
Revealing Prognostic and Immunotherapy-Sensitive Characteristics of a Novel Cuproptosis-Related LncRNA Model in Hepatocellular Carcinoma Patients by Genomic Analysis
 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
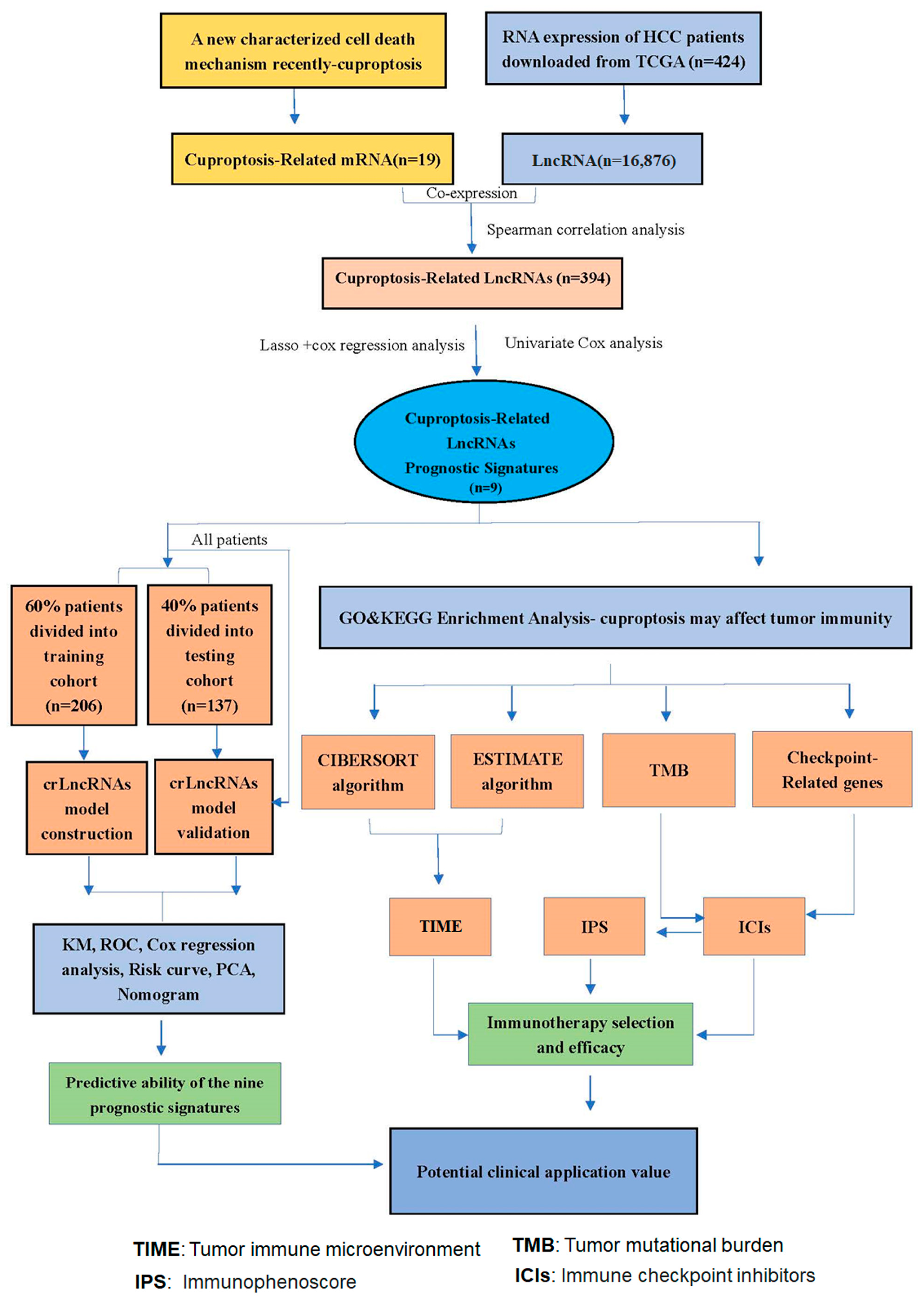
2.1. Data Acquisition and Screening
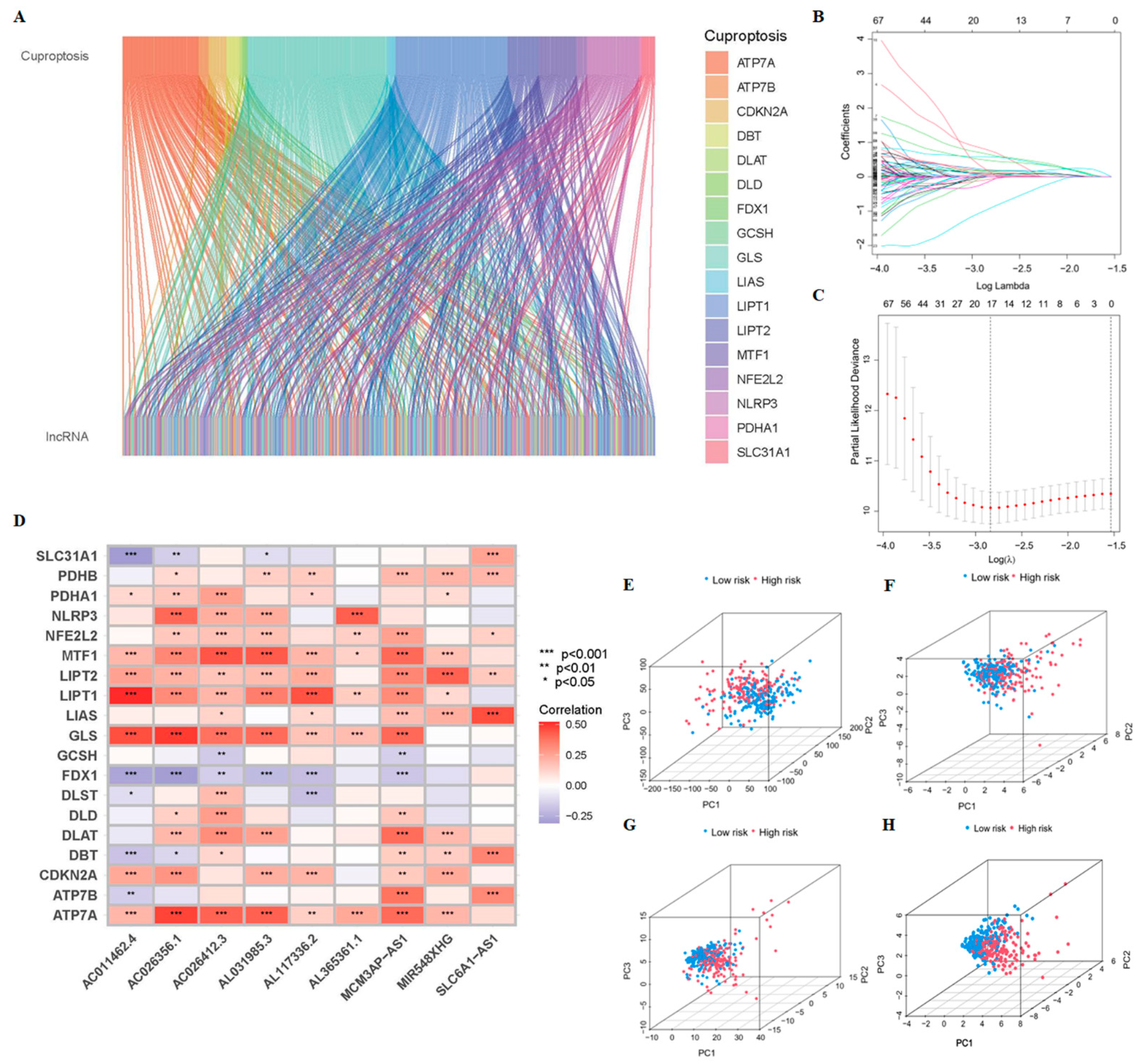
2.2. Identification of CrLncRNAs
2.3. Construction of the Prognostic Model of CrLncRNAs
2.4. Confirmation of the Prognostic Signatures of 9-CrLncRNA
2.5. Nomogram Construction and Assessment
2.6. Enrichment Analysis
2.7. Evaluation of Immune Cell Infiltration and Immune Microenvironment
2.8. Assessment of the Options of Specific ICIs Treatment
2.9. GSVA Analysis
2.10. Prediction of Targeted MiRNAs
2.11. Copy Number Variation (CNV) Analysis
2.12. Drug Sensitivity Analysis
2.13. HCC Tissue Specimens
2.14. Real-Time Quantitative PCR Analysis
2.15. Statistical Analysis
3. Results
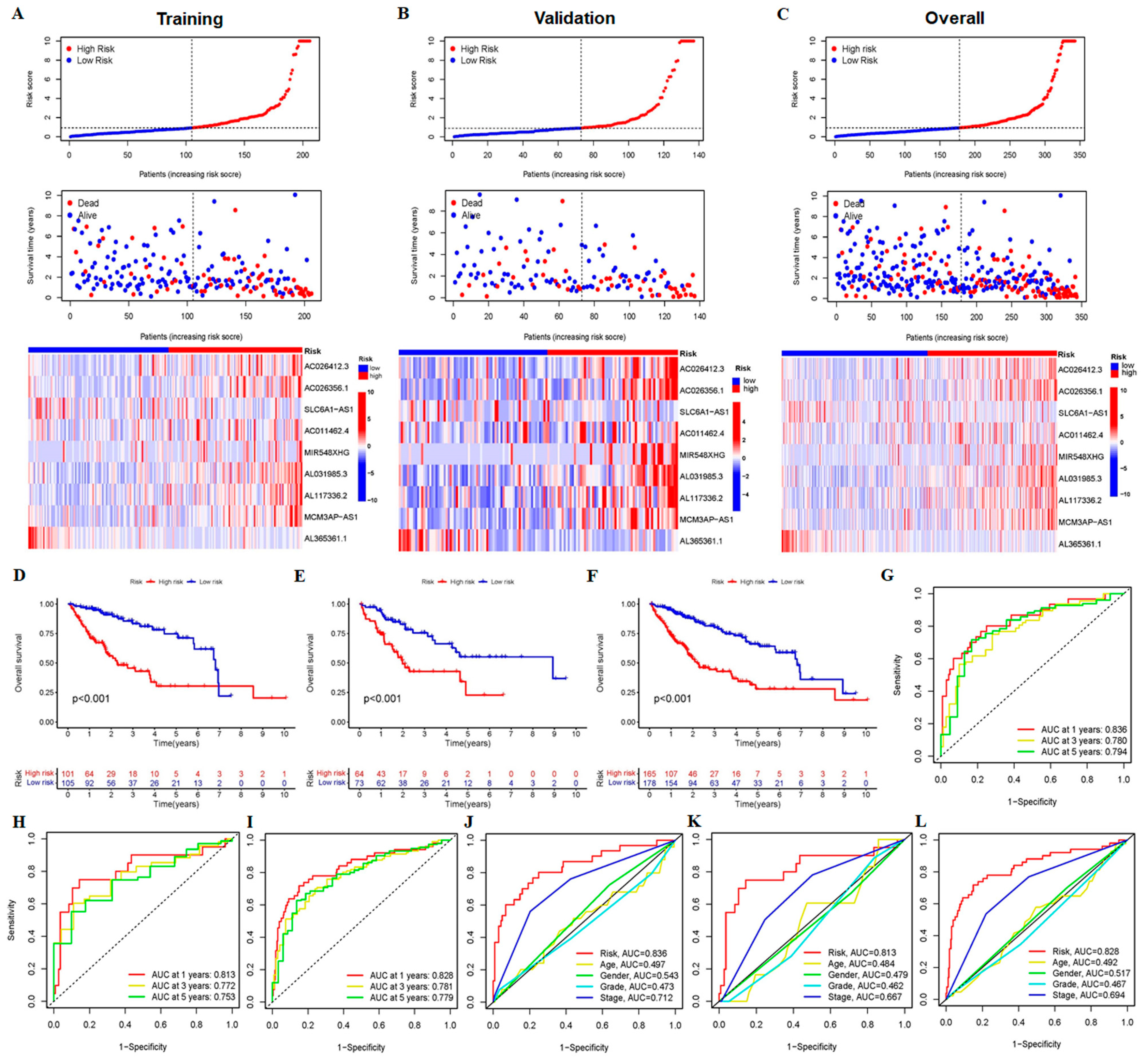
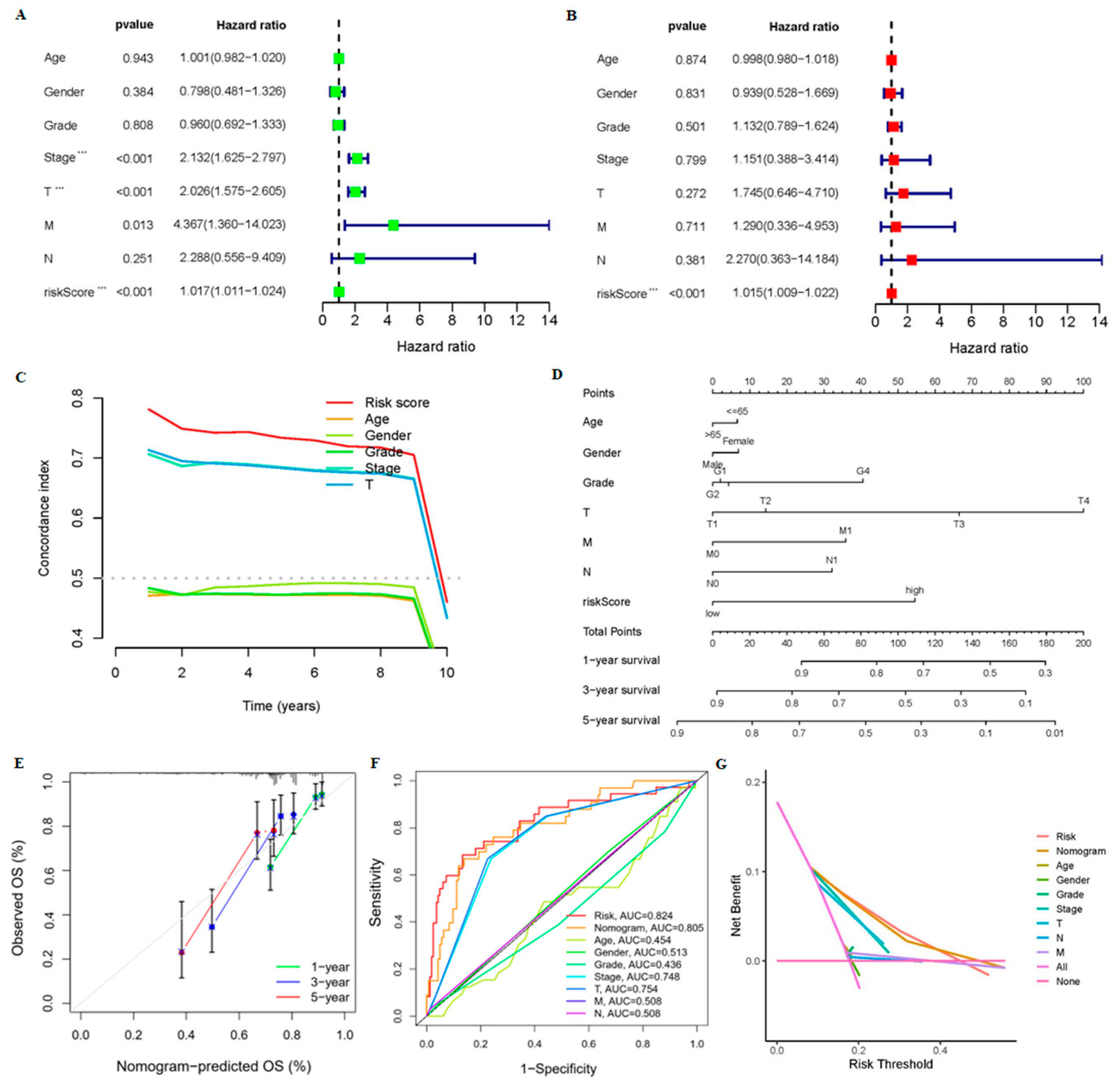
3.1. Construction and Validation of 9-CrLncRNA Prognostic Model in HCC
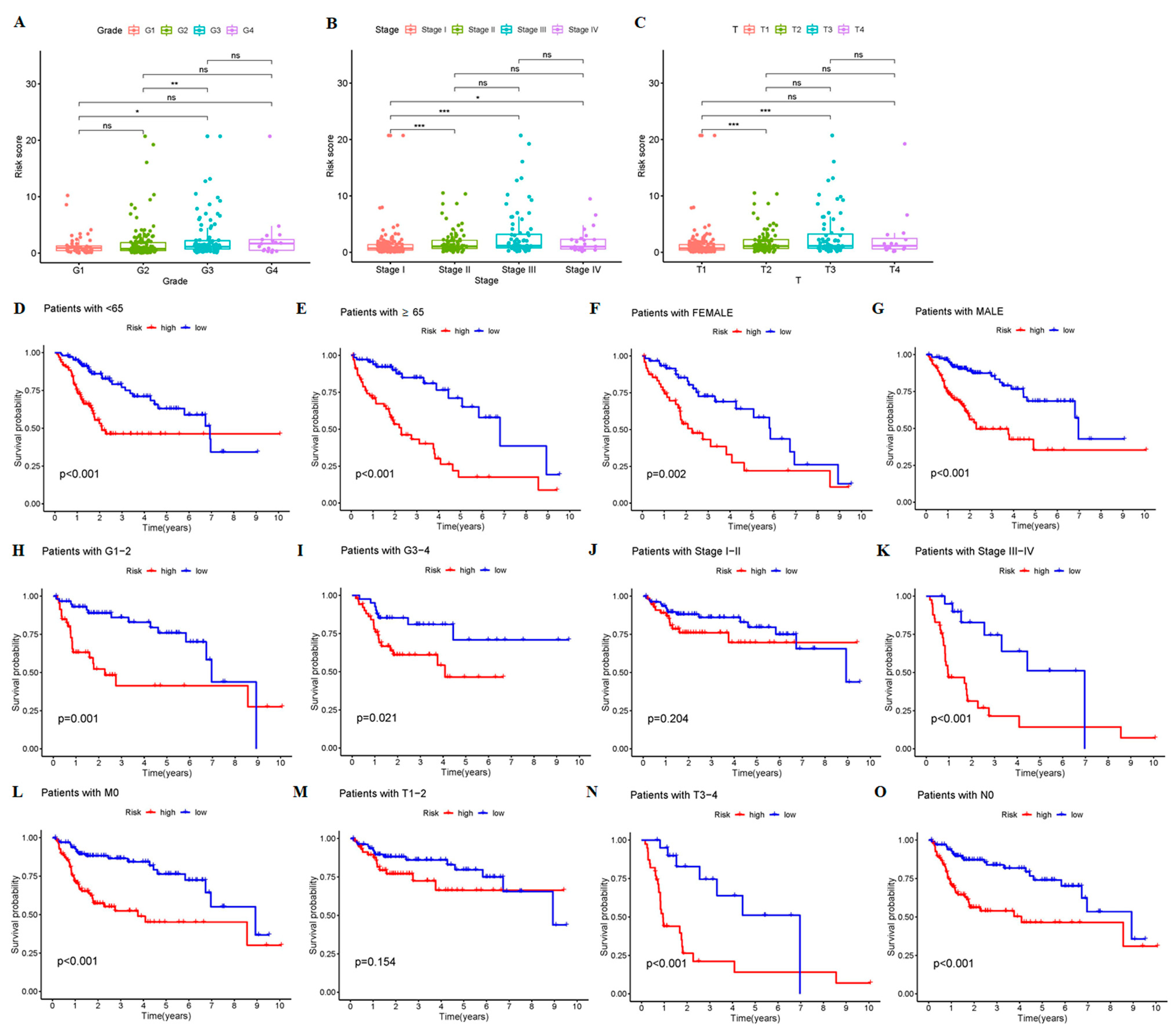
3.2. The 9-CrLncRNA Model Has High Reliability in Predicting the OS and Application Value in Clinical
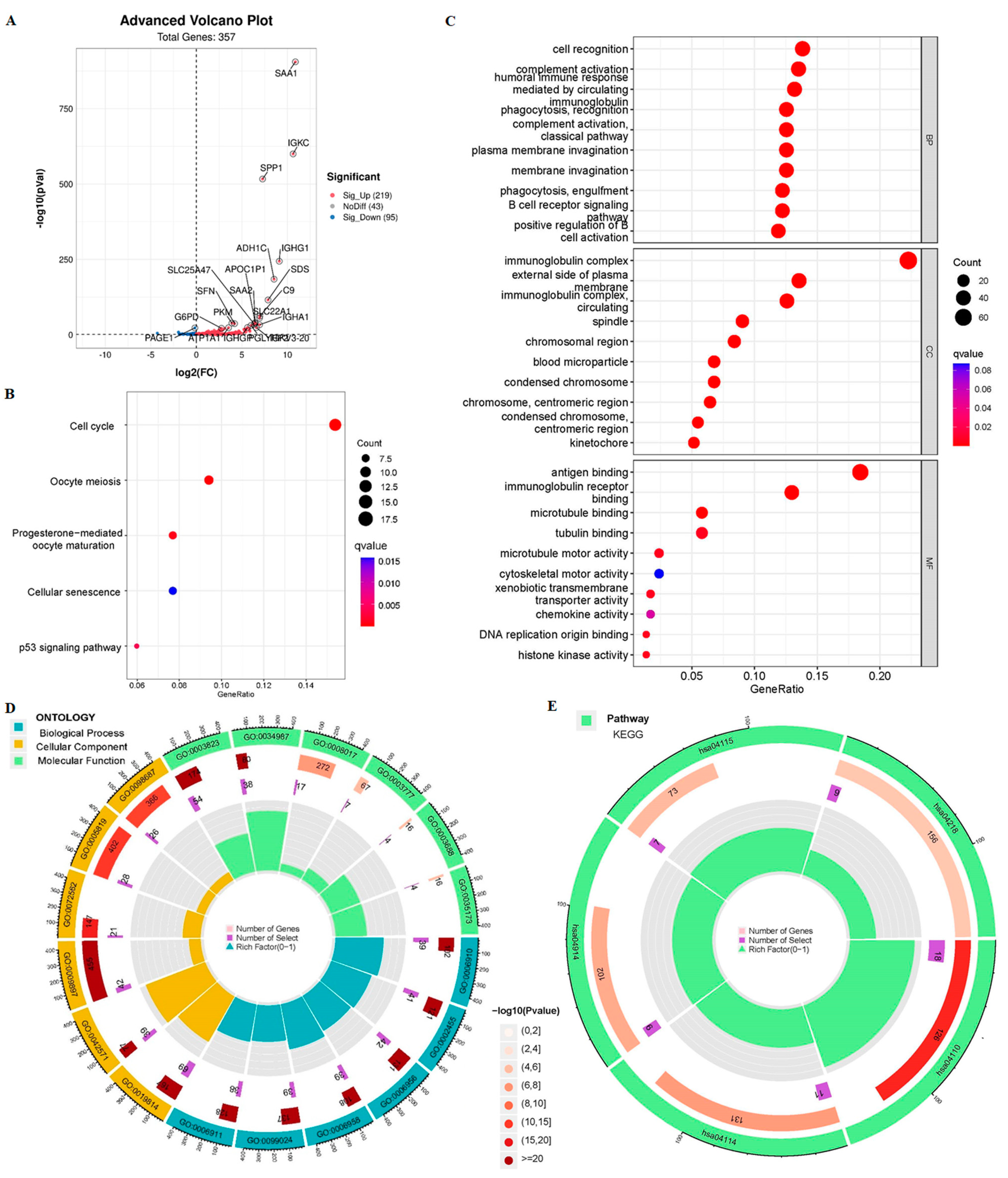
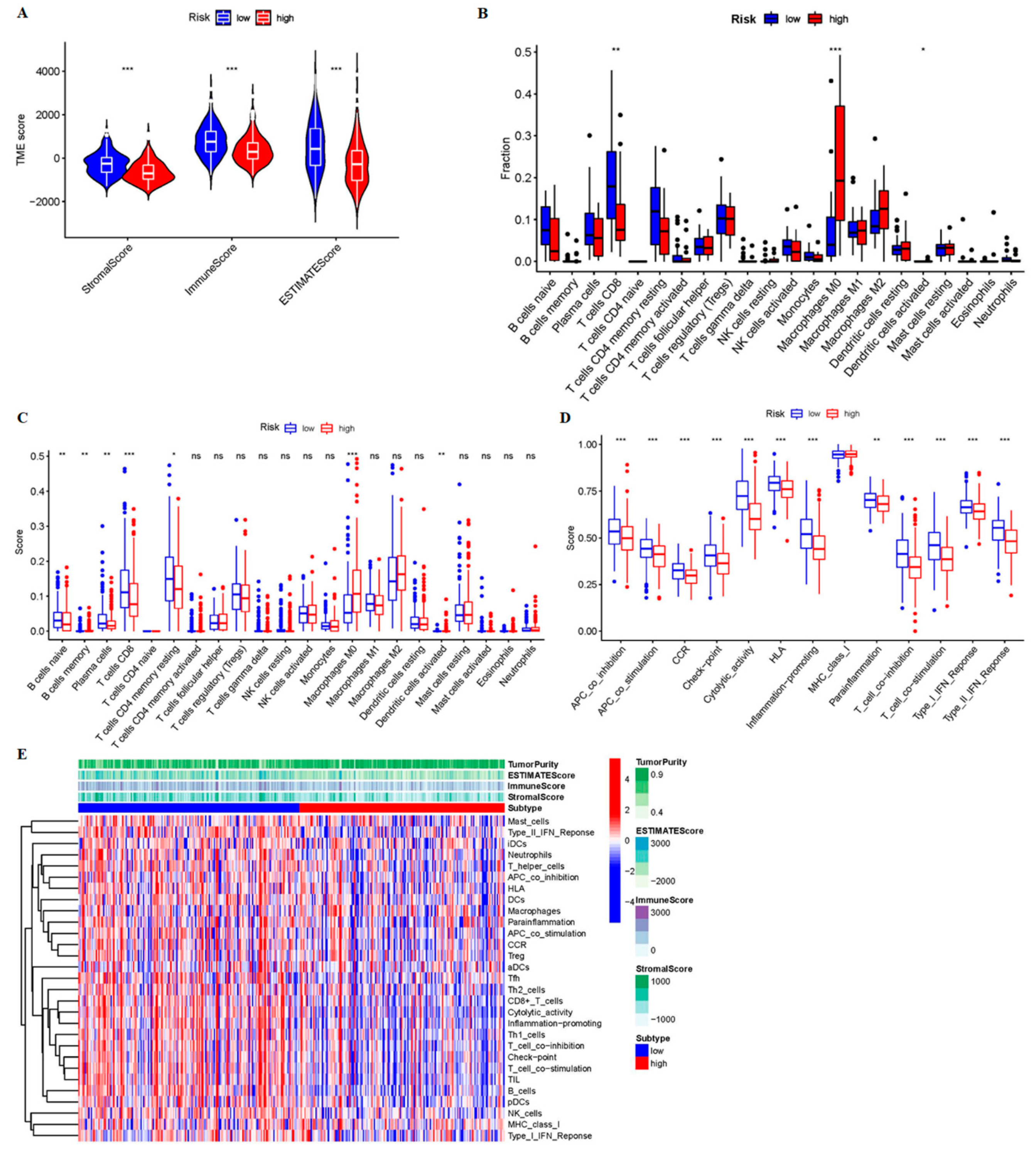
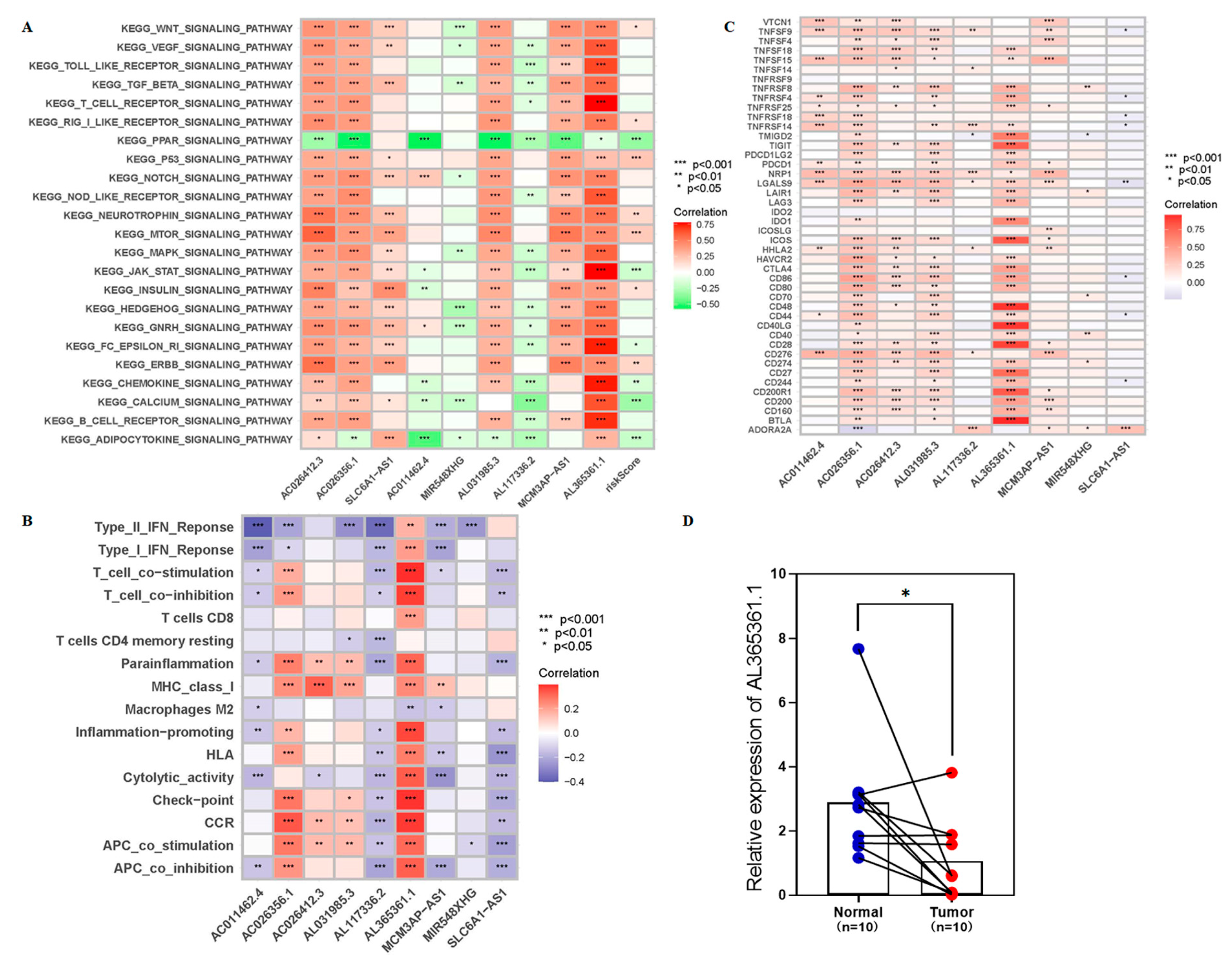
3.3. The 9-CrLncRNA May Be Closely Related to Tumor Immunity
3.4. The Low-Risk Group Was More Likely to Have a Higher Immune Response
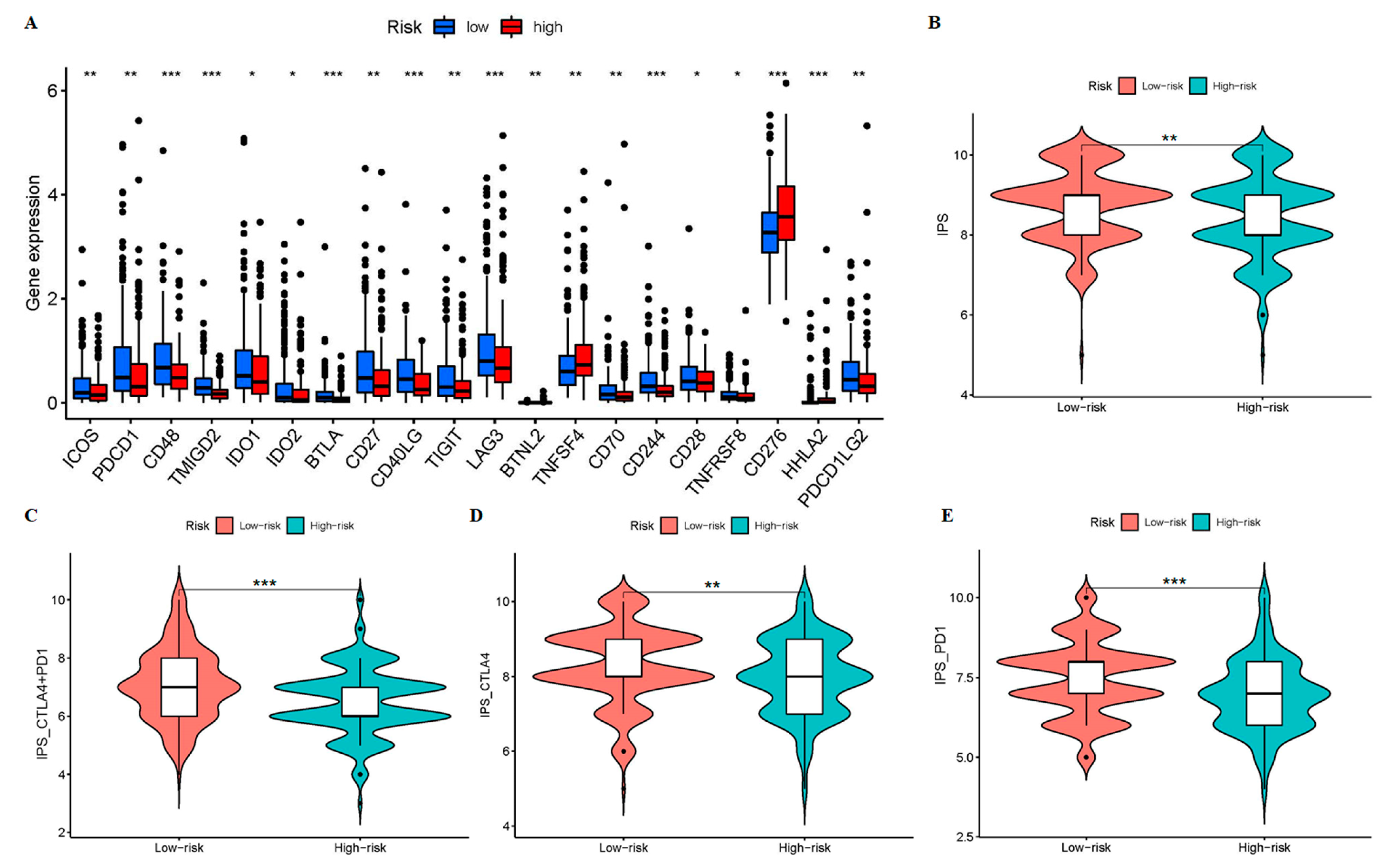
3.5. Patients in the Low-Risk Group May Have a Higher Sensitivity to ICIs
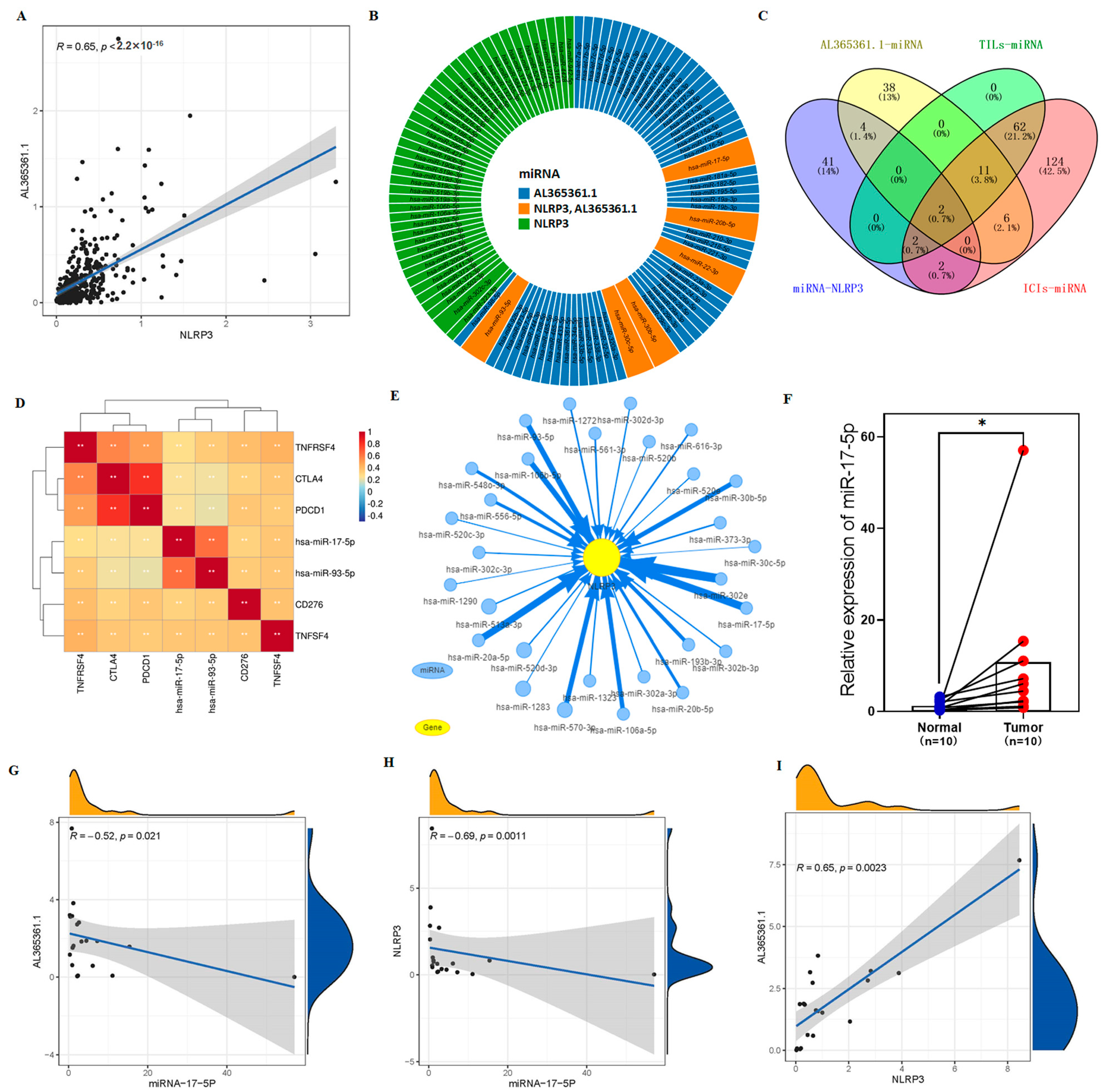
3.6. AL365361.1 May Be the Main Tumor Immune-Related Molecule of the 9-CrLncRNA Model
3.7. Sensitive Drugs for NLRP3 Mutation May Improve OS in High-Risk Group
3.8. The Potential Regulation Axis between AL365361.1 and NLRP3
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | Area under the time-dependent ROC curve |
| crLncRNAs | Cuproptosis-related LncRNAs |
| C-index | Concordance index |
| CNV | Copy number variation |
| CCR | Chemokines and chemokine receptors |
| DEGs | Differentially expressed genes |
