
ASPP2 Is Phosphorylated by CDK1 during Mitosis and Required for Pancreatic Cancer Cell Proliferation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection, and Transduction
2.2. Anti-Mitotic Drugs and Transfection Agents
2.3. Expression Constructs and RNA Interference
- shASPP2-A: GTCTAGTAAATAGGATCATTT.
- shASPP2-B: GAAATCCAGAATCCATATTTA.
2.4. Phos-Tag, Western Blotting, and Peptide Blocking
2.5. Recombinant Protein Purification and In Vitro Kinase Assay
2.6. Antibodies
2.7. Cell Proliferation
2.8. Immunofluorescence Staining and Confocal Microscopy
2.9. RNA Isolation, Reverse Transcription, and RT-PCR
2.10. RNA-Seq
2.11. Xenograft Mouse Model
2.12. Statistical Analysis
3. Results
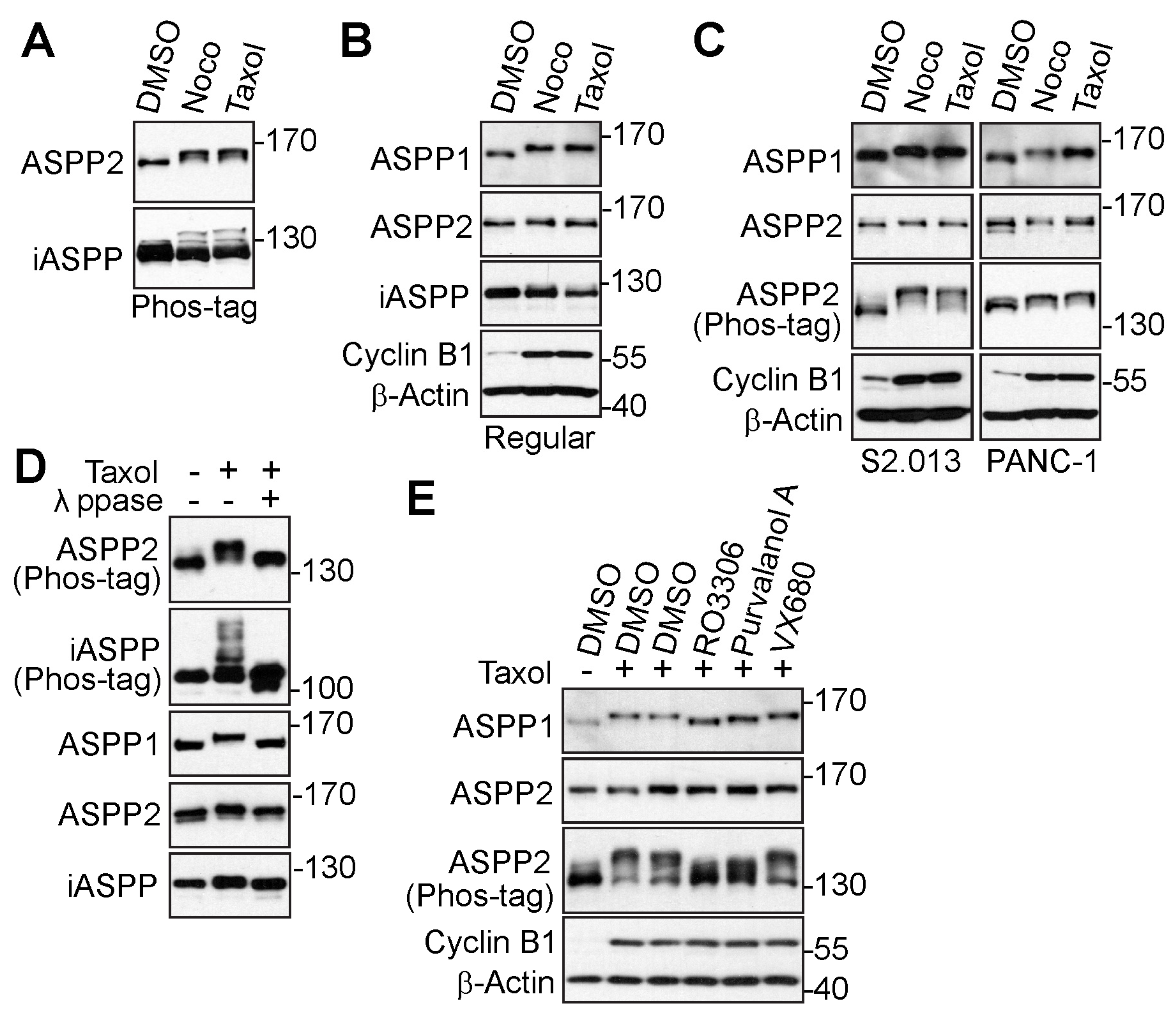
3.1. The ASPP Family of Proteins Are Phosphorylated during Mitosis
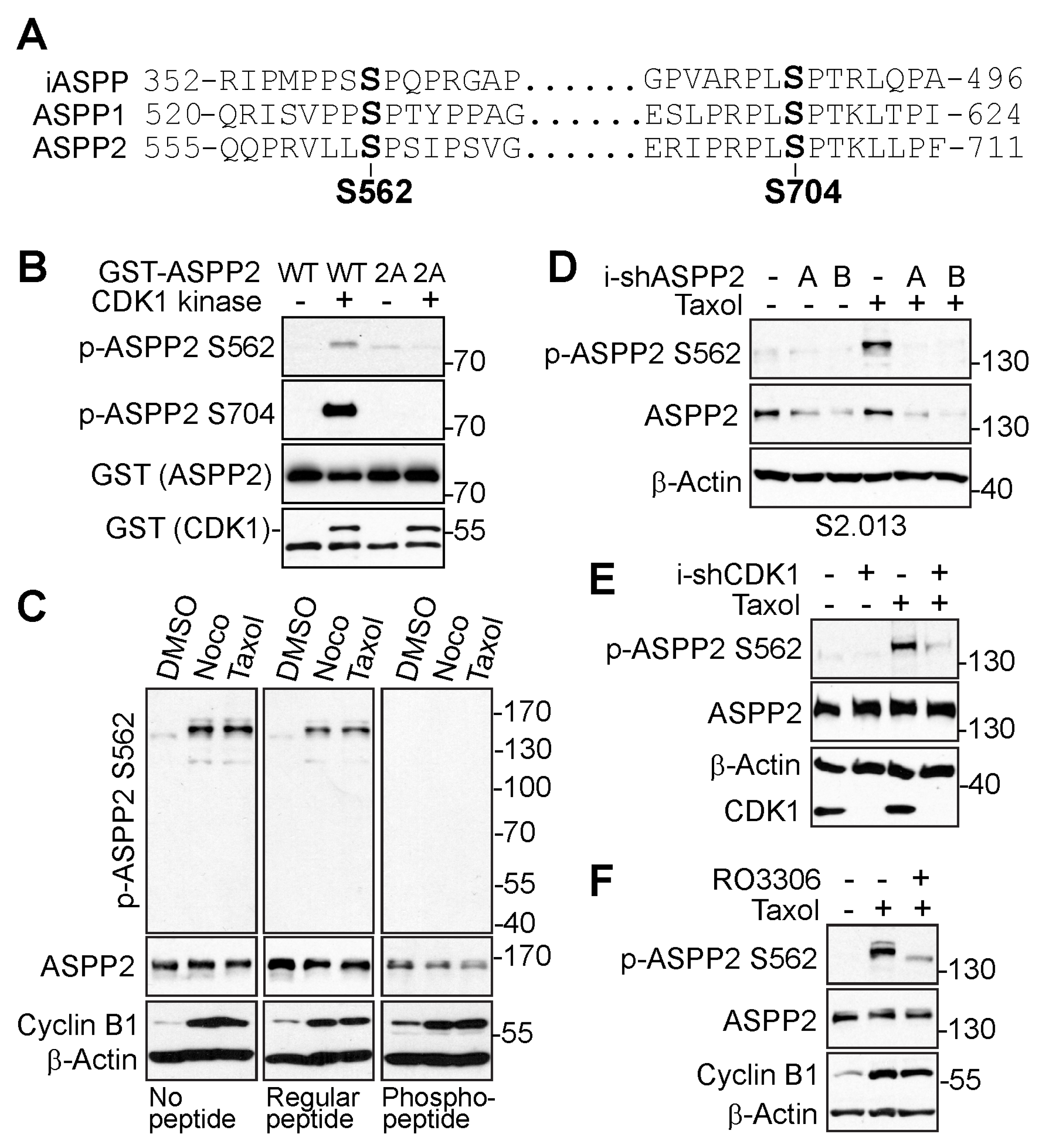
3.2. ASPP2 Is Phosphorylated at S562 and S704 by CDK1 during Mitosis
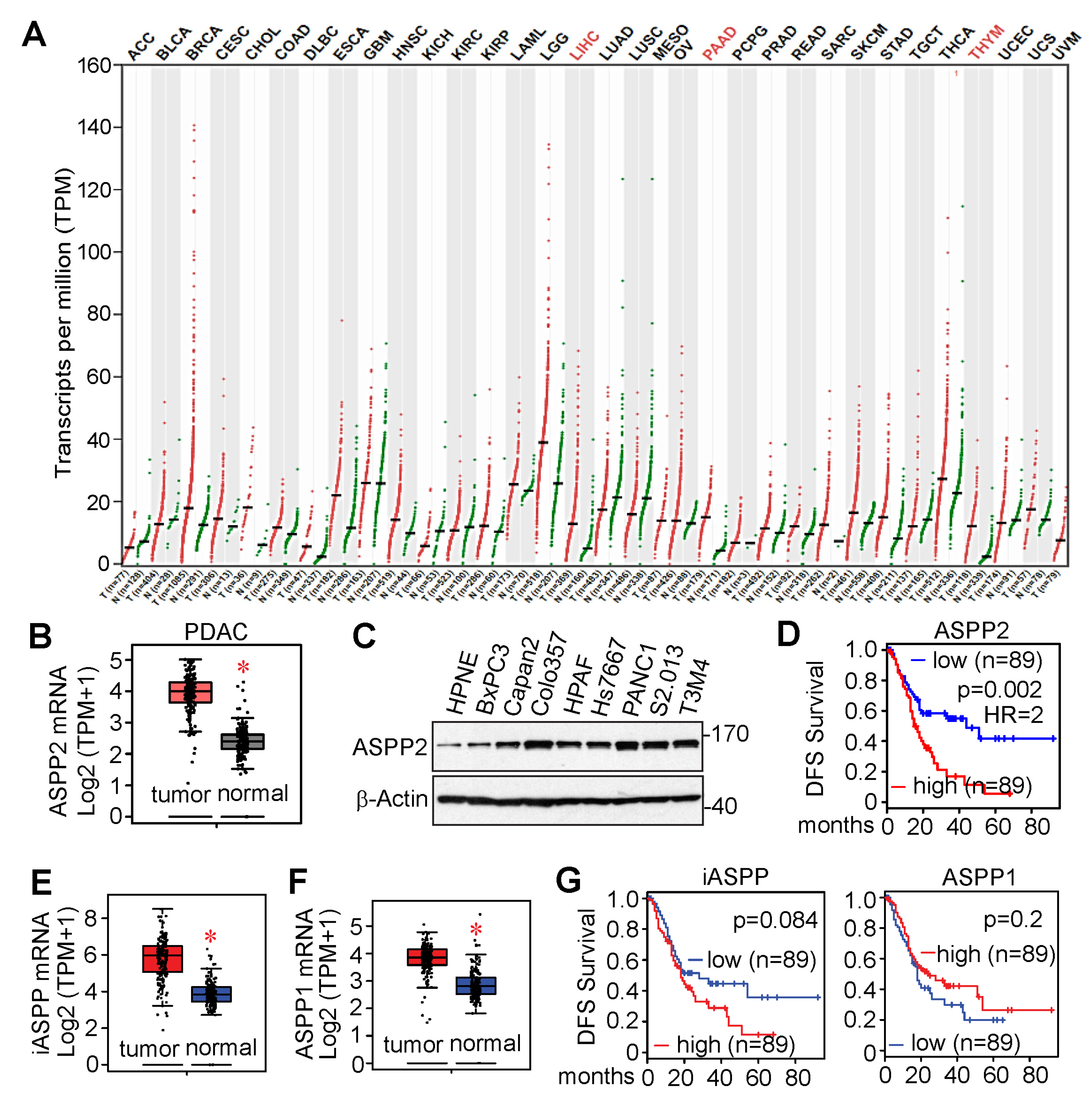
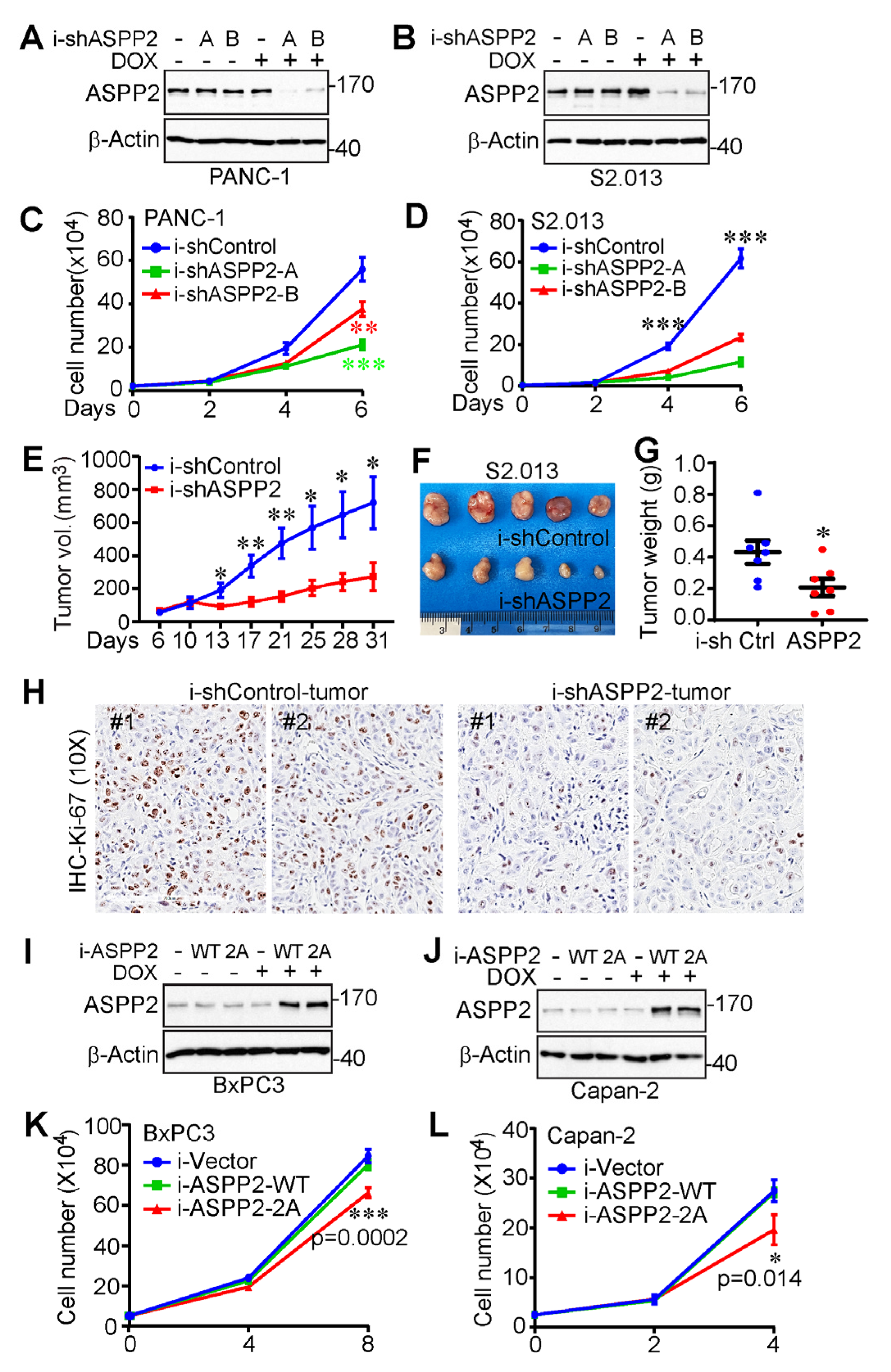
3.3. ASPP2 Is Overexpressed and Required for Pancreatic Cancer Growth
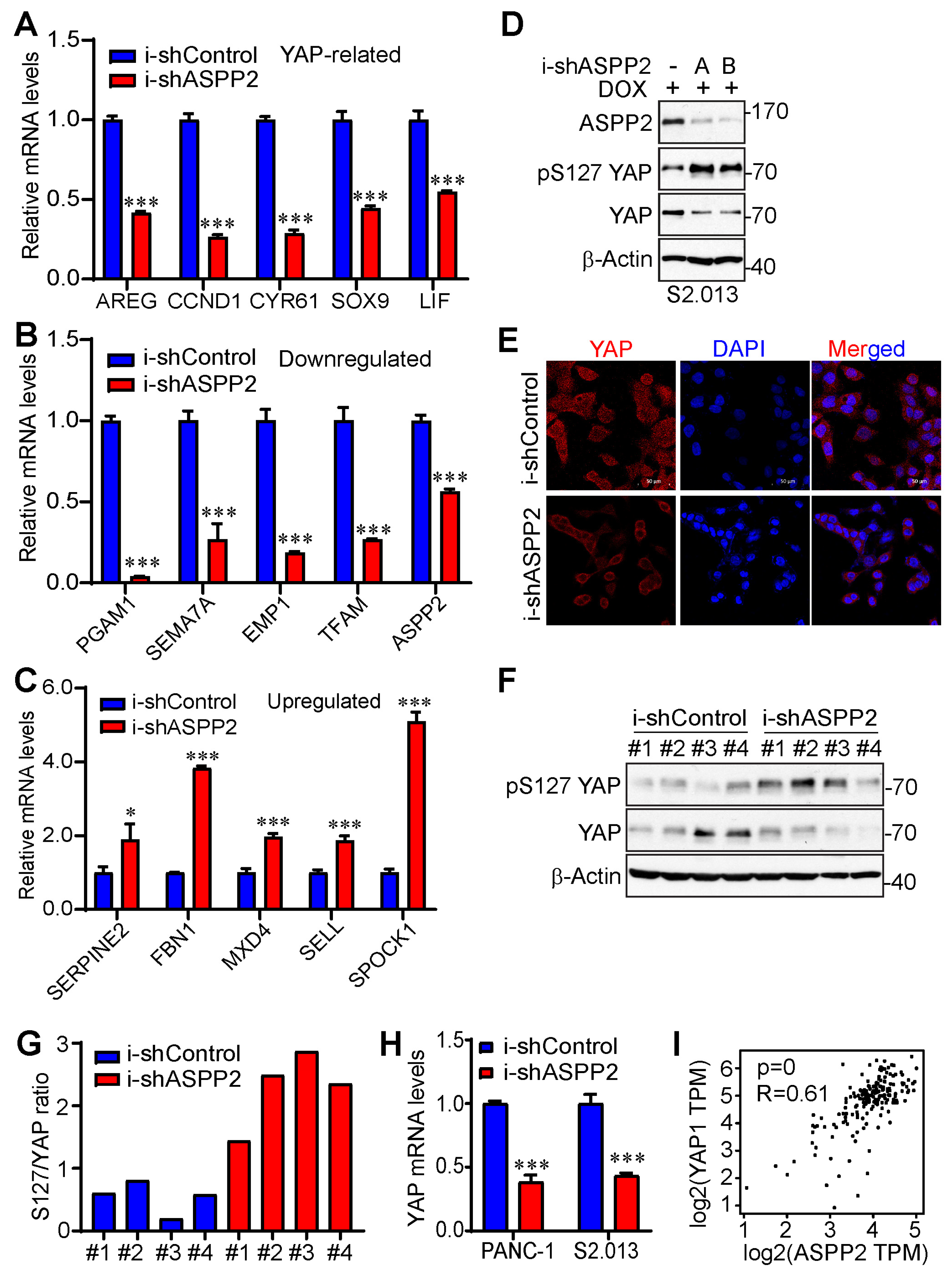
3.4. ASPP2 Regulates the Expression of YAP-Associated Genes
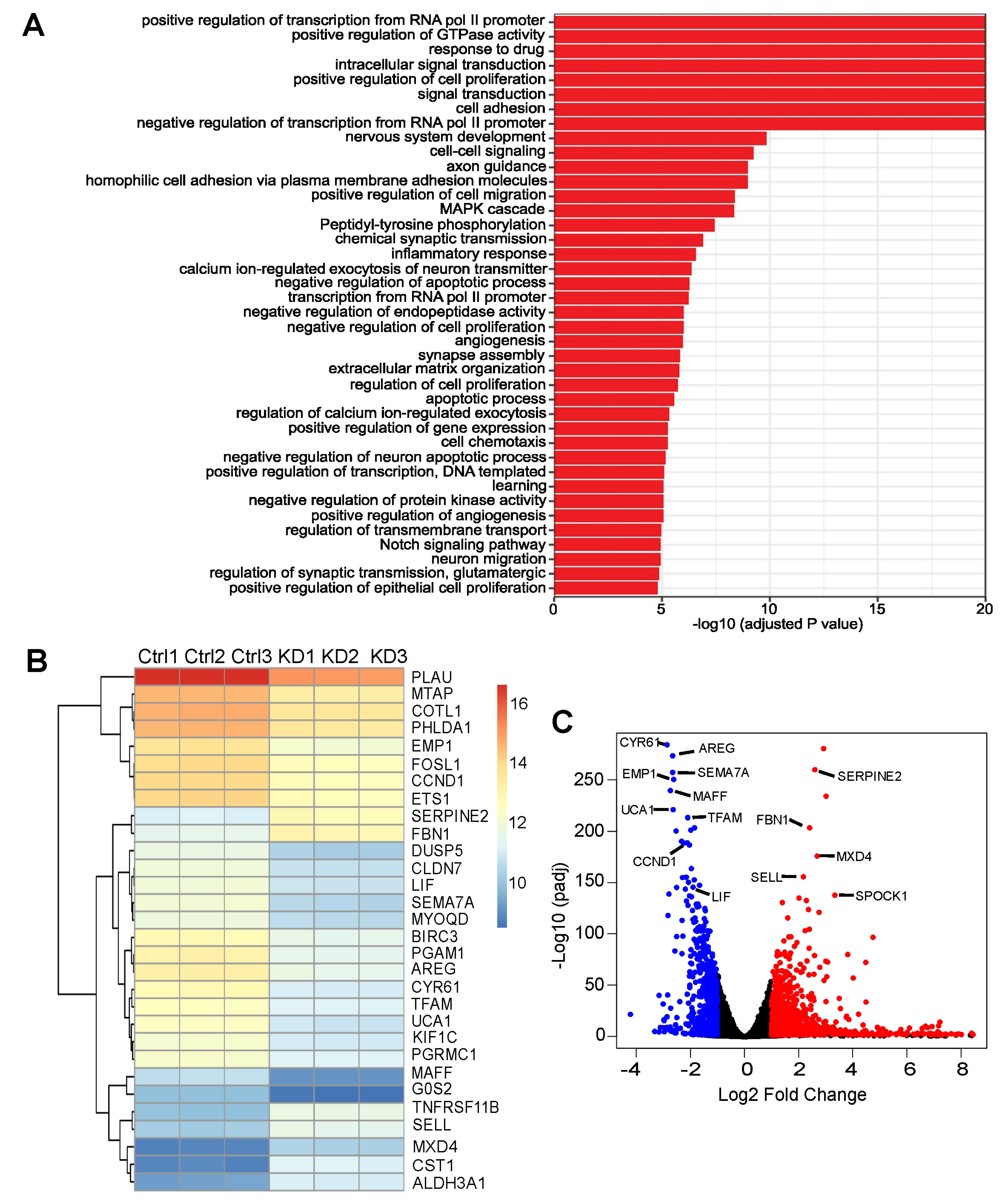
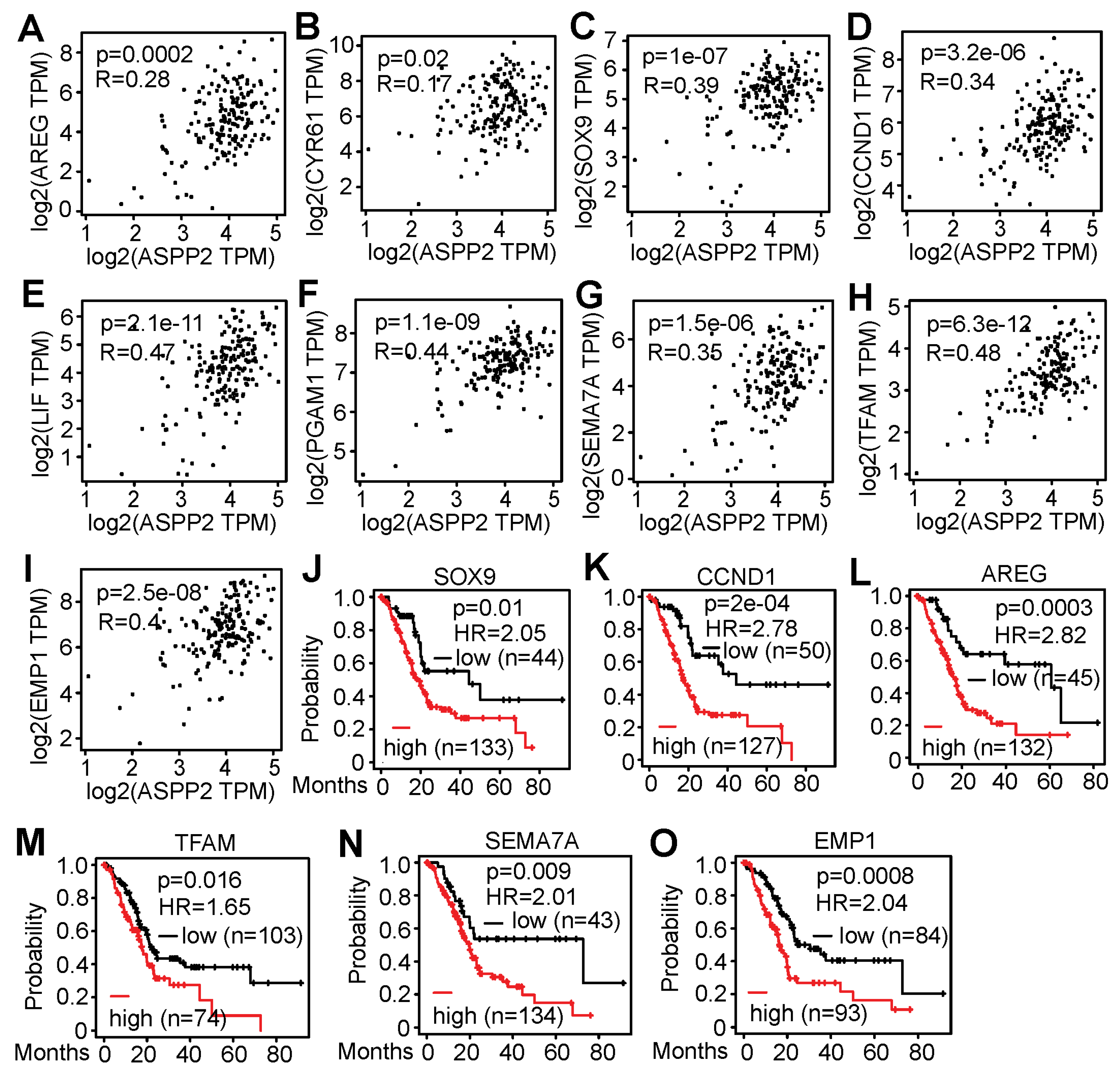
3.5. RNA-Seq Identified Genes Correlating with ASPP2 Expression and Implying Negative Patient Outcome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Lyssiotis, C.A.; Pasca di Magliano, M.; Maitra, A. Pancreatic cancer: Advances and challenges. Cell 2023, 186, 1729–1754. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Kolbeinsson, H.M.; Chandana, S.; Wright, G.P.; Chung, M. Pancreatic Cancer: A Review of Current Treatment and Novel Therapies. J. Investig. Surg. 2023, 36, 2129884. [Google Scholar] [CrossRef] [PubMed]
- Min, H.-Y.; Lee, H.-Y. Molecular targeted therapy for anticancer treatment. Exp. Mol. Med. 2022, 54, 1670–1694. [Google Scholar] [CrossRef]
