
Serum Mass Spectrometry Proteomics and Protein Set Identification in Response to FOLFOX-4 in Drug-Resistant Ovarian Carcinoma
 , , , , , ,
, , , , , ,  , , , , ,
, , , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Serum Immunodepletion of Albumin and IgG’s
2.3. Protein Digestion via Filter-Aided Sample Preparation (FASP)
2.4. LC-MS/MS Label-Free Quantification
2.5. Proteins Network Analysis
2.5.1. Proteins Localization
2.5.2. Local Networks
2.6. Statistical Methods
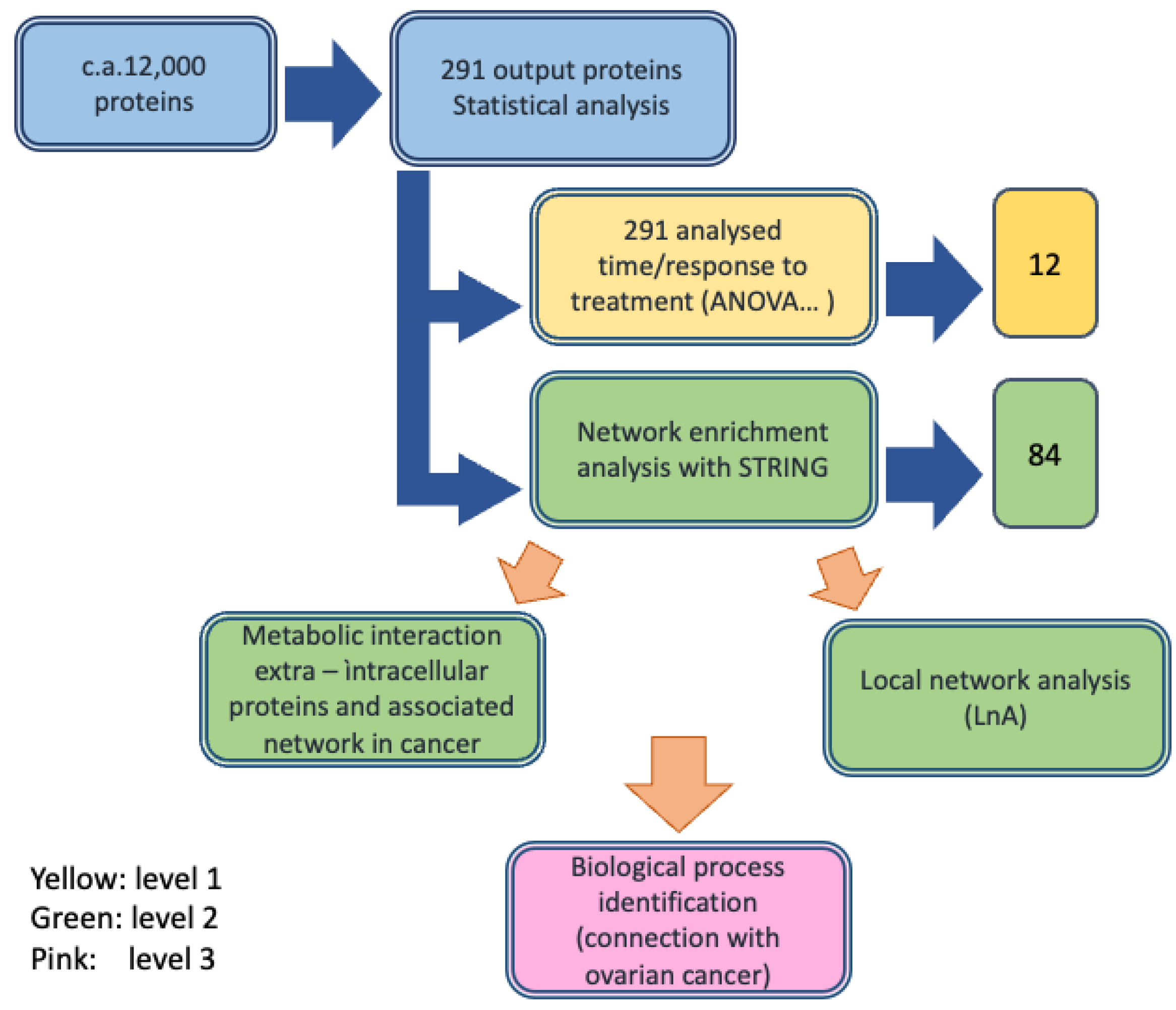
3. Results
3.1. Study Design
3.2. Protein Identification and Characterization (First Analysis Level)
3.2.1. Label-Free MS Proteomic Approach
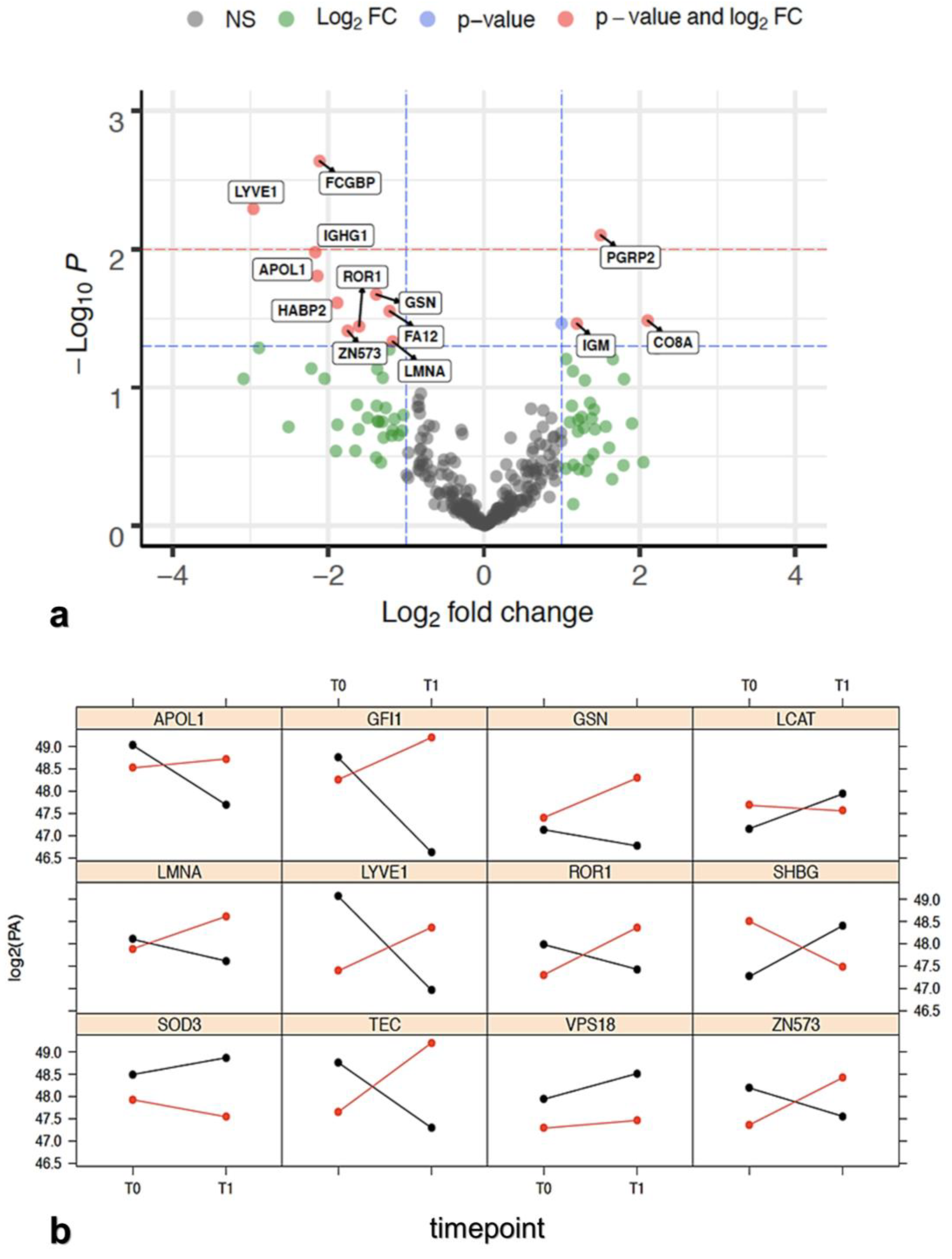
3.2.2. Protein Selection at T0–T1 Timepoints
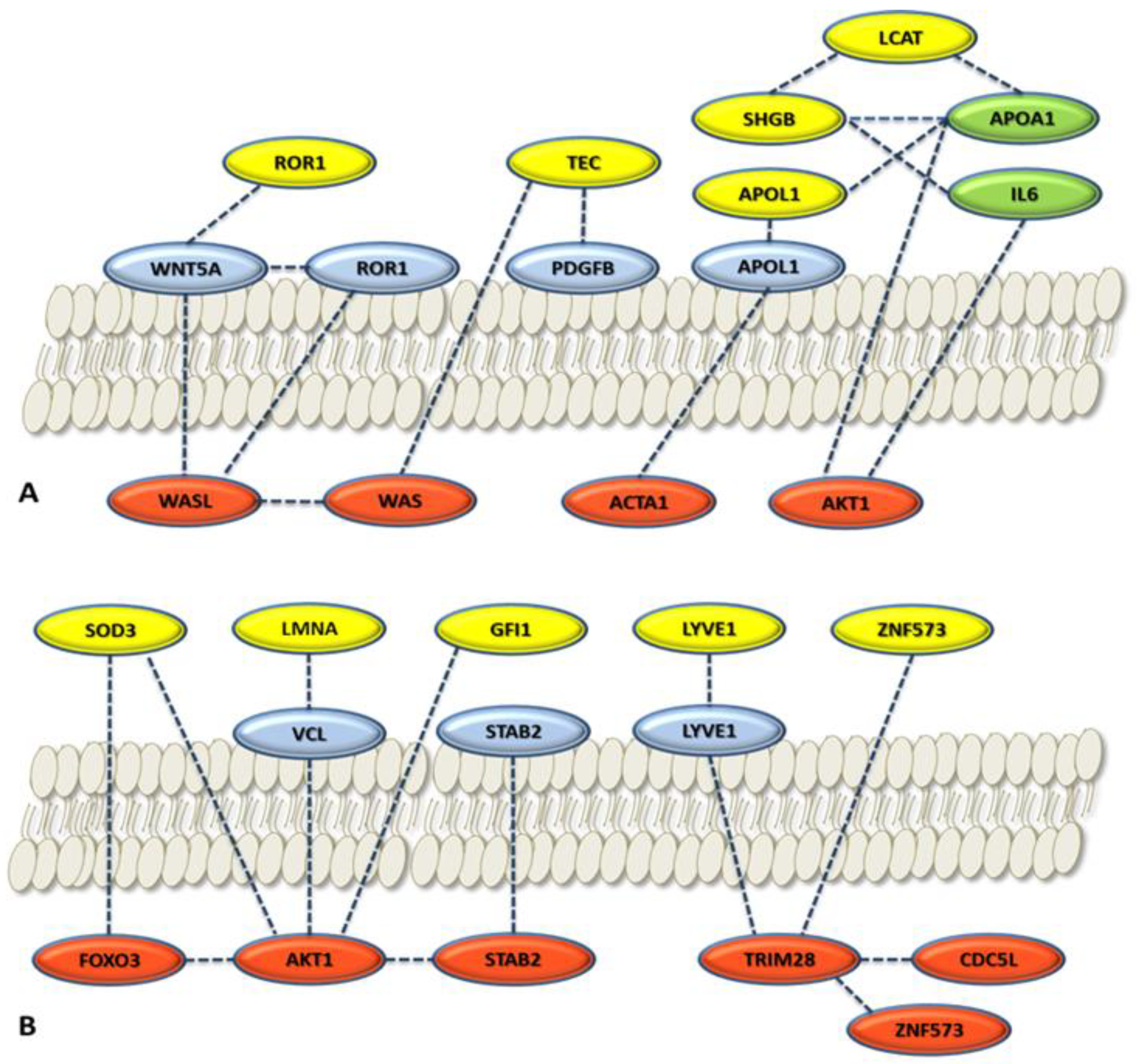
3.2.3. Role of the Selected Proteins in Cancer Processes
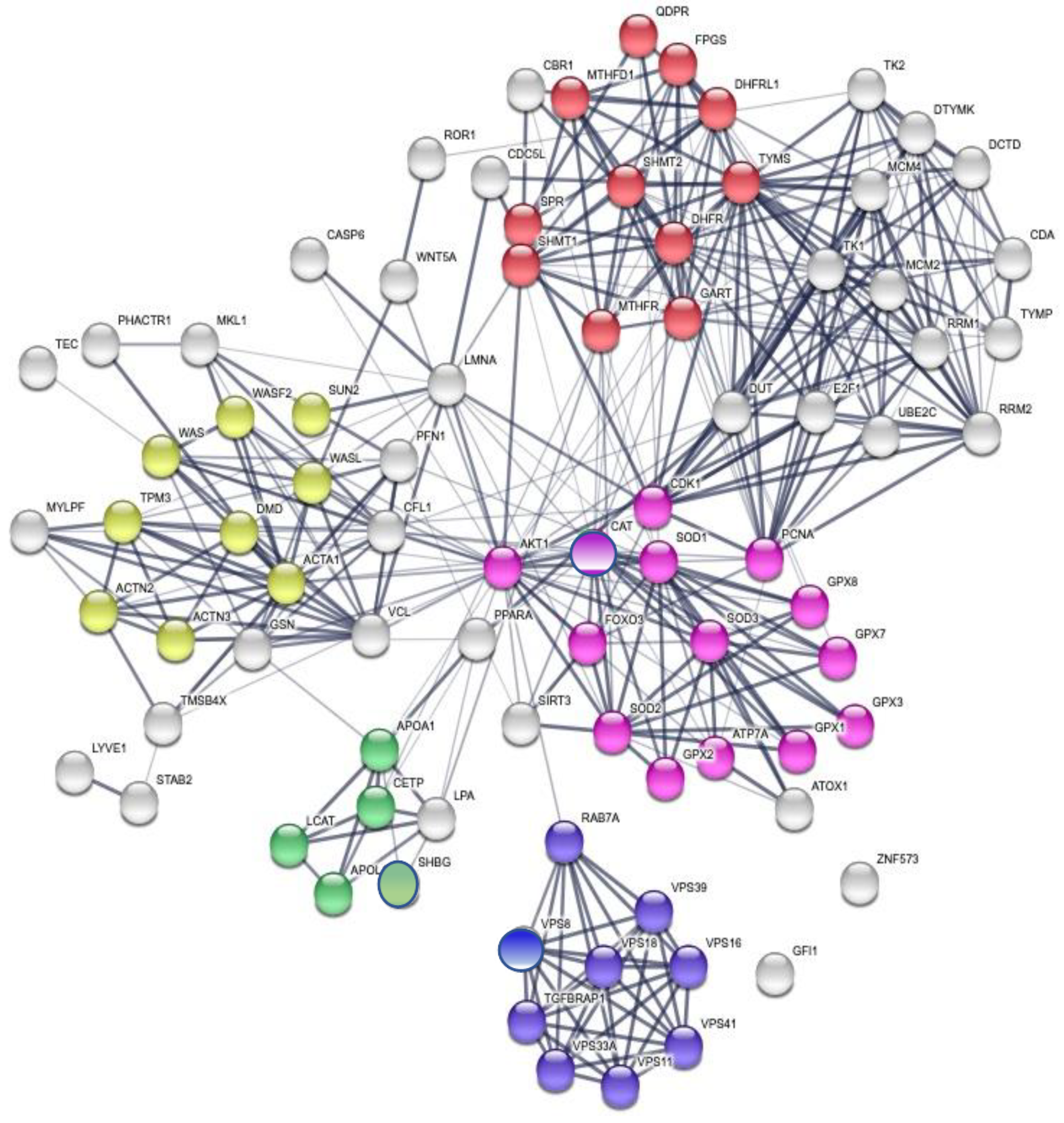
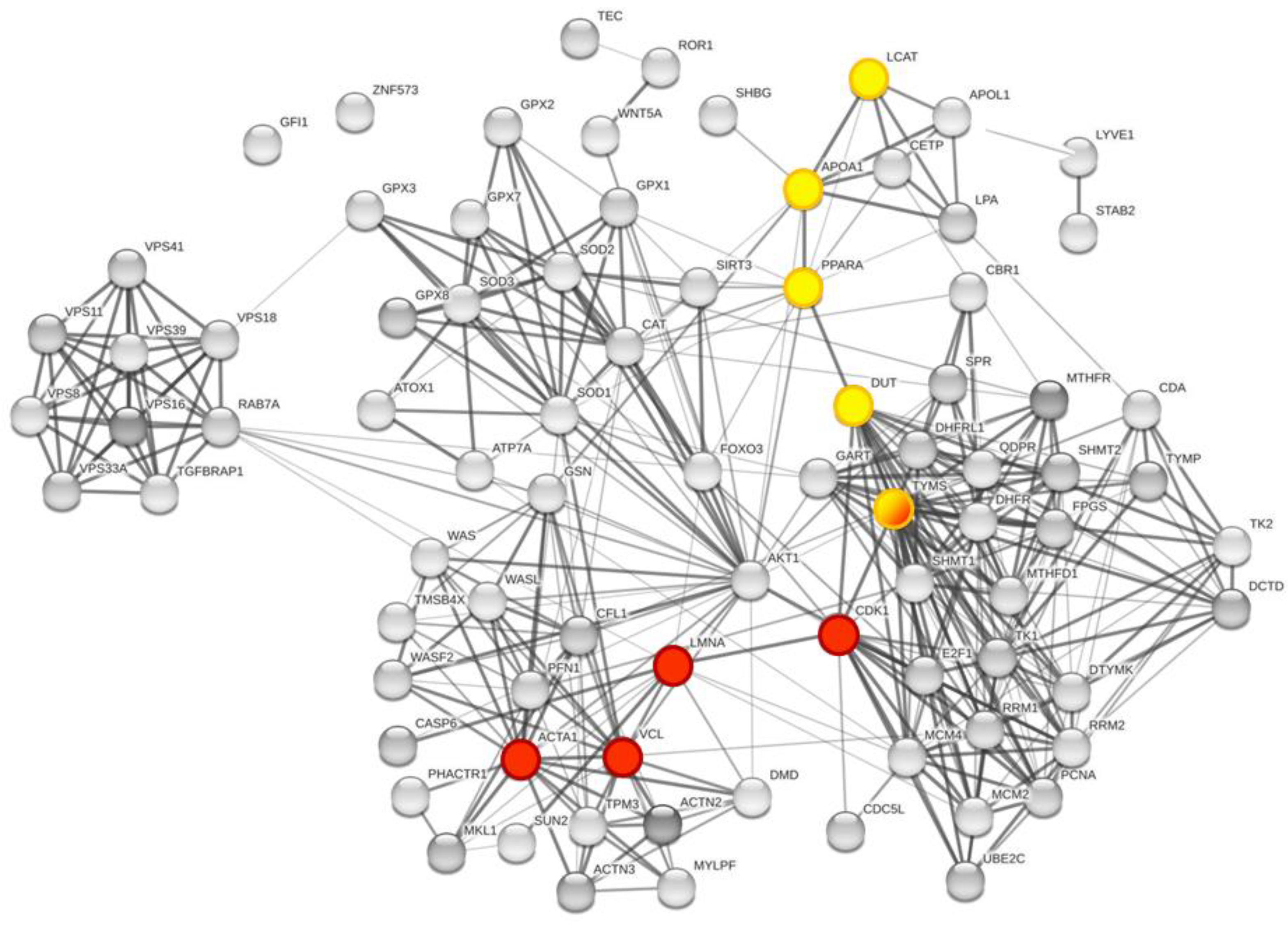
3.3. Enrichment of the Cellular Network of the Selected Protein Panel (Second Analysis Level)
Global Network Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Code (Uniprot) | Protein Function | References |
|---|---|---|
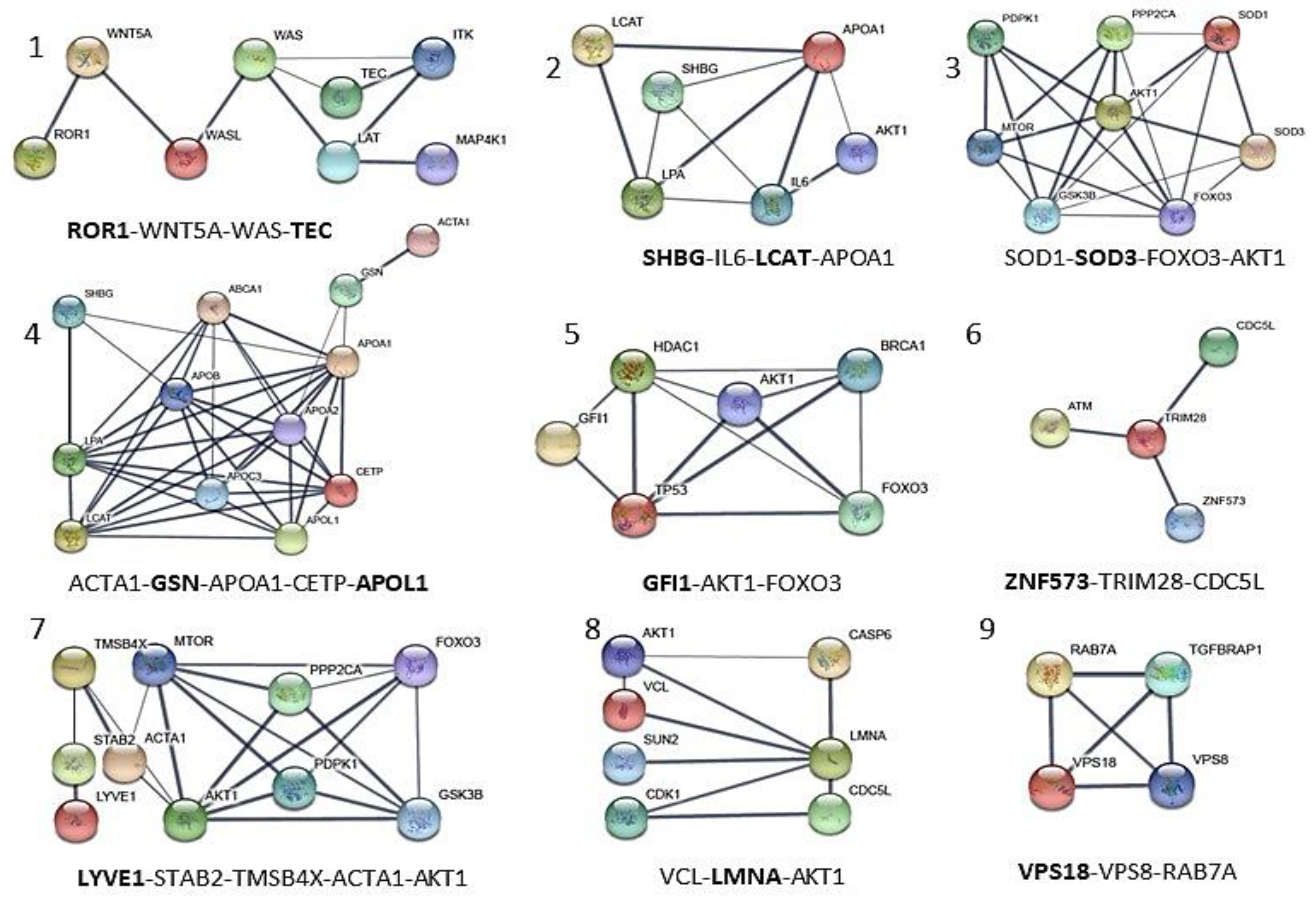
| APOL1 | Apolipoprotein A-I. Local network: APOL1-(CETP-LCAT-LPA-APOA1); APOA1-GSN-ACTA1; ACTA1-GSN-APOA1-CEPT-APOL1 (network 4). | [66] |
| GSN | Gelsolin. Local network: ACTA1-GSN-APOA1-CETP-APOL1 (network 4). | [52,53] |
| GFI1 | Zinc finger protein Gfi-1; Transcription repressor essential for hematopoiesis. Local network: (BRCA1-AKT1-TP53-GFI1-HDAC1); (HDAC1-AKT1-FOXO3-BRCA1); (FOXO3-TP53-GFI1) (network 5). | [56,57] |
| LCAT | Phosphatidylcholine-sterol acyltransferase. Local network: LCAT-LPA-APOA1-SHBG-IL6-AKT1; SHGB-IL6-LCAT-APOA1 (network 2). | [44,45] |
| LMNA | Prelamin-A/C. Local network: VCL-AKT1-LMNA-CDK1; VCL-LMNA-AKT1 (network 8). | [46,47] |
| LYVE1 | Lymphatic vessel endothelial hyaluronic acid receptor 1. Local network: LYVE1-STAB2-TMSB4X-AKT1-MTOR-FOXO3; LYVE1-STAB2-TMSB4X-ACTA1-AKT1 (network 7). | [42,43] |
| ROR1 | Inactive tyrosine-protein kinase transmembrane receptor ROR1. Local network: ROR1-WNT5A-WAS-TEC (network 1). | [49,50,51] |
| SHBG | Sex hormone-binding globulin. Local network: SHBG-IL6-LCAT-APOA1A1 (network 2). | [44,45] |
| SOD3 | (SODE) Extracellular superoxide dismutase [Cu-Zn]. Local network: SOD1-SOD3- AKT1-MTOR; SOD1-SOD3-FOXO3-AKT1 (network 3) | [41,54] |
| TEC | Tyrosine-protein kinase Tec. Local network: TEC-ITK-LAT-WAS-MAP4K1; ROR1-WNT5A-WAS-TEC (network 1). | [48] |
| VPS18 | Vacuolar protein sorting-associated protein 18 homolog. Local network: VPS18-VPS8-RAB-7A (network 9). | [58,59] |
| ZNF573 | Zinc finger protein 573. Local network: ZNF573-TRIM28-CDC5L (network 6). | [55] |