| EMT | Epithelial-mesenchymal transition |
| DCA | Decision curve analysis |
| GSCA | Gene Set Cancer Analysis |
| GDSC | Genomics of Drug Sensitivity |
| GO | Gene Ontology |
| GSVA | Gene set variation analysis |
| HCC | Hepatocellular carcinoma |
| ICIs | Immune checkpoint inhibitors |
| IPS | Immunophenoscore |
| K-M | Kaplan-Meier |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LncRNA | Long noncoding RNA |
| LASSO | Least absolute shrinkage and selection operator |
| OS | Overall survival |
| ORR | Overall response rate |
| PFS | Progression free survival |
| PCA | Principal component analysis |
| ROC | Receiver operating characteristic |
| ssGSEA | Single sample gene set enrichment analysis |
| TCGA | The Cancer Genome Atlas |
| TIME | Tumor immune microenvironment |
| TILs | Tumor infiltrating lymphocytes |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.C.S.; Huang, J.L.W.; George, J.; Huang, J.; Leung, C.; Eslam, M.; Chan, H.L.Y.; Ng, S.C. The changing epidemiology of liver diseases in the Asia-Pacific region. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Papatheodoridis, G.; Sun, J.; Innes, H.; Toyoda, H.; Xie, Q.; Mo, S.; Sypsa, V.; Guha, I.N.; Kumada, T.; et al. aMAP risk score predicts hepatocellular carcinoma development in patients with chronic hepatitis. J. Hepatol. 2020, 73, 1368–1378. [Google Scholar] [CrossRef]
- Huang, A.; Yang, X.R.; Chung, W.Y.; Dennison, A.R.; Zhou, J. Targeted therapy for hepatocellular carcinoma. Signal Transduct. Target. Ther. 2020, 5, 146. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab as Second-Line Therapy in Patients with Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Shek, D.; Read, S.A.; Nagrial, A.; Carlino, M.S.; Gao, B.; George, J.; Ahlenstiel, G. Immune-Checkpoint Inhibitors for Advanced Hepatocellular Carcinoma: A Synopsis of Response Rates. Oncologist 2021, 26, e1216–e1225. [Google Scholar] [CrossRef]
- Genova, C.; Dellepiane, C.; Carrega, P.; Sommariva, S.; Ferlazzo, G.; Paolo, P.; Rosaria, G.; Gilberto, F.; Simona, C.; Michela, C. Therapeutic Implications of Tumor Microenvironment in Lung Cancer: Focus on Immune Checkpoint Blockade. Front. Immunol. 2021, 12, 799455. [Google Scholar] [CrossRef]
