
DNA Damage Response Alterations in Ovarian Cancer: From Molecular Mechanisms to Therapeutic Opportunities

Abstract
:Simple Summary
Abstract
1. Introduction
2. The DNA Damage Response (DDR)
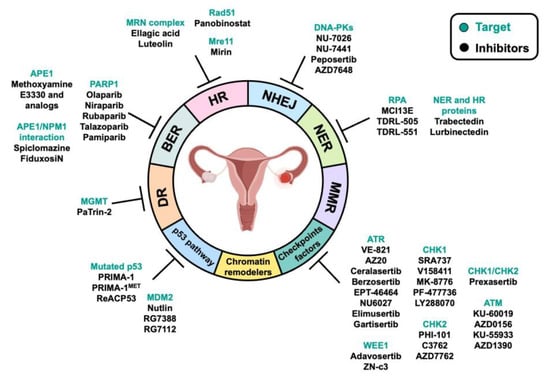
3. DNA Repair Pathways and Their Alteration in OC: Implications for Therapy
3.1. Direct Reversal Repair (DR)
3.2. Mismatch Repair (MMR)
3.3. Nucleotide Excision Repair (NER)
3.4. Base Excision Repair (BER)
3.5. DNA Double-Strand Break Repair by Homologous Recombination (HR)
3.6. DNA Double-Strand Break Repair by Nonhomologous End Joining (NHEJ)
4. DDR-Associated Pathways and Their Alteration in OC: Implications for Therapy
4.1. Chromatin Remodelers
4.2. Checkpoint Factors
4.2.1. ATM Inhibitors
4.2.2. ATR Inhibitors
4.2.3. CHK1 and CHK2 Inhibition
4.2.4. WEE1 Inhibition
4.3. p53 Pathway
5. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ray-Coquard, I.; Morice, P.; Lorusso, D.; Prat, J.; Oaknin, A.; Pautier, P.; Colombo, N. Non-epithelial ovarian cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv1–iv18. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Prim. 2016, 2, 16061. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Dion, L.; Carton, I.; Jaillard, S.; Timoh, K.N.; Henno, S.; Sardain, H.; Foucher, F.; Levêque, J.; de la Motte Rouge, T.; Brousse, S.; et al. The Landscape and Therapeutic Implications of Molecular Profiles in Epithelial Ovarian Cancer. J. Clin. Med. 2020, 9, 2239. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Dyba, T.; Randi, G.; Bray, F.; Martos, C.; Giusti, F.; Nicholson, N.; Gavin, A.; Flego, M.; Neamtiu, L.; Dimitrova, N.; et al. The European cancer burden in 2020: Incidence and mortality estimates for 40 countries and 25 major cancers. Eur. J. Cancer 2021, 157, 308–347. [Google Scholar] [CrossRef] [PubMed]
- Mancari, R.; Cutillo, G.; Bruno, V.; Vincenzoni, C.; Mancini, E.; Baiocco, E.; Bruni, S.; Vocaturo, G.; Chiofalo, B.; Vizza, E. Development of new medical treatment for epithelial ovarian cancer recurrence. Gland. Surg. 2020, 9, 1149–1163. [Google Scholar] [CrossRef]
- Van Zyl, B.; Tang, D.; Bowden, N.A. Biomarkers of platinum resistance in ovarian cancer: What can we use to improve treatment. Endocr. Relat. Cancer 2018, 25, R303–R318. [Google Scholar] [CrossRef]
- Giornelli, G.H. Management of relapsed ovarian cancer: A review. Springerplus 2016, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokhriyal, R.; Hariprasad, R.; Kumar, L.; Hariprasad, G. Chemotherapy Resistance in Advanced Ovarian Cancer Patients. Biomark. Cancer 2019, 11, 1179299X1986081. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darzynkiewicz, Z.; Traganos, F.; Wlodkowic, D. Impaired DNA damage response—An Achilles’ heel sensitizing cancer to chemotherapy and radiotherapy. Eur. J. Pharmacol. 2009, 625, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Kaina, B.; Fritz, G. DNA Damaging Agents. In Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine; Springer: Berlin/Heidelberg, Germany, 2006; pp. 416–423. [Google Scholar] [CrossRef]
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.E.; Malki, M.I. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef]
- De Bont, R.; van Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Moretton, A.; Loizou, J.I. Interplay between Cellular Metabolism and the DNA Damage Response in Cancer. Cancers 2020, 12, 2051. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H.J. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef]
- Wogan, G.N.; Hecht, S.S.; Felton, J.S.; Conney, A.H.; Loeb, L.A. Environmental and chemical carcinogenesis. Semin. Cancer Biol. 2004, 14, 473–486. [Google Scholar] [CrossRef]
- Gutierrez, R.; O’Connor, T.R. DNA direct reversal repair and alkylating agent drug resistance. Cancer Drug Resist 2021, 4, 414–423. [Google Scholar] [CrossRef]
- Pećina-Šlaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef]
- Spivak, G. Nucleotide excision repair in humans. DNA Repair 2015, 36, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Ki, M. The DNA Damage Repair Response. Clin. Oncol. 2020, 3, 1–14. [Google Scholar] [CrossRef]
- Acharya, S.; Wilson, T.; Gradia, S.; Kane, M.F.; Guerrette, S.; Marsischky, G.T.; Kolodner, R.; Fishel, R. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc. Natl. Acad. Sci. USA 1996, 93, 13629–13634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, J.T.; Li, G.-M.; Longley, M.J.; Modrich, P. Isolation of an hMSH2-p160 Heterodimer That Restores DNA Mismatch Repair to Tumor Cells. Science 1995, 268, 1909–1912. [Google Scholar] [CrossRef]
- Palombo, F.; Gallinari, P.; Iaccarino, I.; Lettieri, T.; Hughes, M.; D’Arrigo, A.; Truong, O.; Hsuan, J.J.; Jiricny, J. GTBP, a 160-Kilodalton Protein Essential for Mismatch-binding Activity in Human Cells. Science 1995, 268, 1912–1914. [Google Scholar] [CrossRef]
- Prindle, M.J.; Loeb, L.A. DNA polymerase delta in DNA replication and genome maintenance. Environ. Mol. Mutagen. 2012, 53, 666–682. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Keijzers, G.; Rasmussen, L.J. DNA mismatch repair and its many roles in eukaryotic cells. Mutat. Res. Rev. Mutat. Res. 2017, 773, 174–187. [Google Scholar] [CrossRef]
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [Green Version]
- Lans, H.; Marteijn, J.A.; Vermeulen, W. ATP-dependent chromatin remodeling in the DNA-damage response. Epigenetics Chromatin 2012, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Marintchev, A.; Robertson, A.; Dimitriadis, E.K.; Prasad, R.; Wilson, S.H.; Mullen, G.P. Domain specific interaction in the XRCC1-DNA polymerase beta complex. Nucleic Acids Res. 2000, 28, 2049–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehouse, C.J.; Taylor, R.M.; Thistlethwaite, A.; Zhang, H.; Karimi-Busheri, F.; Lasko, D.D.; Weinfeld, M.; Caldecott, K.W. XRCC1 Stimulates Human Polynucleotide Kinase Activity at Damaged DNA Termini and Accelerates DNA Single-Strand Break Repair. Cell 2001, 104, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, J.M.; Kastan, M.B. 11 - DNA Damage Response Pathways and Cancer. In Abeloff’s Clinical Oncology, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 154–164.e4. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, L.-Y. BRCA1 and homologous recombination: Implications from mouse embryonic development. Cell Biosci. 2020, 10, 49. [Google Scholar] [CrossRef]
- Chen, C.-C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2018, 2, 313–336. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar] [CrossRef]
- Caracciolo, D.; Montesano, M.; Tagliaferri, P.; Tassone, P. Alternative non-homologous end joining repair: A master regulator of genomic instability in cancer. Precis. Cancer Med. 2019, 2, 8. [Google Scholar] [CrossRef]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 2015, 793, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wippo, C.J.; Wal, M.; Ward, E.; Korber, P.; Pugh, B.F. A Packing Mechanism for Nucleosome Organization Reconstituted Across a Eukaryotic Genome. Science 2011, 332, 977–980. [Google Scholar] [CrossRef] [Green Version]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Zannini, L.; Delia, D.; Buscemi, G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef]
- Roh, H.-J.; Suh, D.-S.; Choi, K.-U.; Yoo, H.-J.; Joo, W.-D.; Yoon, M.-S. Inactivation of O6-methyguanine-DNA methyltransferase by promoter hypermethylation: Association of epithelial ovarian carcinogenesis in specific histological types. J. Obstet. Gynaecol. Res. 2011, 37, 851–860. [Google Scholar] [CrossRef]