- Helps, N.R.; Barker, H.M.; Elledge, S.J.; Cohen, P.T. Protein phosphatase 1 interacts with p53BP2, a protein which binds to the tumour suppressor p53. FEBS Lett. 1995, 377, 295–300. [Google Scholar]
- Naumovski, L.; Cleary, M.L. The p53-binding protein 53BP2 also interacts with Bc12 and impedes cell cycle progression at G2/M. Mol. Cell. Biol. 1996, 16, 3884–3892. [Google Scholar] [CrossRef]
- Bergamaschi, D.; Samuels, Y.; O’Neil, N.J.; Trigiante, G.; Crook, T.; Hsieh, J.-K.; O’Connor, D.J.; Zhong, S.; Campargue, I.; Tomlinson, M.L.; et al. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat. Genet. 2003, 33, 162–167. [Google Scholar] [CrossRef]
- Ahn, J.; Byeon, I.L.; Byeon, C.H.; Gronenborn, A.M. Insight into the structural basis of pro- and antiapoptotic p53 modulation by ASPP proteins. J. Biol. Chem. 2009, 284, 13812–13822. [Google Scholar] [CrossRef] [PubMed]
- Iwabuchi, K.; Li, B.; Massa, H.F.; Trask, B.J.; Date, T.; Fields, S. Stimulation of p53-mediated transcriptional activation by the p53-binding proteins, 53BP1 and 53BP2. J. Biol. Chem. 1998, 273, 26061–26068. [Google Scholar] [CrossRef]
- Bergamaschi, D.; Samuels, Y.; Jin, B.; Duraisingham, S.; Crook, T.; Lu, X. ASPP1 and ASPP2: Common activators of p53 family members. Mol. Cell. Biol. 2004, 24, 1341–1350. [Google Scholar] [CrossRef]
- Samuels-Lev, Y.; O’Connor, D.J.; Bergamaschi, D.; Trigiante, G.; Hsieh, J.K.; Zhong, S.; Campargue, I.; Naumovski, L.; Crook, T.; Lu, X. ASPP proteins specifically stimulate the apoptotic function of p53. Mol. Cell 2001, 8, 781–794. [Google Scholar] [CrossRef]
- Braithwaite, A.W.; Del Sal, G.; Lu, X. Some p53-binding proteins that can function as arbiters of life and death. Cell Death Differ. 2006, 13, 984–993. [Google Scholar] [CrossRef]
- Skene-Arnold, T.D.; Luu, H.A.; Uhrig, R.G.; De Wever, V.; Nimick, M.; Maynes, J.; Fong, A.; James, M.N.; Trinkle-Mulcahy, L.; Moorhead, G.B.; et al. Molecular mechanisms underlying the interaction of protein phosphatase-1c with ASPP proteins. Biochem. J. 2013, 449, 649–659. [Google Scholar] [CrossRef]