- Offin, M.; Rizvi, H.; Tenet, M.; Ni, A.; Sanchez-Vega, F.; Li, B.T.; Drilon, A.; Kris, M.G.; Rudin, C.M.; Schultz, N.; et al. Tumor Mutation Burden and Efficacy of EGFR-Tyrosine Kinase Inhibitors in Patients with EGFR-Mutant Lung Cancers. Clin. Cancer Res. 2019, 25, 1063–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinter, M.; Jain, R.K.; Duda, D.G. The Current Landscape of Immune Checkpoint Blockade in Hepatocellular Carcinoma: A Review. JAMA Oncol. 2021, 7, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, Z.; Li, F. Survival prediction and response to immune checkpoint inhibitors: A prognostic immune signature for hepatocellular carcinoma. Transl. Oncol. 2021, 14, 100957. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Hsu, C.; Chan, S.L.; Choo, S.P.; Kudo, M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J. Hepatol. 2020, 72, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhang, S.; Yang, Z.; Lin, H.; Zhu, J.; Liu, L.; Wang, W.; Liu, S.; Liu, W.; Ma, Y.; et al. Self-Recognition of an Inducible Host lncRNA by RIG-I Feedback Restricts Innate Immune Response. Cell 2018, 173, 906–919.e913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; Pan, B.; Zhang, X.; Wang, Z.; Qiu, J.; Wang, X.; Tang, N. Lipopolysaccharide facilitates immune escape of hepatocellular carcinoma cells via m6A modification of lncRNA MIR155HG to upregulate PD-L1 expression. Cell Biol. Toxicol. 2022, 38, 1159–1173. [Google Scholar] [CrossRef]
- Zhang, Z.; Shang, J.; Hu, B.; Shi, H.; Cao, Y.; Li, J.; Jiao, T.; Zhang, W.; Lu, S. Prognosis and Tumour Immune Microenvironment of Patients with Hepatocellular Carcinoma by a Novel Pyroptosis-Related lncRNA Signature. Front. Immunol. 2022, 13, 836576. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, W.; Wang, Y.; Wan, T.; Hu, B.; Li, C.; Ge, X.; Lu, S. Construction and Validation of a Ferroptosis-Related lncRNA Signature as a Novel Biomarker for Prognosis, Immunotherapy and Targeted Therapy in Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2022, 10, 792676. [Google Scholar] [CrossRef]