- Qiao, B.; Zhang, Z.; Li, Y. Association of MGMT promoter methylation with tumorigenesis features in patients with ovarian cancer: A systematic meta-analysis. Mol. Genet. Genom. Med. 2017, 6, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Plazzer, J.P.; Sijmons, R.H.; Woods, M.O.; Peltomäki, P.; Thompson, B.; Dunnen, J.T.D.; Macrae, F. The InSiGHT database: Utilizing 100 years of insights into Lynch Syndrome. Fam. Cancer 2013, 12, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.C.; Mariappan, M.R.; Putcha, G.V.; Husain, A.; Chun, N.; Ford, J.M.; Schrijver, I.; Longacre, T.A. Microsatellite Instability and Mismatch Repair Protein Defects in Ovarian Epithelial Neoplasms in Patients 50 Years of Age and Younger. Am. J. Surg. Pathol. 2008, 32, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.; Bützow, R.; Lynch, H.T.; Mecklin, J.-P.; Järvinen, H.J.; Vasen, H.F.A.; Madlensky, L.; Fidalgo, P.; Bernstein, I. The Clinical Features of Ovarian Cancer in Hereditary Nonpolyposis Colorectal Cancer. Gynecol. Oncol. 2001, 82, 223–228. [Google Scholar] [CrossRef]
- De La Chapelle, A. Microsatellite Instability. N. Engl. J. Med. 2003, 349, 209–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gras, E.; Catasus, L.; Argü, R.; Moreno-Bueno, G.; Palacios, J.; Gamallo, C.; Matias-Guiu, X.; Prat, J. Microsatellite Instability, MLH-1 Promoter Hypermethylation, and Frameshift Mutations at Coding Mononucleotide Repeat Microsatellites in Ovarian Tumors. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2001, 92, 2829–2836. [Google Scholar] [CrossRef]
- Pal, T.; Permuth-Wey, J.; Sellers, T.A. A review of the clinical relevance of mismatch-repair deficiency in ovarian cancer. Cancer 2008, 113, 733–742. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Zhang, A.; Zhao, Y.; Xiang, J.; Yu, D.; Liang, Z.; Xu, C.; Zhang, Q.; Li, J.; Duan, P. The association of polymorphisms in nucleotide excision repair genes with ovarian cancer susceptibility. Biosci. Rep. 2018, 38, 20180114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gee, M.E.; Faraahi, Z.; McCormick, A.; Edmondson, R.J. DNA damage repair in ovarian cancer: Unlocking the heterogeneity. J. Ovarian Res. 2018, 11, 1–12. [Google Scholar] [CrossRef]
- Ceccaldi, R.; O’Connor, K.W.; Mouw, K.W.; Li, A.Y.; Matulonis, U.A.; D’Andrea, A.D.; Konstantinopoulos, P.A. A Unique Subset of Epithelial Ovarian Cancers with Platinum Sensitivity and PARP Inhibitor Resistance. Cancer Res 2015, 75, 628–634. [Google Scholar] [CrossRef] [Green Version]
- D’Errico, M.; Parlanti, E.; Pascucci, B.; Fortini, P.; Baccarini, S.; Simonelli, V.; Dogliotti, E. Single nucleotide polymorphisms in DNA glycosylases: From function to disease. Free. Radic. Biol. Med. 2017, 107, 278–291. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, X.; Wang, J.; Guo, W.; Sun, C.; Cai, Z.; Wu, Q.; Xu, X.; Wang, Y. Functional Polymorphisms of the hOGG1 Gene Confer Risk to Type 2 Epithelial Ovarian Cancer in Chinese. Int. J. Gynecol. Cancer 2011, 21, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Osorio, A.; Milne, R.L.; Kuchenbaecker, K.; Vaclová, T.; Pita, G.; Alonso, R.; Peterlongo, P.; Blanco, I.; de la Hoya, M.; Durán, M.; et al. DNA Glycosylases Involved in Base Excision Repair May Be Associated with Cancer Risk in BRCA1 and BRCA2 Mutation Carriers. PLOS Genet. 2014, 10, e1004256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Li, W. Association between polymorphisms of XRCC1 and ADPRT genes and ovarian cancer survival with platinum-based chemotherapy in Chinese population. Mol. Cell. Biochem. 2013, 372, 27–33. [Google Scholar] [CrossRef]
- Miao, J.; Zhang, X.; Tang, Q.-L.; Wang, X.-Y.; Kai, L. Prediction Value of XRCC 1 Gene Polymorphism on the Survival of Ovarian Cancer Treated by Adjuvant Chemotherapy. Asian Pac. J. Cancer Prev. 2012, 13, 5007–5010. [Google Scholar] [CrossRef] [Green Version]
- Malisic, E.J.; Krivokuca, A.M.; Boljevic, I.Z.; Jankovic, R.N. Impact of RAD51 G135C and XRCC1 Arg399Gln polymorphisms on ovarian carcinoma risk in Serbian women. Cancer Biomarkers 2015, 15, 685–691. [Google Scholar] [CrossRef]
- Zhang, X.; Xin, X.; Zhang, J.; Li, J.; Chen, B.; Zou, W. Apurinic/Apyrimidinic Endonuclease 1 Polymorphisms Are Associated with Ovarian Cancer Susceptibility in a Chinese Population. Int. J. Gynecol. Cancer 2013, 23, 1393–1399. [Google Scholar] [CrossRef]
- Al-Attar, A.; Gossage, L.; Fareed, K.R.; Shehata, M.; Mohammed, M.; Zaitoun, A.M.; Soomro, I.; Lobo, D.N.; Abbotts, R.; Chan, S.; et al. Human apurinic/apyrimidinic endonuclease (APE1) is a prognostic factor in ovarian, gastro-oesophageal and pancreatico-biliary cancers. Br. J. Cancer 2010, 102, 704–709. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Liu, P.; Shi, C.; Qiu, L.; Shang, D.; Lu, Z.; Tu, Z.; Liu, H. Therapeutic targeting of DNA damage repair pathways guided by homologous recombination deficiency scoring in ovarian cancers. Fundam. Clin. Pharmacol. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha Colombo Bonadio, R.R.; Fogace, R.N.; Miranda, V.C.; Diz, M.D.P.E. Homologous recombination deficiency in ovarian cancer: A review of its epidemiology and management. Clinics 2018, 73, e450s. [Google Scholar] [CrossRef]
- Zhao, Q.; Yang, J.; Li, L.; Cao, D.; Yu, M.; Shen, K. Germline and somatic mutations in homologous recombination genes among Chinese ovarian cancer patients detected using next-generation sequencing. J. Gynecol. Oncol. 2017, 28, e39. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczkowska, M.; Krawczynska, N.; Stukan, M.; Kuzniacka, A.; Brozek, I.; Sniadecki, M.; Debniak, J.; Wydra, D.; Biernat, W.; Kozlowski, P.; et al. Spectrum and Prevalence of Pathogenic Variants in Ovarian Cancer Susceptibility Genes in a Group of 333 Patients. Cancers 2018, 10, 442. [Google Scholar] [CrossRef] [Green Version]
- Brandt, S.; Samartzis, E.P.; Zimmermann, A.-K.; Fink, D.; Moch, H.; Noske, A.; Dedes, K.J. Lack of MRE11-RAD50-NBS1 (MRN) complex detection occurs frequently in low-grade epithelial ovarian cancer. BMC Cancer 2017, 17, 44. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Dicks, E.; Ramus, S.J.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Liu, G.; Xue, F.; Edwards, R.; Sood, A.K.; Zhang, W.; Yang, D. Copy number deletion of RAD50 as predictive marker of BRCAness and PARP inhibitor response in BRCA wild type ovarian cancer. Gynecol. Oncol. 2016, 141, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Fatah, T.M.A.; Arora, A.; Moseley, P.; Coveney, C.; Perry, C.; Johnson, K.; Kent, C.; Ball, G.; Chan, S.; Madhusudan, S. ATM, ATR and DNA-PKcs expressions correlate to adverse clinical outcomes in epithelial ovarian cancers. BBA Clin. 2014, 2, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Willis, S.; Villalobos, V.M.; Gevaert, O.; Abramovitz, M.; Williams, C.; Sikic, B.I.; Leyland-Jones, B. Single Gene Prognostic Biomarkers in Ovarian Cancer: A Meta-Analysis. PLoS ONE 2016, 11, e0149183. [Google Scholar] [CrossRef]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A Mutations in Endometriosis-Associated Ovarian Carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Li, Y.; Qian, L.; Deng, S.; Liu, L.; Xiao, W.; Zhou, Y. A Review of the Clinical Characteristics and Novel Molecular Subtypes of Endometrioid Ovarian Cancer. Front Oncol. 2021, 11, 668151. [Google Scholar] [CrossRef] [PubMed]