- Wang, J.; Jia, C.; Gao, Q.; Zhang, J.; Gu, X. iASPP regulates neurite development by interacting with Spectrin proteins. Front. Mol. Neurosci. 2023, 16, 1154770. [Google Scholar] [CrossRef] [PubMed]
- Bertran, M.T.; Mouilleron, S.; Zhou, Y.; Bajaj, R.; Uliana, F.; Kumar, G.S.; van Drogen, A.; Lee, R.; Banerjee, J.J.; Hauri, S.; et al. ASPP proteins discriminate between PP1 catalytic subunits through their SH3 domain and the PP1 C-tail. Nat. Commun. 2019, 10, 771. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, Y.; Gao, K.; Wang, Y.; Jin, X.; Wei, Y.; Saiyin, H.; Wang, D.; Peng, J.; Ma, J.; et al. ASPP1/2-PP1 complexes are required for chromosome segregation and kinetochore-microtubule attachments. Oncotarget 2015, 6, 41550–41565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Wei, Y.; Ma, J.; Peng, J.; Wumaier, R.; Shen, S.; Zhang, P.; Yu, L. The tumor suppressor proteins ASPP1 and ASPP2 interact with C-Nap1 and regulate centrosome linker reassembly. Biochem. Biophys. Res. Commun. 2015, 458, 494–500. [Google Scholar] [CrossRef]
- Gao, K.; Zhang, Y.; Shi, Q.; Zhang, J.; Zhang, L.; Sun, H.; Jiao, D.; Zhao, X.; Tao, H.; Wei, Y.; et al. iASPP–PP1 complex is required for cytokinetic abscission by controlling CEP55 dephosphorylation. Cell Death Dis. 2018, 9, 528. [Google Scholar] [CrossRef]
- Mangon, A.; Salaün, D.; Bouali, M.L.; Kuzmić, M.; Quitard, S.; Thuault, S.; Isnardon, D.; Audebert, S.; Puech, P.H.; Verdier-Pinard, P.; et al. iASPP contributes to cell cortex rigidity, mitotic cell rounding, and spindle positioning. J. Cell Biol. 2021, 220, e202012002. [Google Scholar] [CrossRef]
- Xiao, Y.; Chen, Y.; Peng, A.; Dong, J. The phosphatase CTDSPL2 is phosphorylated in mitosis and a target for restraining tumor growth and motility in pancreatic cancer. Cancer Lett. 2022, 526, 53–65. [Google Scholar] [CrossRef]