- Ge, E.J.; Bush, A.I.; Casini, A.; Cobine, P.A.; Cross, J.R.; DeNicola, G.M.; Dou, Q.P.; Franz, K.J.; Gohil, V.M.; Gupta, S.; et al. Connecting copper and cancer: From transition metal signalling to metalloplasia. Nat. Rev. Cancer. 2022, 22, 102–113. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Oliveri, V. Selective Targeting of Cancer Cells by Copper Ionophores: An Overview. Front. Mol. Biosci. 2022, 9, 841814. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Singh, M.K.; Singh, T.B.; Bhartiya, S.K.; Singh, S.P.; Shukla, V.K. Heavy and trace metals in carcinoma of the gallbladder. World J. Surg. 2013, 37, 2641–2646. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Huo, Z.; Qi, X.; Zuo, T.; Wu, Z. Copper-induced tumor cell death mechanisms and antitumor theragnostic applications of copper complexes. Nanomedicine 2022, 17, 303–324. [Google Scholar] [CrossRef]
- Li, S.R.; Bu, L.L.; Cai, L. Cuproptosis: Lipoylated TCA cycle proteins-mediated novel cell death pathway. Signal Transduct. Target. Ther. 2022, 7, 158. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kroemer, G. Cuproptosis: A copper-triggered modality of mitochondrial cell death. Cell Res. 2022, 32, 417–418. [Google Scholar] [CrossRef]
- Wang, J.; Luo, C.; Shan, C.; You, Q.; Lu, J.; Elf, S.; Zhou, Y.; Wen, Y.; Vinkenborg, J.L.; Fan, J.; et al. Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat. Chem. 2015, 7, 968–979. [Google Scholar] [CrossRef] [Green Version]
- Kahlson, M.A.; Dixon, S.J. Copper-induced cell death. Science 2022, 375, 1231–1232. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [Green Version]
- Olguín-Contreras, L.F.; Mendler, A.N.; Popowicz, G.; Hu, B.; Noessner, E. Double Strike Approach for Tumor Attack: Engineering T Cells Using a CD40L:CD28 Chimeric Co-Stimulatory Switch Protein for Enhanced Tumor Targeting in Adoptive Cell Therapy. Front. Immunol. 2021, 12, 750478. [Google Scholar] [CrossRef]