3.4. Interaction between the 5-Fluorouracil Targeting Pathways and the Protein Set Identified in Serum (Third Analysis Level)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Momenimovahed, Z.; Tiznobaik, A.; Taheri, S.; Salehiniya, H. Ovarian Cancer in the World: Epidemiology and Risk Factors. Int. J. Womens. Health 2019, 11, 287–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowtell, D.D.; Böhm, S.; Ahmed, A.A.; Aspuria, P.-J.; Bast Jr, R.C.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking Ovarian Cancer II: Reducing Mortality from High-Grade Serous Ovarian Cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Lorusso, D.; Fontanella, C.; Maltese, G.; Lepori, S.; Tripodi, E.; Bogani, G.; Raspagliesi, F. The Safety of Antiangiogenic Agents and PARP Inhibitors in Platinum-Sensitive Recurrent Ovarian Cancer. Expert Opin. Drug Saf. 2017, 16, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Pignata, S.; Cecere, S.C.; Du Bois, A.; Harter, P.; Heitz, F. Treatment of Recurrent Ovarian Cancer. Ann. Oncol. 2017, 28, viii51–viii56. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Buquet-Fagot, C.; Djelloul, S.; Mester, J.; Cvitkovic, E.; Allain, P.; Louvet, C.; Gespach, C. Antitumor Activity of Oxaliplatin in Combination with 5-Fluorouracil and the Thymidylate Synthase Inhibitor AG337 in Human Colon, Breast and Ovarian Cancers. Anticancer. Drugs 1997, 8, 876–885. [Google Scholar] [CrossRef]
- Pozzi, C.; Santucci, M.; Marverti, G.; D’Arca, D.; Tagliazucchi, L.; Ferrari, S.; Gozzi, G.; Losi, L.; Tassone, G.; Mangani, S.; et al. Structural Bases for the Synergistic Inhibition of Human Thymidylate Synthase and Ovarian Cancer Cell Growth by Drug Combinations. Cancers (Basel) 2021, 13, 2061. [Google Scholar] [CrossRef]
- Wilson, P.M.; Danenberg, P.V.; Johnston, P.G.; Lenz, H.-J.; Ladner, R.D. Standing the Test of Time: Targeting Thymidylate Biosynthesis in Cancer Therapy. Nat. Rev. Clin. Oncol. 2014, 11, 282–298. [Google Scholar] [CrossRef]
- Goldberg, R.M.; Sargent, D.J.; Morton, R.F.; Fuchs, C.S.; Ramanathan, R.K.; Williamson, S.K.; Findlay, B.P.; Pitot, H.C.; Alberts, S.R. A Randomized Controlled Trial of Fluorouracil Plus Leucovorin, Irinotecan, and Oxaliplatin Combinations in Patients With Previously Untreated Metastatic Colorectal Cancer. J. Clin. Oncol. 2004, 22, 23–30. [Google Scholar] [CrossRef]
- Marverti, G.; Marraccini, C.; Martello, A.; D’Arca, D.; Pacifico, S.; Guerrini, R.; Spyrakis, F.; Gozzi, G.; Lauriola, A.; Santucci, M.; et al. Folic Acid–Peptide Conjugates Combine Selective Cancer Cell Internalization with Thymidylate Synthase Dimer Interface Targeting. J. Med. Chem. 2021, 64, 3204–3221. [Google Scholar] [CrossRef]
- Niedzwiecki, D.; Hasson, R.M.; Lenz, H.-J.; Ye, C.; Redston, M.; Ogino, S.; Fuchs, C.S.; Compton, C.C.; Mayer, R.J.; Goldberg, R.M.; et al. A Study of Thymidylate Synthase Expression as a Biomarker for Resectable Colon Cancer: Alliance (Cancer and Leukemia Group B) 9581 and 89803. Oncologist 2017, 22, 107–114. [Google Scholar] [CrossRef]