- Vives, V.; Su, J.; Zhong, S.; Ratnayaka, I.; Slee, E.; Goldin, R.; Lu, X. ASPP2 is a haploinsufficient tumor suppressor that cooperates with p53 to suppress tumor growth. Genes Dev. 2006, 20, 1262–1267. [Google Scholar] [CrossRef]
- Kampa, K.M.; Acoba, J.D.; Chen, D.; Gay, J.; Lee, H.; Beemer, K.; Padiernos, E.; Boonmark, N.; Zhu, Z.; Fan, A.C.; et al. Apoptosis-stimulating protein of p53 (ASPP2) heterozygous mice are tumor-prone and have attenuated cellular damage-response thresholds. Proc. Natl. Acad. Sci. USA 2009, 106, 4390–4395. [Google Scholar] [CrossRef]
- Wang, S.; Kou, B.; Chai, M.; Gao, Y.; Lin, X.; Yin, L.; Chen, D.; Liu, X. Knockout of ASPP2 promotes DEN-induced hepatocarcinogenesis via the NF-κB pathway in mice. Cancer Gene Ther. 2022, 29, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Jiang, Y.; Song, S.; Jing, W.; Yang, H.; Zhao, L.; Chen, Y.; Tang, Q.; Li, X.; Zhang, L.; et al. ASPP2 suppresses tumour growth and stemness characteristics in HCC by inhibiting Warburg effect via WNT/β-catenin/HK2 axis. J. Cell. Mol. Med. 2023, 27, 659–671. [Google Scholar] [CrossRef]
- Li, H.; Wang, X.; Zhang, C.; Cheng, Y.; Yu, M.; Zhao, K.; Ge, W.; Cai, A.; Zhang, Y.; Han, F.; et al. HDAC1-induced epigenetic silencing of ASPP2 promotes cell motility, tumour growth and drug resistance in renal cell carcinoma. Cancer Lett. 2018, 432, 121–131. [Google Scholar] [CrossRef]
- Wu, T.; Song, H.; Xie, D.; Zhao, B.; Xu, H.; Wu, C.; Hua, K.; Deng, Y.; Ji, C.; Hu, J.; et al. Silencing of ASPP2 promotes the proliferation, migration and invasion of triple-negative breast cancer cells via the PI3K/AKT pathway. Int. J. Oncol. 2018, 52, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Bian, Q.; Zhang, Y.J.; Shao, C.H.; Li, G.; Liu, A.A.; Jing, W.; Liu, R.; Zhou, Y.Q.; Jin, G.; et al. Downregulation of ASPP2 in pancreatic cancer cells contributes to increased resistance to gemcitabine through autophagy activation. Mol. Cancer 2015, 14, 177. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Godin-Heymann, N.; Dan Wang, X.; Bergamaschi, D.; Llanos, S.; Lu, X. ASPP1 and ASPP2 bind active RAS, potentiate RAS signalling and enhance p53 activity in cancer cells. Cell Death Differ. 2013, 20, 525–534. [Google Scholar] [CrossRef]