- Duraiswamy, J.; Turrini, R.; Minasyan, A.; Barras, D.; Crespo, I.; Grimm, A.J.; Casado, J.; Genolet, R.; Benedetti, F.; Wicky, A.; et al. Myeloid antigen-presenting cell niches sustain antitumor T cells and license PD-1 blockade via CD28 costimulation. Cancer Cell 2021, 39, 1623–1642.e1620. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Qu, G.; Wang, D.; Xu, W.; Guo, W. Comprehensive Analysis of the Correlation Between Pyroptosis-Related LncRNAs and Tumor Microenvironment, Prognosis, and Immune Infiltration in Hepatocellular Carcinoma. Front. Genet. 2022, 13, 867627. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lu, Z.; Li, L.; Ma, M.; Long, F.; Wu, R.; Huang, L.; Chou, J.; Yang, K.; Zhang, Y.; et al. Identification and Validation of Ferroptosis-Related LncRNA Signatures as a Novel Prognostic Model for Colon Cancer. Front. Immunol. 2021, 12, 783362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Chen, X.; Fang, J.; Tai, P.; Chen, A.; Cao, K. Cuproptosis status affects treatment options about immunotherapy and targeted therapy for patients with kidney renal clear cell carcinoma. Front. Immunol. 2022, 13, 954440. [Google Scholar] [CrossRef]
- Lv, H.; Liu, X.; Zeng, X.; Liu, Y.; Zhang, C.; Zhang, Q.; Xu, J. Comprehensive Analysis of Cuproptosis-Related Genes in Immune Infiltration and Prognosis in Melanoma. Front. Pharmacol. 2022, 13, 930041. [Google Scholar] [CrossRef]
- Zhang, Z.; Zeng, X.; Wu, Y.; Liu, Y.; Zhang, X.; Song, Z. Cuproptosis-Related Risk Score Predicts Prognosis and Characterizes the Tumor Microenvironment in Hepatocellular Carcinoma. Front. Immunol. 2022, 13, 925618. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, T.; Wang, G.; Zhang, M.; Zhu, Y.; Shi, X.; Ding, Z. Cuproptosis-Related LncRNA Signature for Predicting Prognosis of Hepatocellular Carcinoma: A Comprehensive Analysis. Dis. Mark. 2022, 2022, 3265212. [Google Scholar] [CrossRef]
- Sun, R.; Wang, X.; Chen, J.; Teng, D.; Chan, S.; Tu, X.; Wang, Z.; Zuo, X.; Wei, X.; Lin, L.; et al. Development and validation of a novel cellular senescence-related prognostic signature for predicting the survival and immune landscape in hepatocellular carcinoma. Front. Genet. 2022, 13, 949110. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, L.; Chen, T.; Liu, X.; Guo, Y.; Zhu, Q.; Tong, X.; Yang, W.; Xu, Q.; Huang, D.; et al. A novel lncRNA MCM3AP-AS1 promotes the growth of hepatocellular carcinoma by targeting miR-194-5p/FOXA1 axis. Mol. Cancer. 2019, 18, 28. [Google Scholar] [CrossRef]