- Hollis, R.L.; Gourley, C. Genetic and molecular changes in ovarian cancer. Cancer Biol. Med. 2016, 13, 236–247. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Wang, T.-L.; Shih, I.-M.; Mao, T.-L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent Mutations of Chromatin Remodeling Gene ARID1A in Ovarian Clear Cell Carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okawa, R.; Banno, K.; Iida, M.; Yanokura, M.; Takeda, T.; Iijima, M.; Kunitomi-Irie, H.; Nakamura, K.; Adachi, M.; Umene, K.; et al. Aberrant chromatin remodeling in gynecological cancer. Oncol. Lett. 2017, 14, 5107–5113. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Choi, M.; Overton, J.D.; Bellone, S.; Roque, D.M.; Cocco, E.; Guzzo, F.; English, D.P.; Varughese, J.; Gasparrini, S.; et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 2916–2921. [Google Scholar] [CrossRef] [Green Version]
- Le Gallo, M.; O’Hara, A.J.; Rudd, M.L.; Urick, M.E.; Hansen, N.F.; O’Neil, N.J.; Price, J.C.; Zhang, S.; England, B.M.; Godwin, A.K.; et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat. Genet. 2012, 44, 1310–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaicekauskaitė, I.; Sabaliauskaitė, R.; Lazutka, J.R.; Jarmalaitė, S. The Emerging Role of Chromatin Remodeling Complexes in Ovarian Cancer. Int. J. Mol. Sci. 2022, 23, 13670. [Google Scholar] [CrossRef] [PubMed]
- Oyama, Y.; Shigeta, S.; Tokunaga, H.; Tsuji, K.; Ishibashi, M.; Shibuya, Y.; Shimada, M.; Yasuda, J.; Yaegashi, N. CHD4 regulates platinum sensitivity through MDR1 expression in ovarian cancer: A potential role of CHD4 inhibition as a combination therapy with platinum agents. PLoS ONE 2021, 16, e0251079. [Google Scholar] [CrossRef]
- Xiao, Y.; Lin, F.-T.; Lin, W.-C. ACTL6A promotes repair of cisplatin-induced DNA damage, a new mechanism of platinum resistance in cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2015808118. [Google Scholar] [CrossRef]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [Green Version]
- Kurian, A.W.; Hughes, E.; Handorf, E.A.; Gutin, A.; Allen, B.; Hartman, A.-R.; Hall, M.J. Breast and Ovarian Cancer Penetrance Estimates Derived From Germline Multiple-Gene Sequencing Results in Women. JCO Precis. Oncol. 2017, 1, 1–12. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.J.; Dwight, T.; Gill, A.J.; Dickson, K.-A.; Zhu, Y.; Clarkson, A.; Gard, G.B.; Maidens, J.; Valmadre, S.; Clifton-Bligh, R.; et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci. Rep. 2016, 6, 26191. [Google Scholar] [CrossRef] [Green Version]
- Chien, J.; Sicotte, H.; Fan, J.-B.; Humphray, S.; Cunningham, J.M.; Kalli, K.R.; Oberg, A.L.; Hart, S.N.; Li, Y.; Davila, J.I.; et al. TP53 mutations, tetraploidy and homologous recombination repair defects in early stage high-grade serous ovarian cancer. Nucleic Acids Res. 2015, 43, 6945–6958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barvaux, V.A.; Lorigan, P.; Ranson, M.; Gillum, A.M.; McElhinney, R.S.; McMurry, T.B.H.; Margison, G.P. Sensitization of a human ovarian cancer cell line to temozolomide by simultaneous attenuation of the Bcl-2 antiapoptotic protein and DNA repair by O6-alkylguanine-DNA alkyltransferase. Mol. Cancer Ther. 2004, 3, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Dormi, S.S.; Turchi, A.M.; Woods, D.S.; Turchi, J.J. Chemical inhibitor targeting the replication protein A–DNA interaction increases the efficacy of Pt-based chemotherapy in lung and ovarian cancer. Biochem. Pharmacol. 2015, 93, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Musacchio, L.; Cicala, C.M.; Salutari, V.; Camarda, F.; Carbone, M.V.; Ghizzoni, V.; Giudice, E.; Nero, C.; Perri, M.T.; Ricci, C.; et al. Preclinical and Clinical Evidence of Lurbinectedin in Ovarian Cancer: Current Status and Future Perspectives. Front. Oncol. 2022, 12, 585. [Google Scholar] [CrossRef]
- Soares, D.G.; Machado, M.S.; Rocca, C.J.; Poindessous, V.; Ouaret, D.; Sarasin, A.; Galmarini, C.M.; Henriques, J.A.P.; Escargueil, A.E.; Larsen, A.K. Trabectedin and Its C Subunit Modified Analogue PM01183 Attenuate Nucleotide Excision Repair and Show Activity toward Platinum-Resistant Cells. Mol. Cancer Ther. 2011, 10, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.; Muñoz, C.; Guillén, M.-J.; Moretó, J.; Puertas, S.; Martínez-Iniesta, M.; Figueras, A.; Padullés, L.; García-Rodriguez, F.J.; Berdiel-Acer, M.; et al. Lurbinectedin (PM01183), a New DNA Minor Groove Binder, Inhibits Growth of Orthotopic Primary Graft of Cisplatin-Resistant Epithelial Ovarian Cancer. Clin. Cancer Res. 2012, 18, 5399–5411. [Google Scholar] [CrossRef] [Green Version]
- D’Incalci, M.; Colombo, T.; Ubezio, P.; Nicoletti, I.; Giavazzi, R.; Erba, E.; Ferrarese, L.; Meco, D.; Riccardi, R.; Sessa, C.; et al. The combination of yondelis and cisplatin is synergistic against human tumor xenografts. Eur. J. Cancer 2003, 39, 1920–1926. [Google Scholar] [CrossRef]
- Fishel, M.L.; He, Y.; Smith, M.L.; Kelley, M.R. Manipulation of Base Excision Repair to Sensitize Ovarian Cancer Cells to Alkylating Agent Temozolomide. Clin. Cancer Res. 2007, 13, 260–267. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Delaplane, S.; Jiang, A.; Reed, A.; He, Y.; Fishel, M.; Nyland, R.L.; Borch, R.F.; Qiao, X.; Georgiadis, M.M.; et al. Role of the Multifunctional DNA Repair and Redox Signaling Protein Ape1/Ref-1 in Cancer and Endothelial Cells: Small-Molecule Inhibition of the Redox Function of Ape1. Antioxid. Redox Signal. 2008, 10, 1853–1867. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.R.; Luo, M.; Reed, A.; Su, D.; Delaplane, S.; Borch, R.F.; Nyland, R.L.; Gross, M.L.; Georgiadis, M.M. Functional Analysis of Novel Analogues of E3330 That Block the Redox Signaling Activity of the Multifunctional AP Endonuclease/Redox Signaling Enzyme APE1/Ref-1. Antioxid. Redox Signal. 2011, 14, 1387–1401. [Google Scholar] [CrossRef] [PubMed]
- Poletto, M.; Malfatti, M.C.; Dorjsuren, D.; Scognamiglio, P.L.; Marasco, D.; Vascotto, C.; Jadhav, A.; Maloney, D.J.; Wilson, D.M.; Simeonov, A.; et al. Inhibitors of the apurinic/apyrimidinic endonuclease 1 (APE1)/nucleophosmin (NPM1) interaction that display anti-tumor properties. Mol. Carcinog. 2015, 55, 688–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Ovejero-Sánchez, M.; González-Sarmiento, R.; Herrero, A.B. Synergistic effect of Chloroquine and Panobinostat in ovarian cancer through induction of DNA damage and inhibition of DNA repair. Neoplasia 2021, 23, 515–528. [Google Scholar] [CrossRef]
- Alblihy, A.; Ali, R.; Algethami, M.; Shoqafi, A.; Toss, M.S.; Brownlie, J.; Tatum, N.J.; Hickson, I.; Moran, P.O.; Grabowska, A.; et al. Targeting Mre11 overcomes platinum resistance and induces synthetic lethality in XRCC1 deficient epithelial ovarian cancers. NPJ Precis. Oncol. 2022, 6, 1–12. [Google Scholar] [CrossRef]
- Huang, D.; Chowdhury, S.; Wang, H.; Savage, S.R.; Ivey, R.G.; Kennedy, J.J.; Whiteaker, J.R.; Lin, C.; Hou, X.; Oberg, A.L.; et al. Multiomic analysis identifies CPT1A as a potential therapeutic target in platinum-refractory, high-grade serous ovarian cancer. Cell Rep. Med. 2021, 2, 100471. [Google Scholar] [CrossRef]