- Cong, W.; Hirose, T.; Harita, Y.; Yamashita, A.; Mizuno, K.; Hirano, H.; Ohno, S. ASPP2 regulates epithelial cell polarity through the PAR complex. Curr. Biol. CB 2010, 20, 1408–1414. [Google Scholar] [CrossRef]
- Gen, Y.; Yasui, K.; Kitaichi, T.; Iwai, N.; Terasaki, K.; Dohi, O.; Hashimoto, H.; Fukui, H.; Inada, Y.; Fukui, A.; et al. ASPP2 suppresses invasion and TGF-β1-induced epithelial-mesenchymal transition by inhibiting Smad7 degradation mediated by E3 ubiquitin ligase ITCH in gastric cancer. Cancer Lett. 2017, 398, 52–61. [Google Scholar] [CrossRef]
- Wang, Y.; Bu, F.; Royer, C.; Serres, S.; Larkin, J.R.; Soto, M.S.; Sibson, N.R.; Salter, V.; Fritzsche, F.; Turnquist, C.; et al. ASPP2 controls epithelial plasticity and inhibits metastasis through β-catenin-dependent regulation of ZEB1. Nat. Cell Biol. 2014, 16, 1092–1104. [Google Scholar] [CrossRef] [PubMed]
- Katz, C.; Benyamini, H.; Rotem, S.; Lebendiker, M.; Danieli, T.; Iosub, A.; Refaely, H.; Dines, M.; Bronner, V.; Bravman, T.; et al. Molecular basis of the interaction between the antiapoptotic Bcl-2 family proteins and the proapoptotic protein ASPP2. Proc. Natl. Acad. Sci. USA 2008, 105, 12277–12282. [Google Scholar] [CrossRef] [PubMed]
- Linn, H.; Ermekova, K.S.; Rentschler, S.; Sparks, A.B.; Kay, B.K.; Sudol, M. Using molecular repertoires to identify high-affinity peptide ligands of the WW domain of human and mouse YAP. Biol. Chem. 1997, 378, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Espanel, X.; Sudol, M. Yes-associated protein and p53-binding protein-2 interact through their WW and SH3 domains. J. Biol. Chem. 2001, 276, 14514–14523. [Google Scholar] [CrossRef] [PubMed]