- Zhou, P.; Lu, Y.; Zhang, Y.; Wang, L. Construction of an Immune-Related Six-lncRNA Signature to Predict the Outcomes, Immune Cell Infiltration, and Immunotherapy Response in Patients with Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 661758. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Wei, W.; Huang, Z.; Chen, Z.; Fang, Y.; Pan, L.; Han, X.; Xu, Z. Long non-coding RNA expression profile can predict early recurrence in hepatocellular carcinoma after curative resection. Hepatol. Res. 2018, 48, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Jette, N.R.; Radhamani, S.; Arthur, G.; Ye, R.; Goutam, S.; Bolyos, A.; Petersen, L.F.; Bose, P.; Bebb, D.G. Combined poly-ADP ribose polymerase and ataxia-telangiectasia mutated/Rad3-related inhibition targets ataxia-telangiectasia mutated-deficient lung cancer cells. Br. J. Cancer. 2019, 121, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Manica, M.; Oskooei, A.; Born, J.; Subramanian, V.; Sáez-Rodríguez, J.; Rodríguez Martínez, M. Toward Explainable Anticancer Compound Sensitivity Prediction via Multimodal Attention-Based Convolutional Encoders. Mol. Pharm. 2019, 16, 4797–4806. [Google Scholar] [CrossRef] [PubMed]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Dilek, N.; Poirier, N.; Hulin, P.; Coulon, F.; Mary, C.; Ville, S.; Vie, H.; Clémenceau, B.; Blancho, G.; Vanhove, B. Targeting CD28, CTLA-4 and PD-L1 costimulation differentially controls immune synapses and function of human regulatory and conventional T-cells. PLoS ONE 2013, 8, e83139. [Google Scholar] [CrossRef] [Green Version]
- Frentsch, M.; Stark, R.; Matzmohr, N.; Meier, S.; Durlanik, S.; Schulz, A.R.; Stervbo, U.; Jürchott, K.; Gebhardt, F.; Heine, G.; et al. CD40L expression permits CD8+ T cells to execute immunologic helper functions. Blood 2013, 122, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Fang, P.; Saredy, J.; Xi, H.; Ramon, C.; Yang, W.; Choi, E.T.; Ji, Y.; Mao, W.; Yang, X. Metabolism-associated danger signal-induced immune response and reverse immune checkpoint-activated CD40(+) monocyte differentiation. J. Hematol. Oncol. 2017, 10, 141. [Google Scholar] [CrossRef]