- Pectasides, D.; Pectasides, M.; Farmakis, D.; Bountouroglou, N.; Nikolaou, M.; Koumpou, M.; Mylonakis, N.; Kosmas, C. Oxaliplatin plus High-Dose Leucovorin and 5-Fluorouracil in Pretreated Advanced Breast Cancer: A Phase II Study. Ann. Oncol. 2003, 14, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Hong, J.; Sym, S.J.; Park, S.H.; Park, J.; Cho, E.K.; Lee, J.H.; Shin, D.B. Oxaliplatin, 5-Fluorouracil and Leucovorin (FOLFOX-4) Combination Chemotherapy as a Salvage Treatment in Advanced Gastric Cancer. Cancer Res. Treat. 2010, 42, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Gramont, A.; Figer, A.; Seymour, M.; Homerin, M.; Hmissi, A.; Cassidy, J.; Boni, C.; Cortes-Funes, H.; Cervantes, A.; Freyer, G.; et al. Leucovorin and Fluorouracil With or Without Oxaliplatin as First-Line Treatment in Advanced Colorectal Cancer. J. Clin. Oncol. 2000, 18, 2938–2947. [Google Scholar] [CrossRef]
- Conteduca, V.; Gurioli, G.; Rossi, L.; Scarpi, E.; Lolli, C.; Schepisi, G.; Farolfi, A.; De Lisi, D.; Gallà, V.; Burgio, S.L.; et al. Oxaliplatin plus Leucovorin and 5-Fluorouracil (FOLFOX-4) as a Salvage Chemotherapy in Heavily-Pretreated Platinum-Resistant Ovarian Cancer. BMC Cancer 2018, 18, 1267. [Google Scholar] [CrossRef] [PubMed]
- Wegdam, W.; Argmann, C.A.; Kramer, G.; Vissers, J.P.; Buist, M.R.; Kenter, G.G.; Aerts, J.M.F.G.; Meijer, D.; Moerland, P.D. Label-Free LC-MSe in Tissue and Serum Reveals Protein Networks Underlying Differences between Benign and Malignant Serous Ovarian Tumors. PLoS One 2014, 9, e108046. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Wang, L.; Ni, J.; Beretov, J.; Wasinger, V.; Wu, D.; Duan, W.; Graham, P.; Li, Y. Proteomics Discovery of Chemoresistant Biomarkers for Ovarian Cancer Therapy. Expert Rev. Proteom. 2016, 13, 905–915. [Google Scholar] [CrossRef]
- Agarwal, R.; Kaye, S.B. Ovarian Cancer: Strategies for Overcoming Resistance to Chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef]
- Babačić, H.; Lehtiö, J.; Pico de Coaña, Y.; Pernemalm, M.; Eriksson, H. In-Depth Plasma Proteomics Reveals Increase in Circulating PD-1 during Anti-PD-1 Immunotherapy in Patients with Metastatic Cutaneous Melanoma. J. Immunother. Cancer 2020, 8, e000204. [Google Scholar] [CrossRef]
- Kohli, M.; Oberg, A.L.; Mahoney, D.W.; Riska, S.M.; Sherwood, R.; Zhang, Y.; Zenka, R.M.; Sahasrabudhe, D.; Qin, R.; Zhang, S. Serum Proteomics on the Basis of Discovery of Predictive Biomarkers of Response to Androgen Deprivation Therapy in Advanced Prostate Cancer. Clin. Genitourin. Cancer 2019, 17, 248–253.e7. [Google Scholar] [CrossRef]
- Reymond, M.A.; Schlegel, W. Proteomics in Cancer. Adv Clin Chem. 2007, 44, 103–142. [Google Scholar] [CrossRef]
- Skates, S.J.; Gillette, M.A.; LaBaer, J.; Carr, S.A.; Anderson, L.; Liebler, D.C.; Ransohoff, D.; Rifai, N.; Kondratovich, M.; Težak, Ž.; et al. Statistical Design for Biospecimen Cohort Size in Proteomics-Based Biomarker Discovery and Verification Studies. J. Proteome Res. 2013, 12, 5383–5394. [Google Scholar] [CrossRef]