- Royer, C.; Koch, S.; Qin, X.; Zak, J.; Buti, L.; Dudziec, E.; Zhong, S.; Ratnayaka, I.; Srinivas, S.; Lu, X. ASPP2 links the apical lateral polarity complex to the regulation of YAP activity in epithelial cells. PLoS ONE 2014, 9, e111384. [Google Scholar] [CrossRef]
- Liu, C.Y.; Lv, X.; Li, T.; Xu, Y.; Zhou, X.; Zhao, S.; Xiong, Y.; Lei, Q.Y.; Guan, K.L. PP1 cooperates with ASPP2 to dephosphorylate and activate TAZ. J. Biol. Chem. 2011, 286, 5558–5566. [Google Scholar] [CrossRef]
- Xiao, Y.; Dong, J. The Hippo Signaling Pathway in Cancer: A Cell Cycle Perspective. Cancers 2021, 13, 6214. [Google Scholar] [CrossRef]
- Piccolo, S.; Panciera, T.; Contessotto, P.; Cordenonsi, M. YAP/TAZ as master regulators in cancer: Modulation, function and therapeutic approaches. Nat. Cancer 2023, 4, 9–26. [Google Scholar] [CrossRef]
- Godin-Heymann, N.; Wang, Y.; Slee, E.; Lu, X. Phosphorylation of ASPP2 by RAS/MAPK pathway is critical for its full pro-apoptotic function. PLoS ONE 2013, 8, e82022. [Google Scholar] [CrossRef]
- Lee, K.M.; Nguyen, C.; Ulrich, A.B.; Pour, P.M.; Ouellette, M.M. Immortalization with telomerase of the Nestin-positive cells of the human pancreas. Biochem. Biophys. Res. Commun. 2003, 301, 1038–1044. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, L.; Purohit, V.; Shukla, S.K.; Chen, X.; Yu, F.; Fu, K.; Chen, Y.; Solheim, J.; Singh, P.K.; et al. Active YAP promotes pancreatic cancer cell motility, invasion and tumorigenesis in a mitotic phosphorylation-dependent manner through LPAR3. Oncotarget 2015, 6, 36019–36031. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Chen, Y.; Ji, M.; Dong, J. KIBRA regulates Hippo signaling activity via interactions with large tumor suppressor kinases. J. Biol. Chem. 2011, 286, 7788–7796. [Google Scholar] [CrossRef]
- Yang, S.; Ji, M.; Zhang, L.; Chen, Y.; Wennmann, D.O.; Kremerskothen, J.; Dong, J. Phosphorylation of KIBRA by the extracellular signal-regulated kinase (ERK)-ribosomal S6 kinase (RSK) cascade modulates cell proliferation and migration. Cell. Signal. 