- Awan, F.M.; Naz, A.; Obaid, A.; Ikram, A.; Ali, A.; Ahmad, J.; Naveed, A.K.; Janjua, H.A. MicroRNA pharmacogenomics based integrated model of miR-17-92 cluster in sorafenib resistant HCC cells reveals a strategy to forestall drug resistance. Sci. Rep. 2017, 7, 11448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanmamed, M.F.; Nie, X.; Desai, S.S.; Villaroel-Espindola, F.; Badri, T.; Zhao, D.; Kim, A.W.; Ji, L.; Zhang, T.; Quinlan, E.; et al. A Burned-Out CD8(+) T-cell Subset Expands in the Tumor Microenvironment and Curbs Cancer Immunotherapy. Cancer Discov. 2021, 11, 1700–1715. [Google Scholar] [CrossRef]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2014, 94, 52–62. [Google Scholar] [CrossRef] [Green Version]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer. 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, H.; Sun, M.; He, T.; Liu, Y.; Yang, X.; Shi, X.; Liu, X. Alpinumisoflavone suppresses hepatocellular carcinoma cell growth and metastasis via NLRP3 inflammasome-mediated pyroptosis. Pharmacol. Rep. 2020, 72, 1370–1382. [Google Scholar] [CrossRef]
- Sun, L.; Yang, X.; Yuan, Z.; Wang, H. Metabolic Reprogramming in Immune Response and Tissue Inflammation. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1990–2001. [Google Scholar] [CrossRef]
- Ding, Y.; Yan, Y.; Dong, Y.; Xu, J.; Su, W.; Shi, W.; Zou, Q.; Yang, X. NLRP3 promotes immune escape by regulating immune checkpoints: A pan-cancer analysis. Int. Immunopharmacol. 2022, 104, 108512. [Google Scholar] [CrossRef]
- Gediya, L.K.; Khandelwal, A.; Patel, J.; Belosay, A.; Sabnis, G.; Mehta, J.; Purushottamachar, P.; Njar, V.C. Design, synthesis, and evaluation of novel mutual prodrugs (hybrid drugs) of all-trans-retinoic acid and histone deacetylase inhibitors with enhanced anticancer activities in breast and prostate cancer cells in vitro. J. Med. Chem. 2008, 51, 3895–3904. [Google Scholar] [CrossRef]