- Tanase, C.; Albulescu, R.; Neagu, M. Proteomic Approaches for Biomarker Panels in Cancer. J. Immunoass. Immunochem. 2016, 37, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Haymond, A.; Davis, J.B.; Espina, V. Proteomics for Cancer Drug Design. Expert Rev. Proteom. 2019, 16, 647–664. [Google Scholar] [CrossRef] [PubMed]
- Chinello, C.; Stella, M.; Piga, I.; Smith, A.J.; Bovo, G.; Varallo, M.; Ivanova, M.; Denti, V.; Grasso, M.; Grasso, A.; et al. Proteomics of Liquid Biopsies: Depicting RCC Infiltration into the Renal Vein by MS Analysis of Urine and Plasma. J. Proteom. 2019, 191, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Therasse, P.; Arbuck, S.G.; Eisenhauer, E.A.; Wanders, J.; Kaplan, R.S.; Rubinstein, L.; Verweij, J.; Van Glabbeke, M.; van Oosterom, A.T.; Christian, M.C.; et al. New Guidelines to Evaluate the Response to Treatment in Solid Tumors. JNCI J. Natl. Cancer Inst. 2000, 92, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Rustin, G.J.; Nelstrop, A.E.; McClean, P.; Brady, M.F.; McGuire, W.P.; Hoskins, W.J.; Mitchell, H.; Lambert, H.E. Defining Response of Ovarian Carcinoma to Initial Chemotherapy According to Serum CA 125. J. Clin. Oncol. 1996, 14, 1545–1551. [Google Scholar] [CrossRef]
- Bairoch, A. The SWISS-PROT Protein Sequence Database and Its Supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Liu, X.; Chinello, C.; Musante, L.; Cazzaniga, M.; Tataruch, D.; Calzaferri, G.; James Smith, A.; De Sio, G.; Magni, F.; Zou, H.; et al. Intraluminal Proteome and Peptidome of Human Urinary Extracellular Vesicles. PROTEOMICS—Clin. Appl. 2015, 9, 568–573. [Google Scholar] [CrossRef]
- Raimondo, F.; Chinello, C.; Stella, M.; Santorelli, L.; Magni, F.; Pitto, M. Effects of Hematuria on the Proteomic Profile of Urinary Extracellular Vesicles: Technical Challenges. J. Proteome Res. 2018, 17, 2572–2580. [Google Scholar] [CrossRef]
- Desiere, F. The PeptideAtlas Project. Nucleic Acids Res. 2006, 34, D655–D658. [Google Scholar] [CrossRef]
- Koenig, T.; Menze, B.H.; Kirchner, M.; Monigatti, F.; Parker, K.C.; Patterson, T.; Steen, J.J.; Hamprecht, F.A.; Steen, H. Robust Prediction of the MASCOT Score for an Improved Quality Assessment in Mass Spectrometric Proteomics. J. Proteome Res. 2008, 7, 3708–3717. [Google Scholar] [CrossRef]
- Waters Corporation. Progenesis QI for Proteomics. Licence Code: GLJ-FKGK-A2EP-Z55. Available online: https://www.nonlinear.com/progenesis/qi-for-proteomics/ (accessed on 10 February 2021).
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinforma. 2016, 54, 1–30. [Google Scholar] [CrossRef]
- Orre, L.M.; Vesterlund, M.; Pan, Y.; Arslan, T.; Zhu, Y.; Fernandez Woodbridge, A.; Frings, O.; Fredlund, E.; Lehtiö, J. SubCellBarCode: Proteome-Wide Mapping of Protein Localization and Relocalization. Mol. Cell 2019, 73, 166–182.e7. [Google Scholar] [CrossRef] [Green Version]