- Wilson, A.J.; Sarfo-Kantanka, K.; Barrack, T.; Steck, A.; Saskowski, J.; Crispens, M.A.; Khabele, D. Panobinostat sensitizes cyclin E high, homologous recombination-proficient ovarian cancer to olaparib. Gynecol. Oncol. 2016, 143, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Helland, Ø.; Popa, M.; Bischof, K.; Gjertsen, B.T.; Mc Cormack, E.; Bjørge, L. The HDACi Panobinostat Shows Growth Inhibition Both In Vitro and in a Bioluminescent Orthotopic Surgical Xenograft Model of Ovarian Cancer. PLoS ONE 2016, 11, e0158208. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.-Y.; Lin, H.; Moh, J.-S.; Chen, K.-D.; Wang, I.-W.; Ou, Y.-C.; You, Y.-S.; Lung, C.-C. Low-dose LBH589 increases the sensitivity of cisplatin to cisplatin-resistant ovarian cancer cells. Taiwan. J. Obstet. Gynecol. 2011, 50, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zeng, Z.; Wang, S.; Li, T.; Mastriani, E.; Li, Q.-H.; Bao, H.-X.; Zhou, Y.-J.; Wang, X.; Liu, Y.; et al. Main components of pomegranate, ellagic acid and luteolin, inhibit metastasis of ovarian cancer by down-regulating MMP2 and MMP9. Cancer Biol. Ther. 2017, 18, 990–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovejero-Sánchez, M.; Rubio-Heras, J.; Peña, M.d.C.V.d.l.; San-Segundo, L.; Pérez-Losada, J.; González-Sarmiento, R.; Herrero, A.B. Chloroquine-Induced DNA Damage Synergizes with Nonhomologous End Joining Inhibition to Cause Ovarian Cancer Cell Cytotoxicity. Int. J. Mol. Sci. 2022, 23, 7518. [Google Scholar] [CrossRef] [PubMed]
- Nutley, B.P.; Smith, N.F.; Hayes, A.; Kelland, L.R.; Brunton, L.; Golding, B.T.; Smith, G.C.M.; Martin, N.M.B.; Workman, P.; Raynaud, F.I. Preclinical pharmacokinetics and metabolism of a novel prototype DNA-PK inhibitor NU7026. Br. J. Cancer 2005, 93, 1011–1018. [Google Scholar] [CrossRef] [Green Version]
- Stronach, E.A.; Chen, M.; Maginn, E.N.; Agarwal, R.; Mills, G.B.; Wasan, H.; Gabra, H. DNA-PK Mediates AKT Activation and Apoptosis Inhibition in Clinically Acquired Platinum Resistance. Neoplasia 2011, 13, 1069–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, Y. Development and Evolution of DNA-Dependent Protein Kinase Inhibitors toward Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 4264. [Google Scholar] [CrossRef]
- Haines, E.; Nishida, Y.; Carr, M.I.; Montoya, R.H.; Ostermann, L.B.; Zhang, W.G.; Zenke, F.T.; Blaukat, A.; Andreeff, M.; Vassilev, L.T. DNA-PK inhibitor peposertib enhances p53-dependent cytotoxicity of DNA double-strand break inducing therapy in acute leukemia. Sci Rep-Uk 2021, 11. [Google Scholar] [CrossRef]
- Ramos-Montoya, A.; Fok, J.H.; James, N.; Follia, V.; Vazquez-Chantada, M.; Wijnhoven, P.; O’Connor, L.O.; Karmokar, A.; Staniszewska, A.; Dean, E.; et al. AZD7648, a potent and selective inhibitor of DNA-PK, potentiates activity of the PARP inhibitor olaparib resulting in sustained anti-tumour activity in xenograft and PDX models. Cancer Res 2019, 79. [Google Scholar] [CrossRef]
- Anastasia, A.; Dellavedova, G.; Ramos-Montoya, A.; James, N.; Chiorino, G.; Russo, M.; Baakza, H.; Wilson, J.; Ghilardi, C.; Cadogan, E.B.; et al. The DNA-PK Inhibitor AZD7648 Sensitizes Patient-Derived Ovarian Cancer Xenografts to Pegylated Liposomal Doxorubicin and Olaparib Preventing Abdominal Metastases. Mol. Cancer Ther. 2022, 21, 555–567. [Google Scholar] [CrossRef]
- Wang, N.; Yu, M.; Fu, Y.; Ma, Z. Blocking ATM Attenuates SKOV3 Cell Proliferation and Migration by Disturbing OGT/OGA Expression via Hsa-MiR-542-5p. Front. Oncol. 2022, 12, 2934. [Google Scholar] [CrossRef] [PubMed]
- Teng, P.-N.; Bateman, N.W.; Darcy, K.M.; Hamilton, C.A.; Maxwell, G.L.; Bakkenist, C.J.; Conrads, T.P. Pharmacologic inhibition of ATR and ATM offers clinically important distinctions to enhancing platinum or radiation response in ovarian, endometrial, and cervical cancer cells. Gynecol. Oncol. 2015, 136, 554–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, M.; Bouzid, H.; Soares, D.G.; Selle, F.; Morel, C.; Galmarini, C.M.; Henriques, J.A.P.; Larsen, A.K.; Escargueil, A.E. Dual inhibition of ATR and ATM potentiates the activity of trabectedin and lurbinectedin by perturbing the DNA damage response and homologous recombination repair. Oncotarget 2016, 7, 25885–25901. [Google Scholar] [CrossRef] [PubMed]
- Grimley, E.; Cole, A.J.; Luong, T.T.; McGonigal, S.C.; Sinno, S.; Yang, D.; Bernstein, K.A.; Buckanovich, R.J. Aldehyde dehydrogenase inhibitors promote DNA damage in ovarian cancer and synergize with ATM/ATR inhibitors. Theranostics 2021, 11, 3540–3551. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.; Zenke, F.T.; Curtin, N.J.; Drew, Y. The Role of ATR Inhibitors in Ovarian Cancer: Investigating Predictive Biomarkers of Response. Cells 2022, 11, 2361. [Google Scholar] [CrossRef] [PubMed]
- Huntoon, C.J.; Flatten, K.S.; Hendrickson, A.E.W.; Huehls, A.M.; Sutor, S.L.; Kaufmann, S.H.; Karnitz, L.M. ATR Inhibition Broadly Sensitizes Ovarian Cancer Cells to Chemotherapy Independent of BRCA Status. Cancer Res 2013, 73, 3683–3691. [Google Scholar] [CrossRef] [Green Version]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.L.; Willmore, E.; Mukhopadhyay, A.; Drew, Y.; Curtin, N.J. Differences in Durability of PARP Inhibition by Clinically Approved PARP Inhibitors: Implications for Combinations and Scheduling. cancers 2022, 22, 5559. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.-M.; Nguyen, H.D.; Ho, C.K.; Kwan, T.T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef] [Green Version]
- Hur, J.; Ghosh, M.; Kim, T.H.; Park, N.; Pandey, K.; Cho, Y.B.; Hong, S.D.; Katuwal, N.B.; Kang, M.; An, H.J.; et al. Synergism of AZD6738, an ATR Inhibitor, in Combination with Belotecan, a Camptothecin Analogue, in Chemotherapy-Resistant Ovarian Cancer. Int. J. Mol. Sci. 2021, 22, 1223. [Google Scholar] [CrossRef]
- Kim, H.; George, E.; Ragland, R.L.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.A.; Herlyn, M.; Brown, E.J.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef] [PubMed]
- Wilson, Z.; Odedra, R.; Wallez, Y.; Wijnhoven, P.W.G.; Hughes, A.M.; Gerrard, J.; Jones, G.N.; Bargh-Dawson, H.; Brown, E.; Young, L.A.; et al. ATR Inhibitor AZD6738 (Ceralasertib) Exerts Antitumor Activity as a Monotherapy and in Combination with Chemotherapy and the PARP Inhibitor Olaparib. Cancer Res 2022, 82, 1140–1152. [Google Scholar] [CrossRef]
- Feng, W.; Dean, D.C.; Hornicek, F.J.; Wang, J.; Jia, Y.; Duan, Z.; Shi, H. ATR and p-ATR are emerging prognostic biomarkers and DNA damage response targets in ovarian cancer. Ther. Adv. Med. Oncol. 2020, 12, 1–18. [Google Scholar] [CrossRef]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef] [Green Version]
- Peasland, A.; Wang, L.-Z.; Rowling, E.; Kyle, S.; Chen, T.; Hopkins, A.; Cliby, W.A.; Sarkaria, J.; Beale, G.; Edmondson, R.J.; et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer 2011, 105, 372–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siemeister, G.; Mengel, A.; Fernández-Montalván, A.E.; Bone, W.; Schröder, J.; Zitzmann-Kolbe, S.; Briem, H.; Prechtl, S.; Holton, S.J.; Mönning, U.; et al. Inhibition of BUB1 Kinase by BAY 1816032 Sensitizes Tumor Cells toward Taxanes, ATR, and PARP Inhibitors In Vitro and In Vivo. Clin. Cancer Res. 2019, 25, 1404–1414. [Google Scholar] [CrossRef] [Green Version]
- Wengner, A.M.; Siemeister, G.; Lücking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, L.P.B.; Bömer, U.; Moosmayer, D.; Eberspächer, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage–Inducing or Repair–Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef]