2014, 26, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zeng, Y.; Cui, L.; Chen, X.; Stauffer, S.; Wang, Z.; Yu, F.; Lele, S.M.; Talmon, G.A.; Black, A.R.; et al. Zyxin promotes colon cancer tumorigenesis in a mitotic phosphorylation-dependent manner and through CDK8-mediated YAP activation. Proc. Natl. Acad. Sci. USA 2018, 115, E6760–E6769. [Google Scholar] [CrossRef]
- Stauffer, S.; Zeng, Y.; Zhou, J.; Chen, X.; Chen, Y.; Dong, J. CDK1-mediated mitotic phosphorylation of PBK is involved in cytokinesis and inhibits its oncogenic activity. Cell. Signal. 2017, 39, 74–83. [Google Scholar] [CrossRef]
- Stauffer, S.; Zeng, Y.; Santos, M.; Zhou, J.; Chen, Y.; Dong, J. Cyclin-dependent kinase 1-mediated AMPK phosphorylation regulates chromosome alignment and mitotic progression. J. Cell Sci. 2019, 132, jcs236000. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Kong, Y.; Shi, W.; Kuss, M.; Liao, K.; Hu, G.; Xiao, P.; Sankarasubramanian, J.; Guda, C.; Wang, X.; et al. Exosomes derived from differentiated human ADMSC with the Schwann cell phenotype modulate peripheral nerve-related cellular functions. Bioact. Mater. 2022, 14, 61–75. [Google Scholar] [CrossRef]
- Nigg, E.A. Cellular substrates of p34(cdc2) and its companion cyclin-dependent kinases. Trends Cell Biol. 1993, 3, 296–301. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Byrne, D.P.; Burgess, S.G.; Bormann, J.; Baković, J.; Huang, Y.; Zhyvoloup, A.; Yu, B.Y.K.; Peak-Chew, S.; Tran, T.; et al. Covalent Aurora A regulation by the metabolic integrator coenzyme A. Redox Biol. 2020, 28, 101318. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.B.; Yang, Y.; Liang, W.Q.; Zhang, K.C.; Chen, L.; Zhang, Z.T. Leukemia inhibitory factor promotes gastric cancer cell proliferation, migration, and invasion via the LIFR-Hippo-YAP pathway. Ann. N. Y. Acad. Sci. 2021, 1484, 74–89. [Google Scholar] [CrossRef]