- Binder, J.X.; Pletscher-Frankild, S.; Tsafou, K.; Stolte, C.; O’Donoghue, S.I.; Schneider, R.; Jensen, L.J. COMPARTMENTS: Unification and Visualization of Protein Subcellular Localization Evidence. Database (Oxford) 2014, 2014, bau012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A Subcellular Map of the Human Proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.-P.; Mushayamaha, T.; Thomas, P.D. PANTHER Version 16: A Revised Family Classification, Tree-Based Classification Tool, Enhancer Regions and Extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef]
- Mira, E.; Carmona-Rodríguez, L.; Pérez-Villamil, B.; Casas, J.; Fernández-Aceñero, M.J.; Martínez-Rey, D.; Martín-González, P.; Heras-Murillo, I.; Paz-Cabezas, M.; Tardáguila, M.; et al. SOD3 Improves the Tumor Response to Chemotherapy by Stabilizing Endothelial HIF-2α. Nat. Commun. 2018, 9, 575. [Google Scholar] [CrossRef]
- Sundar, S.S.; Ganesan, T.S. Role of Lymphangiogenesis in Cancer. J. Clin. Oncol. 2007, 25, 4298–4307. [Google Scholar] [CrossRef] [PubMed]
- Nunomiya, K.; Shibata, Y.; Abe, S.; Inoue, S.; Igarashi, A.; Yamauchi, K.; Kimura, T.; Aida, Y.; Nemoto, T.; Sato, M.; et al. Relationship between Serum Level of Lymphatic Vessel Endothelial Hyaluronan Receptor-1 and Prognosis in Patients with Lung Cancer. J. Cancer 2014, 5, 242–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ose, J.; Poole, E.M.; Schock, H.; Lehtinen, M.; Arslan, A.A.; Zeleniuch-Jacquotte, A.; Visvanathan, K.; Helzlsouer, K.; Buring, J.E.; Lee, I.-M.; et al. Androgens Are Differentially Associated with Ovarian Cancer Subtypes in the Ovarian Cancer Cohort Consortium. Cancer Res. 2017, 77, 3951–3960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, M.R.; Graham, C.; D’Amato, A.; Gentry-Maharaj, A.; Ryan, A.; Kalsi, J.K.; Ainley, C.; Whetton, A.D.; Menon, U.; Jacobs, I.; et al. A Combined Biomarker Panel Shows Improved Sensitivity for the Early Detection of Ovarian Cancer Allowing the Identification of the Most Aggressive Type II Tumours. Br. J. Cancer 2017, 117, 666–674. [Google Scholar] [CrossRef]
- Bertacchini, J.; Beretti, F.; Cenni, V.; Guida, M.; Gibellini, F.; Mediani, L.; Marin, O.; Maraldi, N.M.; de Pol, A.; Lattanzi, G.; et al. The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression. FASEB J. 2013, 27, 2145–2155. [Google Scholar] [CrossRef]
- Hudson, M.E.; Pozdnyakova, I.; Haines, K.; Mor, G.; Snyder, M. Identification of Differentially Expressed Proteins in Ovarian Cancer Using High-Density Protein Microarrays. Proc. Natl. Acad. Sci. USA 2007, 104, 17494–17499. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Gao, M.; Chu, S.; Su, Y.; Ye, C.; Wang, Y.; Pan, Z.; Wang, Z.; Zhang, H.; Tong, H.; et al. Antitumor Activity of a Newly Developed Monoclonal Antibody against ROR1 in Ovarian Cancer Cells. Oncotarget 2017, 8, 94210–94222. [Google Scholar] [CrossRef] [Green Version]
- Kipps, T. UC-961 (Cirmtuzumab) in Relapsed or Refractory Chronic Lymphocytic Leukemia; University of California: San Diego, CA, USA, 2020. [Google Scholar]
- National Cancer Institute (NCI). Genetically Modified T-Cell Therapy in Treating Patients With Advanced ROR1+ Malignancies; National Cancer Institute (NCI): Bethesda, MD, USA, 2022. [Google Scholar]