- Wickstroem, K.; Hagemann, U.B.; Cruciani, V.; Wengner, A.M.; Kristian, A.; Ellingsen, C.; Siemeister, G.; Bjerke, R.M.; Karlsson, J.; Ryan, O.B.; et al. Synergistic Effect of a Mesothelin-Targeted 227Th Conjugate in Combination with DNA Damage Response Inhibitors in Ovarian Cancer Xenograft Models. J. Nucl. Med. 2019, 60, 1293–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, U.; Senatorov, I.S.; Zimmermann, A.; Saha, L.K.; Murai, Y.; Kim, S.H.; Rajapakse, V.N.; Elloumi, F.; Takahashi, N.; Schultz, C.W.; et al. Novel and Highly Potent ATR Inhibitor M4344 Kills Cancer Cells With Replication Stress, and Enhances the Chemotherapeutic Activity of Widely Used DNA Damaging Agents. Mol. Cancer Ther. 2021, 20, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.; Poklepovic, A.; Dent, P. The CHK1 inhibitor SRA737 synergizes with PARP1 inhibitors to kill carcinoma cells. Cancer Biol. Ther. 2018, 19, 786–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, C.; Rawlinson, R.; Massey, A.J. Chk1 Inhibition as a novel therapeutic strategy for treating triple-negative breast and ovarian cancers. BMC Cancer 2014, 14, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, B.T.; Anderson, A.M.; McCorkle, J.R.; Wu, J.; Ueland, F.R.; Kolesar, J.M. Olaparib Combined with an ATR or Chk1 Inhibitor as a Treatment Strategy for Acquired Olaparib-Resistant BRCA1 Mutant Ovarian Cells. Diagnostics 2020, 10, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.K.; James, J.; Annunziata, C.M. Topotecan synergizes with CHEK1 (CHK1) inhibitor to induce apoptosis in ovarian cancer cells. BMC Cancer 2015, 15, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobson, A.G.; Lountos, G.T.; Lorenzi, P.L.; Llamas, J.; Connelly, J.; Cerna, D.; Tropea, J.E.; Onda, A.; Zoppoli, G.; Kondapaka, S.; et al. Cellular Inhibition of Checkpoint Kinase 2 (Chk2) and Potentiation of Camptothecins and Radiation by the Novel Chk2 Inhibitor PV1019 [7-Nitro-1H-indole-2-carboxylic acid {4-[1-(guanidinohydrazone)-ethyl]-phenyl}-amide]. J. Pharmacol. Exp. Ther. 2009, 331, 816–826. [Google Scholar] [CrossRef] [Green Version]
- Han, J.H.-J.; Kim, K.-T.; Im, J.; Park, S.; Choi, M.K.; Kim, I.; Nam, K.-Y.; Yoon, J. Abstract 1461: PHI-101, a potent and novel inhibitor of CHK2 in ovarian and breast cancer cells. Cancer Res 2021, 81, 1461. [Google Scholar] [CrossRef]
- Liang, X.; Guo, Y.; Figg, W.D.; Fojo, A.T.; Mueller, M.D.; Yu, J.J. The Role of Wild-Type p53 in Cisplatin-Induced Chk2 Phosphorylation and the Inhibition of Platinum Resistance with a Chk2 Inhibitor. Chemother. Res. Pr. 2011, 2011, 1–8. [Google Scholar] [CrossRef]
- Itamochi, H.; Nishimura, M.; Oumi, N.; Kato, M.; Oishi, T.; Shimada, M.; Sato, S.; Naniwa, J.; Sato, S.; Kudoh, A.; et al. Checkpoint Kinase Inhibitor AZD7762 Overcomes Cisplatin Resistance in Clear Cell Carcinoma of the Ovary. Int. J. Gynecol. Cancer 2014, 24, 61–69. [Google Scholar] [CrossRef]
- Pillay, N.; Tighe, A.; Nelson, L.; Littler, S.; Coulson-Gilmer, C.; Bah, N.; Golder, A.; Bakker, B.; Spierings, D.C.; James, D.I.; et al. DNA Replication Vulnerabilities Render Ovarian Cancer Cells Sensitive to Poly(ADP-Ribose) Glycohydrolase Inhibitors. Cancer Cell 2019, 35, 519–533.e8. [Google Scholar] [CrossRef] [Green Version]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.-B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Färkkilä, A.; Reznichenko, E.; et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef]
- Brill, E.; Yokoyama, T.; Nair, J.; Yu, M.; Ahn, Y.-R.; Lee, J.-M. Prexasertib, a cell cycle checkpoint kinases 1 and 2 inhibitor, increases in vitro toxicity of PARP inhibition by preventing Rad51 foci formation in BRCA wild type high-grade serous ovarian cancer. Oncotarget 2017, 8, 111026–111040. [Google Scholar] [CrossRef]
- Roering, P.; Siddiqui, A.; Heuser, V.D.; Potdar, S.; Mikkonen, P.; Oikkonen, J.; Li, Y.; Pikkusaari, S.; Wennerberg, K.; Hynninen, J.; et al. Effects of Wee1 inhibitor adavosertib on patient-derived high-grade serous ovarian cancer cells are multiple and independent of homologous recombination status. Front. Oncol. 2022, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Kang, X.; Zhang, X.; Xie, W.; Su, Y.; Liu, X.; Guo, L.; Guo, E.; Li, F.; Hu, D.; et al. WEE1 inhibitor and ataxia telangiectasia and RAD3-related inhibitor trigger stimulator of interferon gene-dependent immune response and enhance tumor treatment efficacy through programmed death-ligand 1 blockade. Cancer Sci. 2021, 112, 4444–4456. [Google Scholar] [CrossRef] [PubMed]
- Carrassa, L.; Chilà, R.; Lupi, M.; Ricci, F.; Celenza, C.; Mazzoletti, M.; Broggini, M.; Damia, G. Combined inhibition of Chk1 and Wee1: In vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle 2012, 11, 2507–2517. [Google Scholar] [CrossRef] [Green Version]
- Lindenblatt, D.; Terraneo, N.; Pellegrini, G.; Cohrs, S.; Spycher, P.R.; Vukovic, D.; Béhé, M.; Schibli, R.; Grünberg, J. Combination of lutetium-177 labelled anti-L1CAM antibody chCE7 with the clinically relevant protein kinase inhibitor MK1775: A novel combination against human ovarian carcinoma. BMC Cancer 2018, 18, 922. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Abedini, M.; Sakuragi, N.; Tsang, B.K. PRIMA-1 increases cisplatin sensitivity in chemoresistant ovarian cancer cells with p53 mutation: A requirement for Akt down-regulation. J. Ovarian Res. 2013, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bykov, V.J.N.; Issaeva, N.; Selivanova, G.; Wiman, K.G. Mutant p53-dependent growth suppression distinguishes PRIMA-1 from known anticancer drugs: A statistical analysis of information in the National Cancer Institute database. Carcinogenesis 2002, 23, 2011–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohell, N.; Alfredsson, J.; Fransson, A.; Uustalu, M.; Bystrom, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.N.; Bjorklund, U.; Wiman, K.G. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef] [PubMed]
- Fransson, Å.; Glaessgen, D.; Alfredsson, J.; Wiman, K.G.; Bajalica-Lagercrantz, S.; Mohell, N. Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with TP53 mutant High-Grade Serous ovarian cancer. J. Ovarian Res. 2016, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Nguyen, A.T.-Q.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Neal, A.; Lai, T.; Singh, T.; Rahseparian, N.; Grogan, T.; Elashoff, D.; Scott, P.; Pellegrini, M.; Memarzadeh, S. Combining ReACp53 with Carboplatin to Target High-Grade Serous Ovarian Cancers. Cancers 2021, 13, 5908. [Google Scholar] [CrossRef]
- Crane, E.K.; Kwan, S.-Y.; Izaguirre, D.I.; Tsang, Y.T.M.; Mullany, L.K.; Zu, Z.; Richards, J.S.; Gershenson, D.M.; Wong, K.-K. Nutlin-3a: A Potential Therapeutic Opportunity for TP53 Wild-Type Ovarian Carcinomas. PLoS ONE 2015, 10, e0135101. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Kruyt, F.A.E.; Van Der Zee, A.G.J.; Hollema, H.; Le, P.; Hoor, K.A.T.; Groothuis, G.M.M.; Quax, W.J.; de Vries, E.G.E.; De Jong, S. Nutlin-3 preferentially sensitises wild-type p53-expressing cancer cells to DR5-selective TRAIL over rhTRAIL. Br. J. Cancer 2013, 109, 2685–2695. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; He, G.; Siddik, Z.H. Cisplatin in Combination with MDM2 Inhibition Downregulates Rad51 Recombinase in a Bimodal Manner to Inhibit Homologous Recombination and Augment Tumor Cell Kill. Mol. Pharmacol. 2020, 97, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Zanjirband, M.; Edmondson, R.J.; Lunec, J. Pre-clinical efficacy and synergistic potential of the MDM2-p53 antagonists, Nutlin-3 and RG7388, as single agents and in combined treatment with cisplatin in ovarian cancer. Oncotarget 2016, 7, 40115–40134. [Google Scholar] [CrossRef] [Green Version]