- Qi, X.; Yang, M.; Ma, L.; Sauer, M.; Avella, D.; Kaifi, J.T.; Bryan, J.; Cheng, K.; Staveley-O’Carroll, K.F.; Kimchi, E.T.; et al. Synergizing sunitinib and radiofrequency ablation to treat hepatocellular cancer by triggering the antitumor immune response. J. Immunother. Cancer 2020, 8, e001038. [Google Scholar] [CrossRef]
- Ahonen, C.; Manning, E.; Erickson, L.D.; O’Connor, B.; Lind, E.F.; Pullen, S.S.; Kehry, M.R.; Noelle, R.J. The CD40-TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nat. Immunol. 2002, 3, 451–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turpin, A.; de Baere, T.; Heurgué, A.; Le Malicot, K.; Ollivier-Hourmand, I.; Lecomte, T.; Perrier, H.; Vergniol, J.; Sefrioui, D.; Rinaldi, Y.; et al. Liver transarterial chemoembolization and sunitinib for unresectable hepatocellular carcinoma: Results of the PRODIGE 16 study. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101464. [Google Scholar] [CrossRef] [PubMed]
- Cainap, C.; Qin, S.; Huang, W.T.; Chung, I.J.; Pan, H.; Cheng, Y.; Kudo, M.; Kang, Y.K.; Chen, P.J.; Toh, H.C.; et al. Linifanib versus Sorafenib in patients with advanced hepatocellular carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2015, 33, 172–179. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, Z.; Nie, Y.; Jia, W.; Wang, Y.; Li, J.; Zhang, T.; Lei, X.; Shi, W.; Song, W.; Zhang, X. Revealing Prognostic and Immunotherapy-Sensitive Characteristics of a Novel Cuproptosis-Related LncRNA Model in Hepatocellular Carcinoma Patients by Genomic Analysis. Cancers 2023, 15, 544. https://doi.org/10.3390/cancers15020544
Mao Z, Nie Y, Jia W, Wang Y, Li J, Zhang T, Lei X, Shi W, Song W, Zhang X. Revealing Prognostic and Immunotherapy-Sensitive Characteristics of a Novel Cuproptosis-Related LncRNA Model in Hepatocellular Carcinoma Patients by Genomic Analysis. Cancers. 2023; 15(2):544. https://doi.org/10.3390/cancers15020544
Chicago/Turabian StyleMao, Zhenzhen, Ye Nie, Weili Jia, Yanfang Wang, Jianhui Li, Tianchen Zhang, Xinjun Lei, Wen Shi, Wenjie Song, and Xiao Zhang. 2023. "Revealing Prognostic and Immunotherapy-Sensitive Characteristics of a Novel Cuproptosis-Related LncRNA Model in Hepatocellular Carcinoma Patients by Genomic Analysis" Cancers 15, no. 2: 544. https://doi.org/10.3390/cancers15020544