- Minkenberg, R. Result Disclosure on ClinicalTrials. Gov—First Experiences and Challenges. Pharm. Program. 2010, 3, 51–56. [Google Scholar] [CrossRef]
- Abedini, M.R.; Wang, P.-W.; Huang, Y.-F.; Cao, M.; Chou, C.-Y.; Shieh, D.-B.; Tsang, B.K. Cell Fate Regulation by Gelsolin in Human Gynecologic Cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 14442–14447. [Google Scholar] [CrossRef] [Green Version]
- Asare-Werehene, M.; Communal, L.; Carmona, E.; Le, T.; Provencher, D.; Mes-Masson, A.-M.; Tsang, B.K. Pre-Operative Circulating Plasma Gelsolin Predicts Residual Disease and Detects Early Stage Ovarian Cancer. Sci. Rep. 2019, 9, 13924. [Google Scholar] [CrossRef]
- Griess, B.; Tom, E.; Domann, F.; Teoh-Fitzgerald, M. Extracellular Superoxide Dismutase and Its Role in Cancer. Free Radic. Biol. Med. 2017, 112, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Qin, X.; Li, L.; Zhou, J.; Zhou, M.; Li, X.; Xu, Y.; Wang, X.; Xing, H. The Transcriptome Profiles and Methylation Status Revealed the Potential Cancer-related LncRNAs in Patients with Cervical Cancer. J. Cell Physiol. 2018, 234, 9756–9763. [Google Scholar] [CrossRef] [PubMed]
- Vadnais, C.; Chen, R.; Fraszczak, J.; Hamard, P.-J.; Manfredi, J.J.; Möröy, T. A Novel Regulatory Circuit between P53 and GFI1 Controls Induction of Apoptosis in T Cells. Sci. Rep. 2019, 9, 6304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Rudneva, V.A.; Erkek, S.; Zapatka, M.; Chau, L.Q.; Tacheva-Grigorova, S.K.; Garancher, A.; Rusert, J.M.; Aksoy, O.; Lea, R.; et al. Lsd1 as a Therapeutic Target in Gfi1-Activated Medulloblastoma. Nat. Commun. 2019, 10, 332. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.S.; Vats, S.; Chia, A.Y.-Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual Role of Autophagy in Hallmarks of Cancer. Oncogene 2017, 37, 1142–1158. [Google Scholar] [CrossRef]
- Segala, G.; Bennesch, M.A.; Ghahhari, N.M.; Pandey, D.P.; Echeverria, P.C.; Karch, F.; Maeda, R.K.; Picard, D. Vps11 and Vps18 of Vps-C Membrane Traffic Complexes Are E3 Ubiquitin Ligases and Fine-Tune Signalling. Nat. Commun. 2019, 10, 1833. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [Green Version]
- Wan, G.; Zhaorigetu, S.; Liu, Z.; Kaini, R.; Jiang, Z.; Hu, C.A. Apolipoprotein L1, a Novel Bcl-2 Homology Domain 3-Only Lipid-Binding Protein, Induces Autophagic Cell Death. J. Biol. Chem. 2008, 283, 21540–21549. [Google Scholar] [CrossRef] [Green Version]
- Taddia, L.; D’Arca, D.; Ferrari, S.; Marraccini, C.; Severi, L.; Ponterini, G.; Assaraf, Y.G.; Marverti, G.; Costi, M.P. Inside the Biochemical Pathways of Thymidylate Synthase Perturbed by Anticancer Drugs: Novel Strategies to Overcome Cancer Chemoresistance. Drug Resist. Updat. 2015, 23, 20–54. [Google Scholar] [CrossRef]