- Mir, R.; Tortosa, A.; Martinez-Soler, F.; Vidal, A.; Condom, E.; Pérez-Perarnau, A.; Ruiz-Larroya, T.; Gil, J.; Giménez-Bonafé, P. Mdm2 antagonists induce apoptosis and synergize with cisplatin overcoming chemoresistance inTP53wild-type ovarian cancer cells. Int. J. Cancer 2013, 132, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Zanjirband, M.; Curtin, N.; Edmondson, R.J.; Lunec, J. Combination treatment with rucaparib (Rubraca) and MDM2 inhibitors, Nutlin-3 and RG7388, has synergistic and dose reduction potential in ovarian cancer. Oncotarget 2017, 8, 69779–69796. [Google Scholar] [CrossRef]
- Makii, C.; Oda, K.; Ikeda, Y.; Sone, K.; Hasegawa, K.; Uehara, Y.; Nishijima, A.; Asada, K.; Koso, T.; Fukuda, T.; et al. MDM2 is a potential therapeutic target and prognostic factor for ovarian clear cell carcinomas with wild type TP53. Oncotarget 2016, 7, 75328–75338. [Google Scholar] [CrossRef]
- Ranson, M.; Middleton, M.R.; Bridgewater, J.; Lee, S.M.; Dawson, M.; Jowle, D.; Halbert, G.; Waller, S.; McGrath, H.; Gumbrell, L.; et al. Lomeguatrib, a Potent Inhibitor of O6-Alkylguanine-DNA-Alkyltransferase: Phase I Safety, Pharmacodynamic, and Pharmacokinetic Trial and Evaluation in Combination with Temozolomide in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2006, 12, 1577–1584. [Google Scholar] [CrossRef] [Green Version]
- Del Campo, J.M.; Roszak, A.; Bidzinski, M.; Ciuleanu, T.E.; Hogberg, T.; Wojtukiewicz, M.Z.; Poveda, A.; Boman, K.; Westermann, A.M.; Lebedinsky, C. Phase II randomized study of trabectedin given as two different every 3 weeks dose schedules (1.5 mg/m2 24 h or 1.3 mg/m2 3 h) to patients with relapsed, platinum-sensitive, advanced ovarian cancer. Ann. Oncol. 2009, 20, 1794–1802. [Google Scholar] [CrossRef]
- Thertulien, R.; Manikhas, G.M.; Dirix, L.Y.; Vermorken, J.B.; Park, K.; Jain, M.M.; Jiao, J.J.; Natarajan, J.; Parekh, T.; Zannikos, P.; et al. Effect of trabectedin on the QT interval in patients with advanced solid tumor malignancies. Cancer Chemother. Pharmacol. 2012, 69, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Yovine, A.; Riofrio, M.; Blay, J.-Y.; Brain, E.; Alexandre, J.; Kahatt, C.; Taamma, A.; Jimeno, J.; Martin, C.; Salhi, Y.; et al. Phase II Study of Ecteinascidin-743 in Advanced Pretreated Soft Tissue Sarcoma Patients. J. Clin. Oncol. 2004, 22, 890–899. [Google Scholar] [CrossRef]
- Lorusso, D.; Pignata, S.; Tamberi, S.; Mangili, G.; Bologna, A.; Nicoloso, M.S.; Giolitto, S.; Salutari, V.; Mantero, M.; Pisano, C.; et al. Efficacy and safety of trabectedin for the treatment of advanced uterine or ovarian carcinosarcoma: Results of a phase II multicenter clinical trial (MITO-26). Gynecol. Oncol. 2022, 167, 436–443. [Google Scholar] [CrossRef]
- Lorusso, D.; Scambia, G.; Pignata, S.; Sorio, R.; Amadio, G.; Lepori, S.; Mosconi, A.; Pisano, C.; Mangili, G.; Maltese, G.; et al. Prospective phase II trial of trabectedin in BRCA-mutated and/or BRCAness phenotype recurrent ovarian cancer patients: The MITO 15 trial. Ann. Oncol. 2016, 27, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Scambia, G.; Raspagliesi, F.; Valabrega, G.; Colombo, N.; Pisano, C.; Cassani, C.; Tognon, G.; Tamberi, S.; Mangili, G.; Mammoliti, S.; et al. Randomized phase III trial on trabectedin (ET-743) single agent versus clinician’s choice chemotherapy in recurrent ovarian, primary peritoneal, or fallopian tube cancers of BRCA-mutated or BRCAness phenotype patients (MITO23). J. Clin. Oncol. 2022, 40, LBA5504. [Google Scholar] [CrossRef]
- Colombo, N.; Gadducci, A.; Sehouli, J.; Biagioli, E.; Nyvang, G.-B.; Riniker, S.; Montes, A.; Ottevanger, N.; Zeimet, A.G.G.; Vergote, I.B.; et al. LBA30 INOVATYON study: Randomized phase III international study comparing trabectedin/PLD followed by platinum at progression vs carboplatin/PLD in patients with recurrent ovarian cancer progressing within 6–12 months after last platinum line. Ann. Oncol. 2020, 31, S1161. [Google Scholar] [CrossRef]
- Jones, R.L.; Herzog, T.J.; Patel, S.R.; von Mehren, M.; Schuetze, S.M.; Van Tine, B.A.; Coleman, R.L.; Knoblauch, R.; Triantos, S.; Hu, P.; et al. Cardiac safety of trabectedin monotherapy or in combination with pegylated liposomal doxorubicin in patients with sarcomas and ovarian cancer. Cancer Med. 2021, 10, 3565–3574. [Google Scholar] [CrossRef]
- Krasner, C.N.; Poveda, A.; Herzog, T.J.; Vermorken, J.B.; Kaye, S.B.; Nieto, A.; Claret, P.L.; Park, Y.C.; Parekh, T.; Monk, B.J. Patient-reported outcomes in relapsed ovarian cancer: Results from a randomized Phase III study of trabectedin with pegylated liposomal doxorubicin (PLD) versus PLD Alone. Gynecol. Oncol. 2012, 127, 161–167. [Google Scholar] [CrossRef]
- Monk, B.J.; Herzog, T.J.; Wang, G.; Triantos, S.; Maul, S.; Knoblauch, R.; McGowan, T.; Shalaby, W.S.W.; Coleman, R.L. A phase 3 randomized, open-label, multicenter trial for safety and efficacy of combined trabectedin and pegylated liposomal doxorubicin therapy for recurrent ovarian cancer. Gynecol. Oncol. 2020, 156, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Herzog, T.J.; Kaye, S.B.; Krasner, C.N.; Vermorken, J.B.; Muggia, F.M.; Pujade-Lauraine, E.; Park, Y.C.; Parekh, T.V.; Poveda, A.M. Trabectedin plus pegylated liposomal doxorubicin (PLD) versus PLD in recurrent ovarian cancer: Overall survival analysis. Eur. J. Cancer 2012, 48, 2361–2368. [Google Scholar] [CrossRef]
- Chekerov, R.; Deryal, M.; Aktas, B.; Röhle, R.; Stürzebecher, A.; Battista, M.J.; Kurbacher, C.M.; Wimberger, P.; Lorenz, R.; Barinoff, J.; et al. 2022-RA-672-ESGO Comparison of quality of life in patients with platinum-sensitive recurrent ovarian, fallopian tube and peritoneal cancer treated with trabectedin plus pegylated liposomal doxorubicin (PLD) or standard platinum-based therapy: Data look of the NOGGO S16/COMPASS trial. Int. J. Gynecol. Cancer 2022, 32, A252–A253. [Google Scholar] [CrossRef]
- Romero, I.; Mallol, P.; Santaballa, A.; Del Campo, J.M.; Mori, M.; González-Santiago, S.; Casado, A.; Vicente, D.; Ortega, E.; Herrero, A.; et al. Multicenter retrospective study to evaluate the impact of trabectedin plus pegylated liposomal doxorubicin on the subsequent treatment in women with recurrent, platinum-sensitive ovarian cancer. Anti-Cancer Drugs 2019, 30, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Pignata, S.; Scambia, G.; Villanucci, A.; Naglieri, E.; Ibarbia, M.A.; Brusa, F.; Bourgeois, H.; Sorio, R.; Casado, A.; Reichert, D.; et al. A European, Observational, Prospective Trial of Trabectedin Plus Pegylated Liposomal Doxorubicin in Patients with Platinum-Sensitive Ovarian Cancer. Oncologist 2021, 26, e658–e668. [Google Scholar] [CrossRef] [PubMed]
- Selle, F.; Heudel, P.-E.; Hardy-Bessard, A.-C.; Pozet, A.; Meunier, J.; Gladieff, L.; Lotz, J.-P.; Provansal, M.; Augereau, P.; Berton, D.; et al. GINECO Prospective Non-interventional PROSPECTYON Study: Trabectedin Plus Pegylated Liposomal Doxorubicin for Platinum-sensitive Recurrent Ovarian Cancer. Anticancer. Res. 2020, 40, 3939–3945. [Google Scholar] [CrossRef] [PubMed]
