muller, H.L.; Rosenfeld, R.G. Antiproliferative actions of insulin-like growth factor binding protein (IGFBP)-3 in human breast cancer cells. Prog. Growth Factor. Res. 1995, 6, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Moon, H.; Chun, K.H.; Chang, Y.S.; Hassan, K.; Ji, L.; Lotan, R.; Khuri, F.R.; Hong, W.K. Effects of insulin-like growth factor binding protein-3 and farnesyltransferase inhibitor SCH66336 on Akt expression and apoptosis in non-small-cell lung cancer cells. J. Natl. Cancer Inst. 2004, 96, 1536–1548. [Google Scholar] [CrossRef]
- Wu, C.; Liu, X.; Wang, Y.; Tian, H.; Xie, Y.; Li, Q.; Zhang, X.; Liu, F. Insulin-like factor binding protein-3 promotes the G1 cell cycle arrest in several cancer cell lines. Gene 2013, 512, 127–133. [Google Scholar] [CrossRef]
- Johnson, M.A.; Firth, S.M. IGFBP-3: A cell fate pivot in cancer and disease. Growth Horm. IGF Res. 2014, 24, 164–173. [Google Scholar] [CrossRef]
- Chen, C.H.; Chen, P.Y.; Lin, Y.Y.; Feng, L.Y.; Chen, S.H.; Chen, C.Y.; Huang, Y.C.; Huang, C.Y.; Jung, S.M.; Chen, L.Y.; et al. Suppression of tumor growth via IGFBP3 depletion as a potential treatment in glioma. J. Neurosurg. 2019, 132, 168–179. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, G.; Gong, Y.; Zhao, L.; Song, P.; Zhang, H.; Zhang, Y.; Ju, H.; Wang, X.; Wang, B.; et al. IGFBP3 induced by the TGF-beta/EGFRvIII transactivation contributes to the malignant phenotype of glioblastoma. iScience 2023, 26, 106639. [Google Scholar] [CrossRef]
- Liu, X.; Lan, Y.; Zhang, D.; Wang, K.; Wang, Y.; Hua, Z.C. SPRY1 promotes the degradation of uPAR and inhibits uPAR-mediated cell adhesion and proliferation. Am. J. Cancer Res. 2014, 4, 683–697. [Google Scholar]
- Mao, X.H.; Chen, M.; Wang, Y.; Cui, P.G.; Liu, S.B.; Xu, Z.Y. MicroRNA-21 regulates the ERK/NF-kappaB signaling pathway to affect the proliferation, migration, and apoptosis of human melanoma A375 cells by targeting SPRY1, PDCD4, and PTEN. Mol. Carcinog. 2017, 56, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Lo, T.L.; Yusoff, P.; Fong, C.W.; Guo, K.; McCaw, B.J.; Phillips, W.A.; Yang, H.; Wong, E.S.; Leong, H.F.; Zeng, Q.; et al. The ras/mitogen-activated protein kinase pathway inhibitor and likely tumor suppressor proteins, sprouty 1 and sprouty 2 are deregulated in breast cancer. Cancer Res. 2004, 64, 6127–6136. [Google Scholar] [CrossRef] [PubMed]
- Mekkawy, A.H.; Pourgholami, M.H.; Morris, D.L. Human Sprouty1 suppresses growth, migration, and invasion in human breast cancer cells. Tumour Biol. 2014, 35, 5037–5048. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Jing, H.; Liaw, L.; Gower, L.; Vary, C.; Hua, S.; Yang, X. Suppression of Spry1 inhibits triple-negative breast cancer malignancy by decreasing EGF/EGFR mediated mesenchymal phenotype. Sci. Rep. 2016, 6, 23216. [Google Scholar] [CrossRef]
- Chen, Y.; Ning, J.; Cao, W.; Wang, S.; Du, T.; Jiang, J.; Feng, X.; Zhang, B. Research Progress of TXNIP as a Tumor Suppressor Gene Participating in the Metabolic Reprogramming and Oxidative Stress of Cancer Cells in Various Cancers. Front. Oncol. 2020, 10, 568574. [Google Scholar] [CrossRef]