- Genovese, F.; Gualandi, A.; Taddia, L.; Marverti, G.; Pirondi, S.; Marraccini, C.; Perco, P.; Pelà, M.; Guerrini, R.; Amoroso, M.R.; et al. Mass Spectrometric/Bioinformatic Identification of a Protein Subset That Characterizes the Cellular Activity of Anticancer Peptides. J. Proteome Res. 2014, 13, 5250–5261. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Q.; Huang, H.; Huang, W.; Chen, Y.; McGarvey, P.B.; Wu, C.H.; Arighi, C.N. A Crowdsourcing Open Platform for Literature Curation in UniProt. PLOS Biol. 2021, 19, e3001464. [Google Scholar] [CrossRef]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway Enrichment Analysis and Visualization of Omics Data Using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef] [PubMed]
- Georgila, K.; Vyrla, D.; Drakos, E. Apolipoprotein A-I (ApoA-I), Immunity, Inflammation and Cancer. Cancers (Basel) 2019, 11, 1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Angulo, A.M.; Hennessy, B.T.; Meric-Bernstam, F.; Sahin, A.; Liu, W.; Ju, Z.; Carey, M.S.; Myhre, S.; Speers, C.; Deng, L.; et al. Functional Proteomics Can Define Prognosis and Predict Pathologic Complete Response in Patients with Breast Cancer. Clin. Proteom. 2011, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Angulo, A.M.; Liu, S.; Chen, H.; Chavez-MacGregor, M.; Sahin, A.; Hortobagyi, G.N.; Mills, G.B.; Do, K.-A.; Meric-Bernstam, F. Functional Proteomics Characterization of Residual Breast Cancer after Neoadjuvant Systemic Chemotherapy. Ann. Oncol. 2013, 24, 909–916. [Google Scholar] [CrossRef]
- Stearns, V.; Singh, B.; Tsangaris, T.; Crawford, J.G.; Novielli, A.; Ellis, M.J.; Isaacs, C.; Pennanen, M.; Tibery, C.; Farhad, A.; et al. A Prospective Randomized Pilot Study to Evaluate Predictors of Response in Serial Core Biopsies to Single Agent Neoadjuvant Doxorubicin or Paclitaxel for Patients with Locally Advanced Breast Cancer. Clin. Cancer Res. 2003, 9, 124–133. [Google Scholar]
- Asem, M.S.; Buechler, S.; Wates, R.B.; Miller, D.L.; Stack, M.S. Wnt5a Signaling in Cancer. Cancers (Basel) 2016, 8, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Mao, Y.; Zhang, N.; Su, Y.; Zhu, J.; Tong, H.; Zhang, H. A Fully Chimeric IgG Antibody for ROR1 Suppresses Ovarian Cancer Growth in Vitro and in Vivo. Biomed. Pharmacother. 2019, 119, 109420. [Google Scholar] [CrossRef]
- Timms, J.F.; Arslan-Low, E.; Kabir, M.; Worthington, J.; Camuzeaux, S.; Sinclair, J.; Szaub, J.; Afrough, B.; Podust, V.N.; Fourkala, E.-O.; et al. Discovery of Serum Biomarkers of Ovarian Cancer Using Complementary Proteomic Profiling Strategies. Proteomics. Clin. Appl. 2014, 8, 982–993. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Bergami, P.; Barbero, G. The Emerging Role of Wnt5a in the Promotion of a Pro-Inflammatory and Immunosuppressive Tumor Microenvironment. Cancer Metastasis Rev. 2020, 39, 933–952. [Google Scholar] [CrossRef]