- Zhang, H.; Pasolli, H.A.; Fuchs, E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc. Natl. Acad. Sci. USA 2011, 108, 2270–2275. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, J.Y.; Yu, M.; Overholtzer, M.; Smolen, G.A.; Wang, R.; Brugge, J.S.; Dyson, N.J.; Haber, D.A. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat. Cell Biol. 2009, 11, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Murakami, H.; Fujii, M.; Ishiguro, F.; Tanaka, I.; Kondo, Y.; Akatsuka, S.; Toyokuni, S.; Yokoi, K.; Osada, H.; et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 2012, 31, 5117–5122. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Z.; Yu, X.; Huang, X.; Liu, Z.; Chai, Y.; Yang, L.; Wang, Q.; Li, M.; Zhao, J.; et al. Unbalanced YAP–SOX9 circuit drives stemness and malignant progression in esophageal squamous cell carcinoma. Oncogene 2019, 38, 2042–2055. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, A.M.; Ludwig, R.L.; Vousden, K.H. Cytoplasmic ASPP1 inhibits apoptosis through the control of YAP. Genes Dev. 2010, 24, 2430–2439. [Google Scholar] [CrossRef]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef]
- Escobar-Hoyos, L.F.; Penson, A.; Kannan, R.; Cho, H.; Pan, C.-H.; Singh, R.K.; Apken, L.H.; Hobbs, G.A.; Luo, R.; Lecomte, N.; et al. Altered RNA Splicing by Mutant p53 Activates Oncogenic RAS Signaling in Pancreatic Cancer. Cancer Cell 2020, 38, 198–211.e8. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Y.; Chen, Y.; Chen, J.; Dong, J. ASPP2 Is Phosphorylated by CDK1 during Mitosis and Required for Pancreatic Cancer Cell Proliferation. Cancers 2023, 15, 5424. https://doi.org/10.3390/cancers15225424
Xiao Y, Chen Y, Chen J, Dong J. ASPP2 Is Phosphorylated by CDK1 during Mitosis and Required for Pancreatic Cancer Cell Proliferation. Cancers. 2023; 15(22):5424. https://doi.org/10.3390/cancers15225424
Chicago/Turabian StyleXiao, Yi, Yuanhong Chen, Jianan Chen, and Jixin Dong. 2023. "ASPP2 Is Phosphorylated by CDK1 during Mitosis and Required for Pancreatic Cancer Cell Proliferation" Cancers 15, no. 22: 5424. https://doi.org/10.3390/cancers15225424