- Runnebaum, I.B.; Reichert, D.; Ringsdorf, U.; Kuther, M.; Hesse, T.; Sehouli, J.; Wimberger, P. Trabectedin plus pegylated liposomal doxorubicin (PLD) for patients with platinum-sensitive recurrent ovarian cancer: A prospective, observational, multicenter study. J. Cancer Res. Clin. Oncol. 2018, 144, 1185–1195. [Google Scholar] [CrossRef] [Green Version]
- Toulmonde, M.; Brahmi, M.; Giraud, A.; Chakiba, C.; Bessede, A.; Kind, M.; Toulza, E.; Pulido, M.; Albert, S.; Guégan, J.-P.; et al. Trabectedin plus Durvalumab in Patients with Advanced Pretreated Soft Tissue Sarcoma and Ovarian Carcinoma (TRAMUNE): An Open-Label, Multicenter Phase Ib Study. Clin. Cancer Res. 2022, 28, 1765–1772. [Google Scholar] [CrossRef]
- Monk, B.J.; Sill, M.W.; Hanjani, P.; Edwards, R.; Rotmensch, J.; De Geest, K.; Bonebrake, A.J.; Walker, J.L. Docetaxel plus trabectedin appears active in recurrent or persistent ovarian and primary peritoneal cancer after up to three prior regimens: A phase II study of the Gynecologic Oncology Group. Gynecol. Oncol. 2011, 120, 459–463. [Google Scholar] [CrossRef]
- Colombo, N.; Zaccarelli, E.; Baldoni, A.; Frezzini, S.; Scambia, G.; Palluzzi, E.; Tognon, G.; Lissoni, A.A.; Rubino, D.; Ferrero, A.; et al. Multicenter, randomised, open-label, non-comparative phase 2 trial on the efficacy and safety of the combination of bevacizumab and trabectedin with or without carboplatin in women with partially platinum-sensitive recurrent ovarian cancer. Br. J. Cancer 2019, 121, 744–750. [Google Scholar] [CrossRef]
- Calvo, E.; Sessa, C.; Harada, G.; de Miguel, M.; Kahatt, C.; Luepke-Estefan, X.E.; Siguero, M.; Fernandez-Teruel, C.; Cullell-Young, M.; Stathis, A.; et al. Phase I study of lurbinectedin in combination with weekly paclitaxel with or without bevacizumab in patients with advanced solid tumors. Investig. New Drugs 2022, 40, 1263–1273. [Google Scholar] [CrossRef]
- Poveda, A.; Oaknin, A.; Romero, I.; Guerrero, A.; Fariñas-Madrid, L.; Rodriguez-Freixinos, V.; Soto-Matos, A.; Peris, C.; Teruel, M.; Lopez-Reig, R.; et al. Phase I study to evaluate the tolerability, pharmacokinetics (PK) and pharmacodynamic (PD) of PM01183 (Lurbinectedin) in combination with olaparib in patients with advanced solid tumors. J. Clin. Oncol. 2017, 35, 5573. [Google Scholar] [CrossRef]
- Cortesi, L.; Venturelli, M.; Barbieri, E.; Baldessari, C.; Bardasi, C.; Coccia, E.; Baglio, F.; Rimini, M.; Greco, S.; Napolitano, M.; et al. Exceptional response to lurbinectedin and irinotecan in BRCA-mutated platinum-resistant ovarian cancer patient: A case report. Ther. Adv. Chronic Dis. 2022, 13, 20406223211063023. [Google Scholar] [CrossRef]
- Gaillard, S.; Oaknin, A.; Ray-Coquard, I.; Vergote, I.; Scambia, G.; Colombo, N.; Fernandez, C.; Alfaro, V.; Kahatt, C.; Nieto, A.; et al. Lurbinectedin versus pegylated liposomal doxorubicin or topotecan in patients with platinum-resistant ovarian cancer: A multicenter, randomized, controlled, open-label phase 3 study (CORAIL). Gynecol. Oncol. 2021, 163, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Coyne, G.O.; Kummar, S.; Meehan, R.S.; Do, K.; Collins, J.M.; Anderson, L.; Ishii, K.; Takebe, N.; Zlott, J.; Juwara, L.; et al. Phase I trial of TRC102 (methoxyamine HCl) in combination with temozolomide in patients with relapsed solid tumors and lymphomas. Oncotarget 2020, 11, 3959–3971. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Oizumi, S.; Minami, H.; Kitagawa, K.; Komatsu, Y.; Fujiwara, Y.; Inada, M.; Yuki, S.; Kiyota, N.; Mitsuma, A.; et al. Phase I dose-escalating study of panobinostat (LBH589) Administered intravenously to Japanese patients with advanced solid tumors. Investig. New Drugs 2012, 30, 1950–1957. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.F.; Infante, J.R.; Thompson, D.S.; Mohyuddin, A.; Bendell, J.C.; Yardley, D.A.; Burris, H.A. A phase I trial of oral administration of panobinostat in combination with paclitaxel and carboplatin in patients with solid tumors. Cancer Chemother. Pharmacol. 2012, 70, 471–475. [Google Scholar] [CrossRef]
- Shah, P.D.; Wethington, S.L.; Pagan, C.; Latif, N.; Tanyi, J.; Martin, L.P.; Morgan, M.; Burger, R.A.; Haggerty, A.; Zarrin, H.; et al. Combination ATR and PARP Inhibitor (CAPRI): A phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol. Oncol. 2021, 163, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, H.; Hafez, N.; Doroshow, D.; Sohal, D.; Keedy, V.; Do, K.T.; LoRusso, P.; Jürgensmeier, J.; Avedissian, M.; Sklar, J.; et al. Ceralasertib-Mediated ATR Inhibition Combined With Olaparib in Advanced Cancers Harboring DNA Damage Response and Repair Alterations (Olaparib Combinations). JCO Precis. Oncol. 2021, 5, 1432–1442. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Vergotte, I.; Colombo, N.; Barve, M.; Grisham, R.; Mehr, K.T.; Falk, M.; Beier, F.; Hennessy, M.; Schroeder, A.; et al. Randomized, phase Ib/II study of M6620 + avelumab + carboplatin vs standard care (sc) in patients (pts) with platinum-sensitive poly (ADP-ribose) polymerase inhibitor-(PARPi)-resistant ovarian cancer. Ann. Oncol. 2019, 30, v431–v432. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Wesolowski, R.; Devoe, C.; Lord, S.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R.; Yap, T.A. Phase 1 study of the ATR inhibitor berzosertib in combination with cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; Luken, M.J.D.M.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination With Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef]
- Thomas, A.; Takahashi, N.; Rajapakse, V.N.; Zhang, X.; Sun, Y.; Ceribelli, M.; Wilson, K.M.; Zhang, Y.; Beck, E.; Sciuto, L.; et al. Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell 2021, 39, 566–579.e7. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Cheng, S.-C.; Hendrickson, A.E.W.; Penson, R.T.; Schumer, S.T.; Doyle, L.A.; Lee, E.K.; Kohn, E.C.; Duska, L.R.; Crispens, M.A.; et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Plummer, E.R.; Kristeleit, R.S.; Cojocaru, E.; Haris, N.M.; Carter, L.; Jones, R.H.; Blagden, S.P.; Evans, T.R.J.; Arkenau, H.-T.; Sarker, D.; et al. A first-in-human phase I/II trial of SRA737 (a Chk1 Inhibitor) in subjects with advanced cancer. J. Clin. Oncol. 2019, 37, 3094. [Google Scholar] [CrossRef]
- Banerji, U.; Plummer, E.R.; Moreno, V.; Ang, J.E.; Quinton, A.; Drew, Y.; Hernández, T.; Roda, D.; Carter, L.; Navarro, A.; et al. A phase I/II first-in-human trial of oral SRA737 (a Chk1 inhibitor) given in combination with low-dose gemcitabine in subjects with advanced cancer. J. Clin. Oncol. 2019, 37, 3095. [Google Scholar] [CrossRef]
- Chu, Q.S.; Jonker, D.J.; Provencher, D.M.; Miller, W.H.; Bouganim, N.; Shields, A.F.; Shapiro, G.; Sawyer, M.B.; Lheureux, S.; Samouelian, V.; et al. A phase Ib study of oral Chk1 inhibitor LY2880070 in combination with gemcitabine in patients with advanced or metastatic cancer. J. Clin. Oncol. 2020, 38, 3581. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Lee, J.-M.; Gao, B.; Miller, R.; Lee, J.Y.; Colombo, N.; Vergote, I.; Credille, K.M.; Young, S.R.; McNeely, S.; et al. A Phase 2 study of prexasertib (LY2606368) in platinum resistant or refractory recurrent ovarian cancer. Gynecol. Oncol. 2022, 167, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Do, K.T.; Kochupurakkal, B.S.; Kelland, S.; de Jonge, A.; Hedglin, J.; Powers, A.; Quinn, N.; Gannon, C.; Vuong, L.; Parmar, K.; et al. Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-grade Serous Ovarian Cancer and Other Solid Tumors. Clin. Cancer Res. 2021, 27, 4710–4716. [Google Scholar] [CrossRef]