- Vermeulen, K.; Berneman, Z.N.; Van Bockstaele, D.R. Cell cycle and apoptosis. Cell Prolif. 2003, 36, 165–175. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Shah, M.A. Targeting the cell cycle: A new approach to cancer therapy. J. Clin. Oncol. 2005, 23, 9408–9421. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Zhang, W.; Lu, Y.; Li, X.; Zhang, J.; Zheng, L.; Zhang, W.; Lin, C.; Lin, W.; Li, X. CDCA3 promotes cell proliferation by activating the NF-kappaB/cyclin D1 signaling pathway in colorectal cancer. Biochem. Biophys. Res. Commun. 2018, 500, 196–203. [Google Scholar] [CrossRef]
- Liu, J.; Sun, J.; Zhang, Q.; Zeng, Z. shRNA knockdown of DNA helicase ERCC6L expression inhibits human breast cancer growth. Mol. Med. Rep. 2018, 18, 3490–3496. [Google Scholar] [CrossRef]
- Yu, Q.; Pu, S.Y.; Wu, H.; Chen, X.Q.; Jiang, J.J.; Gu, K.S.; He, Y.H.; Kong, Q.P. TICRR Contributes to Tumorigenesis Through Accelerating DNA Replication in Cancers. Front. Oncol. 2019, 9, 516. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Peng, X.; Zhang, Y. Downregulation of cell division cycle-associated protein 7 (CDCA7) suppresses cell proliferation, arrests cell cycle of ovarian cancer, and restrains angiogenesis by modulating enhancer of zeste homolog 2 (EZH2) expression. Bioengineered 2021, 12, 7007–7019. [Google Scholar] [CrossRef]
- Mu, Y.; Zou, H.; Chen, B.; Fan, Y.; Luo, S. FAM83D knockdown regulates proliferation, migration and invasion of colorectal cancer through inhibiting FBXW7/Notch-1 signalling pathway. Biomed. Pharmacother. 2017, 90, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Shang, C.; Zhao, J.; Han, Y.; Liu, J.; Chen, K.; Shi, W. Knockdown of response gene to complement 32 (RGC32) induces apoptosis and inhibits cell growth, migration, and invasion in human lung cancer cells. Mol. Cell Biochem. 2014, 394, 109–118. [Google Scholar] [CrossRef]
- Ding, X.; Duan, H.; Luo, H. Identification of Core Gene Expression Signature and Key Pathways in Colorectal Cancer. Front. Genet. 2020, 11, 45. [Google Scholar] [CrossRef]
- He, S.B.; Yuan, Y.; Wang, L.; Yu, M.J.; Zhu, Y.B.; Zhu, X.G. Effects of cyclin-dependent kinase 8 specific siRNA on the proliferation and apoptosis of colon cancer cells. J. Exp. Clin. Cancer Res. 2011, 30, 109. [Google Scholar] [CrossRef]
- Zhang, X.X.; Yan, Y.Y.; Ma, X.; Xiao, Y.; Lei, C.C.; Wang, Y.M.; Liu, C.; Wang, Q.; Zhang, X.T.; Cheng, W.D.; et al. Discovery of a novel oral type I CDK8 inhibitor against acute myeloid leukemia. Eur. J. Med. Chem. 2023, 251, 115214. [Google Scholar] [CrossRef]
- Cheriyath, V.; Leaman, D.W.; Borden, E.C. Emerging roles of FAM14 family members (G1P3/ISG 6-16 and ISG12/IFI27) in innate immunity and cancer. J. Interferon Cytokine Res. 2011, 31, 173–181. [Google Scholar] [CrossRef]
- Zeng, Y.; Chen, H.Q.; Zhang, Z.; Fan, J.; Li, J.Z.; Zhou, S.M.; Wang, N.; Yan, S.P.; Cao, J.; Liu, J.Y.; et al. IFI44L as a novel epigenetic silencing tumor suppressor promotes apoptosis through JAK/STAT1 pathway during lung carcinogenesis. Environ. Pollut. 2023, 319, 120943. [Google Scholar] [CrossRef]
- Stawowczyk, M.; Van Scoy, S.; Kumar, K.P.; Reich, N.C. The interferon stimulated gene 54 promotes apoptosis. J. Biol. Chem. 2011, 286, 7257–7266. [Google Scholar] [CrossRef]