- Peng, C.; Zhang, X.; Yu, H.; Wu, D.; Zheng, J. Wnt5a as a Predictor in Poor Clinical Outcome of Patients and a Mediator in Chemoresistance of Ovarian Cancer. Int. J. Gynecol. Cancer 2011, 21, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Varma, R.; Hector, S.; Clark, K.; Greco, W.; Hawthorn, L.; Pendyala, L. Gene Expression Profiling of a Clonal Isolate of Oxaliplatin-Resistant Ovarian Carcinoma Cell Line A2780/C10. Oncol. Rep. 2005, 14, 925–932. [Google Scholar] [CrossRef] [PubMed]







| Protein | PR Group log2(T1/T0) Mean FC | NR Group log2(T1/T0) Mean FC | t Test p-Value | PR/NR Regulation |
|---|---|---|---|---|
| APOL1 | −1.5447 | 0.5949 | 0.0156 | Downregulation |
| CO8A | −0.0363 | −2.1393 | 0.0328 | Upregulation |
| FA12 | −0.2436 | 0.9692 | 0.028 | Downregulation |
| FCGBP | −0.79 | 1.3221 | 0.0023 | Downregulation |
| GELS | −0.5374 | 0.8473 | 0.0212 | Downregulation |
| HABP2 | −0.6816 | 1.2037 | 0.0244 | Downregulation |
| IGHG1 | −1.2833 | 0.8845 | 0.0105 | Downregulation |
| IGM | 0.6886 | −0.504 | 0.0345 | Upregulation |
| LCAT | 0.8256 | −0.1726 | 0.0345 | Upregulation |
| LMNA | −0.509 | 0.6646 | 0.0465 | Downregulation |
| LYVE1 | −1.8428 | 1.1198 | 0.0051 | Downregulation |
| PGRP2 | 0.5958 | −0.902 | 0.0079 | Upregulation |
| ROR1 | −0.5573 | 1.0462 | 0.0361 | Downregulation |
| ZN573 | −0.684 | 1.0665 | 0.0389 | Downregulation |
| Interaction | ||||||
|---|---|---|---|---|---|---|
| Response to Treatment | Timepoint | Response × Timepoint | ||||
| Proteins | F Value | p-Value | F Value | p-Value | F Value | p-Value |
| APOL1 | 0.06 | 0.8230 | 1.2 | 0.3241 | 12.92 | 0.0156 |
| GELS | 4.47 | 0.0881 | 1.5 | 0.2746 | 10.96 | 0.0212 |
| GFI1 | 4.06 | 0.1001 | 0.27 | 0.6267 | 8.6 | 0.0325 |
| LCAT | 0.17 | 0.6939 | 2.22 | 0.1967 | 8.3 | 0.0345 |
| LMNA | 2.57 | 0.1699 | 0.54 | 0.4969 | 6.92 | 0.0465 |
| LYVE1 | 0.07 | 0.8054 | 0.24 | 0.6471 | 22.65 | 0.0051 |
| ROR1 | 0.3 | 0.6056 | 2.15 | 0.2024 | 10.51 | 0.0229 |
| SHBG | 0.01 | 0.9347 | 0.16 | 0.7064 | 6.74 | 0.0485 |
| TEC | 0.1 | 0.7751 | 9.81 | 0.0520 | 25.57 | 0.0149 |
| ZNF573 | 0.05 | 0.8327 | 1.12 | 0.3380 | 8.41 | 0.0338 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Arca, D.; Severi, L.; Ferrari, S.; Dozza, L.; Marverti, G.; Magni, F.; Chinello, C.; Pagani, L.; Tagliazucchi, L.; Villani, M.; et al. Serum Mass Spectrometry Proteomics and Protein Set Identification in Response to FOLFOX-4 in Drug-Resistant Ovarian Carcinoma. Cancers 2023, 15, 412. https://doi.org/10.3390/cancers15020412
D’Arca D, Severi L, Ferrari S, Dozza L, Marverti G, Magni F, Chinello C, Pagani L, Tagliazucchi L, Villani M, et al. Serum Mass Spectrometry Proteomics and Protein Set Identification in Response to FOLFOX-4 in Drug-Resistant Ovarian Carcinoma. Cancers. 2023; 15(2):412. https://doi.org/10.3390/cancers15020412
Chicago/Turabian StyleD’Arca, Domenico, Leda Severi, Stefania Ferrari, Luca Dozza, Gaetano Marverti, Fulvio Magni, Clizia Chinello, Lisa Pagani, Lorenzo Tagliazucchi, Marco Villani, and et al. 2023. "Serum Mass Spectrometry Proteomics and Protein Set Identification in Response to FOLFOX-4 in Drug-Resistant Ovarian Carcinoma" Cancers 15, no. 2: 412. https://doi.org/10.3390/cancers15020412