- Mullan, P.B.; Hosey, A.M.; Buckley, N.E.; Quinn, J.E.; Kennedy, R.D.; Johnston, P.G.; Harkin, D.P. The 2,5 oligoadenylate synthetase/RNaseL pathway is a novel effector of BRCA1- and interferon-gamma-mediated apoptosis. Oncogene 2005, 24, 5492–5501. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, P.; Wang, X.; Wu, Y.; Xia, K.; Mu, X.; Xuan, Q.; Xiao, J.; He, Y.; Liu, W.; et al. piR-36249 and DHX36 together inhibit testicular cancer cells progression by upregulating OAS2. Noncoding RNA Res. 2023, 8, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, J.; Sambade, M.J.; Sather, S.; Moschos, S.J.; Tan, A.C.; Winges, A.; DeRyckere, D.; Carson, C.C.; Trembath, D.G.; Tentler, J.J.; et al. MERTK receptor tyrosine kinase is a therapeutic target in melanoma. J. Clin. Investig. 2013, 123, 2257–2267. [Google Scholar] [CrossRef] [PubMed]
- Sinik, L.; Minson, K.A.; Tentler, J.J.; Carrico, J.; Bagby, S.M.; Robinson, W.A.; Kami, R.; Burstyn-Cohen, T.; Eckhardt, S.G.; Wang, X.; et al. Inhibition of MERTK Promotes Suppression of Tumor Growth in BRAF Mutant and BRAF Wild-Type Melanoma. Mol. Cancer Ther. 2019, 18, 278–288. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, D.; Peng, H.; Chen, X.; Han, X.; Yu, J.; Wang, W.; Liang, L.; Liu, Z.; Zheng, Y.; et al. Epigenetically upregulated oncoprotein PLCE1 drives esophageal carcinoma angiogenesis and proliferation via activating the PI-PLCepsilon-NF-kappaB signaling pathway and VEGF-C/Bcl-2 expression. Mol. Cancer 2019, 18, 1. [Google Scholar] [CrossRef]
- Yue, Q.Y.; Zhao, W.; Tan, Y.; Deng, X.L.; Zhang, Y.H. PLCE1 inhibits apoptosis of non-small cell lung cancer via promoting PTEN methylation. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6211–6216. [Google Scholar]
- Geering, B. Death-associated protein kinase 2: Regulator of apoptosis, autophagy and inflammation. Int. J. Biochem. Cell Biol. 2015, 65, 151–154. [Google Scholar] [CrossRef]
- Plenchette, S.; Cheung, H.H.; Fong, W.G.; LaCasse, E.C.; Korneluk, R.G. The role of XAF1 in cancer. Curr. Opin. Investig. Drugs 2007, 8, 469–476. [Google Scholar]
- Borhani, S.; Gartel, A.L. FOXM1: A potential therapeutic target in human solid cancers. Expert. Opin. Ther. Targets 2020, 24, 205–217. [Google Scholar] [CrossRef]
- Khan, M.A.; Khan, P.; Ahmad, A.; Fatima, M.; Nasser, M.W. FOXM1: A small fox that makes more tracks for cancer progression and metastasis. Semin. Cancer Biol. 2023, 92, 1–15. [Google Scholar] [CrossRef]
- Li, G.; Li, W.; Angelastro, J.M.; Greene, L.A.; Liu, D.X. Identification of a novel DNA binding site and a transcriptional target for activating transcription factor 5 in c6 glioma and mcf-7 breast cancer cells. Mol. Cancer Res. 2009, 7, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Melber, A.; Haynes, C.M. UPR(mt) regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018, 28, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Madarampalli, B.; Yuan, Y.; Liu, D.; Lengel, K.; Xu, Y.; Li, G.; Yang, J.; Liu, X.; Lu, Z.; Liu, D.X. ATF5 Connects the Pericentriolar Materials to the Proximal End of the Mother Centriole. Cell 2015, 162, 580–592. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q.; Greene, L.A. Dpep Inhibits Cancer Cell Growth and Survival via Shared and Context-Dependent Transcriptome Perturbations. Cancers 2023, 15, 5318. https://doi.org/10.3390/cancers15225318
Zhou Q, Greene LA. Dpep Inhibits Cancer Cell Growth and Survival via Shared and Context-Dependent Transcriptome Perturbations. Cancers. 2023; 15(22):5318. https://doi.org/10.3390/cancers15225318
Chicago/Turabian StyleZhou, Qing, and Lloyd A. Greene. 2023. "Dpep Inhibits Cancer Cell Growth and Survival via Shared and Context-Dependent Transcriptome Perturbations" Cancers 15, no. 22: 5318. https://doi.org/10.3390/cancers15225318