- Moore, K.N.; Chambers, S.K.; Hamilton, E.P.; Chen, L.-M.; Oza, A.M.; Ghamande, S.A.; Konecny, G.E.; Plaxe, S.C.; Spitz, D.L.; Geenen, J.J.J.; et al. Adavosertib with Chemotherapy in Patients with Primary Platinum-Resistant Ovarian, Fallopian Tube, or Peritoneal Cancer: An Open-Label, Four-Arm, Phase II Study. Clin. Cancer Res. 2022, 28, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; van Geel, R.M.J.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [Green Version]
- Lheureux, S.; Cristea, M.C.; Bruce, J.P.; Garg, S.; Cabanero, M.; Mantia-Smaldone, G.; Olawaiye, A.B.; Ellard, S.L.; Weberpals, J.I.; Hendrickson, A.E.W.; et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 281–292. [Google Scholar] [CrossRef]
- Oza, A.M.; Estevez-Diz, M.D.P.; Grischke, E.-M.; Hall, M.; Marmé, F.; Provencher, D.M.; Uyar, D.S.; Weberpals, J.I.; Wenham, R.M.; Laing, N.; et al. A Biomarker-enriched, Randomized Phase II Trial of Adavosertib (AZD1775) Plus Paclitaxel and Carboplatin for Women with Platinum-sensitive TP53-mutant Ovarian Cancer. Clin. Cancer Res. 2020, 26, 4767–4776. [Google Scholar] [CrossRef]
- Westin, S.N.; Coleman, R.L.; Fellman, B.M.; Yuan, Y.; Sood, A.K.; Soliman, P.T.; Wright, A.A.; Horowitz, N.S.; Campos, S.M.; Konstantinopoulos, P.A.; et al. EFFORT: EFFicacy Of adavosertib in parp ResisTance: A randomized two-arm non-comparative phase II study of adavosertib with or without olaparib in women with PARP-resistant ovarian cancer. J. Clin. Oncol. 2021, 39, 5505. [Google Scholar] [CrossRef]
- Hamilton, E.P.; Wang, J.S.-Z.; Falchook, G.; Jones, S.F.; Cook, C.; Mugundu, G.; Jewsbury, P.J.; O’Connor, M.J.; Pierce, A.J.; Li, B.T.; et al. A phase Ib study of AZD1775 and olaparib combination in patients with refractory solid tumors. J. Clin. Oncol. 2016, 34, 5562. [Google Scholar] [CrossRef]
- Hamilton, E.; Falchook, G.S.; Wang, J.S.; Fu, S.; Oza, A.; Karen, S.; Imedio, E.R.; Kumar, S.; Ottesen, L.; Mugundu, G.M.; et al. Abstract CT025: Phase Ib study of adavosertib in combination with olaparib in patients with refractory solid tumors: Dose escalation. Cancer Res 2019, 79, CT025. [Google Scholar] [CrossRef]
- Basu, B.; Gourley, C.; Gabra, H.; Vergote, I.B.; Brenton, J.D.; Abrahmsen, L.; Smith, A.; Von Euler, M.; Green, J.A. PISARRO: A EUTROC phase 1b study of APR-246 with carboplatin (C) and pegylated liposomal doxorubicin (PLD) in relapsed platinum-sensitive high grade serous ovarian cancer (HGSOC). Ann. Oncol. 2016, 27, vi123. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Gerson, S.L. MGMT: Its role in cancer aetiology and cancer therapeutics. Nat. Rev. Cancer 2004, 4, 296–307. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Luo, Q.; Zhao, P.; Chang, W.; Wang, Y.; Shu, T.; Ding, F.; Li, B.; Liu, Z. MGMT-activated DUB3 stabilizes MCL1 and drives chemoresistance in ovarian cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 2961–2966. [Google Scholar] [CrossRef] [Green Version]
- Toss, A.; Tomasello, C.; Razzaboni, E.; Contu, G.; Grandi, G.; Cagnacci, A.; Schilder, R.J.; Cortesi, L. Hereditary Ovarian Cancer: Not Only BRCA 1 and 2 Genes. BioMed Res. Int. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Cicek, M.S.; Dicks, E.; Harrington, P.; Ramus, S.J.; Cunningham, J.M.; Fridley, B.L.; Tyrer, J.P.; Alsop, J.; Jimenez-Linan, M.; et al. The contribution of deleterious germline mutations in BRCA1, BRCA2 and the mismatch repair genes to ovarian cancer in the population. Hum. Mol. Genet. 2014, 23, 4703–4709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begum, R.; Martin, S.A. Targeting Mismatch Repair defects: A novel strategy for personalized cancer treatment. DNA Repair 2016, 38, 135–139. [Google Scholar] [CrossRef]
- Tomasova, K.; Cumova, A.; Seborova, K.; Horák, J.; Koucka, K.; Vodickova, L.; Vaclavikova, R.; Vodicka, P. DNA Repair and Ovarian Carcinogenesis: Impact on Risk, Prognosis and Therapy Outcome. Cancers 2020, 12, 1713. [Google Scholar] [CrossRef]
- He, Y.; Zhang, L.; Zhou, R.; Wang, Y.; Chen, H. The role of DNA mismatch repair in immunotherapy of human cancer. Int. J. Biol. Sci. 2022, 18, 2821–2832. [Google Scholar] [CrossRef] [PubMed]
- Leary, A.; Tan, D.; Ledermann, J. Immune checkpoint inhibitors in ovarian cancer: Where do we stand? Ther. Adv. Med Oncol. 2021, 13, 17588359211039899. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.R.; Fishel, M.L. Overview of DNA repair pathways, current targets, and clinical trials bench to clinic. In DNA Repair in Cancer Therapy: Molecular Targets and Clinical Applications, 2nd ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 1–54. [Google Scholar] [CrossRef]
- Topka, S.; Steinsnyder, Z.; Ravichandran, V.; Tkachuk, K.; Kemel, Y.; Bandlamudi, C.; Madsen, M.W.; Furberg, H.; Ouerfelli, O.; Rudin, C.M.; et al. Targeting Germline- and Tumor-Associated Nucleotide Excision Repair Defects in Cancer. Clin. Cancer Res. 2021, 27, 1997–2010. [Google Scholar] [CrossRef]
- Ishibashi, M.; Toyoshima, M.; Zhang, X.; Hasegawa-Minato, J.; Shigeta, S.; Usui, T.; Kemp, C.J.; Grandori, C.; Kitatani, K.; Yaegashi, N. Tyrosine kinase receptor TIE-1 mediates platinum resistance by promoting nucleotide excision repair in ovarian cancer. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Duan, M.R.; Ulibarri, J.; Liu, K.J.; Mao, P. Role of Nucleotide Excision Repair in Cisplatin Resistance. Int J Mol Sci 2020, 21, 9248. [Google Scholar] [CrossRef]
- Gentile, F.; Tuszynski, J.A.; Barakat, K.H. New design of nucleotide excision repair (NER) inhibitors for combination cancer therapy. J. Mol. Graph. Model. 2016, 65, 71–82. [Google Scholar] [CrossRef]
- Herrero, A.B.; Martín-Castellanos, C.; Marco, E.; Gago, F.; Moreno, S. Cross-Talk between Nucleotide Excision and Homologous Recombination DNA Repair Pathways in the Mechanism of Action of Antitumor Trabectedin. Cancer Res 2006, 66, 8155–8162. [Google Scholar] [CrossRef] [Green Version]
- Casado, A.; Callata, H.R.; Manzano, A.; Marquina, G.; Alonso, T.; Gajate, P.; Sotelo, M.; Cabezas, S.; Fernández, C.; Díaz-Rubio, E. Trabectedin for reversing platinum resistance and resensitization to platinum in patients with recurrent ovarian cancer. Future Oncol. 2019, 15, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Mabuchi, S.; Kawano, M.; Sasano, T.; Matsumoto, Y.; Kuroda, H.; Kozasa, K.; Hashimoto, K.; Sawada, K.; Kimura, T. Preclinical Investigations of PM01183 (Lurbinectedin) as a Single Agent or in Combination with Other Anticancer Agents for Clear Cell Carcinoma of the Ovary. PLoS ONE 2016, 11, e0151050. [Google Scholar] [CrossRef]
- Kawano, M.; Mabuchi, S.; Kishimoto, T.; Hisamatsu, T.; Matsumoto, Y.; Sasano, T.; Takahashi, R.; Sawada, K.; Takahashi, K.; Takahashi, T.; et al. Combination Treatment With Trabectedin and Irinotecan or Topotecan Has Synergistic Effects Against Ovarian Clear Cell Carcinoma Cells. Int. J. Gynecol. Cancer 2014, 24, 829–837. [Google Scholar] [CrossRef]
- Ventriglia, J.; Paciolla, I.; Cecere, S.C.; Pisano, C.; Di Napoli, M.; Arenare, L.; Setola, S.V.; Losito, N.S.; Califano, D.; Orditura, M.; et al. Trabectedin in Ovarian Cancer: Is it now a Standard of Care? Clin. Oncol. 2018, 30, 498–503. [Google Scholar] [CrossRef]
- Monk, B.J.; Herzog, T.J.; Kaye, S.B.; Krasner, C.N.; Vermorken, J.B.; Muggia, F.M.; Pujade-Lauraine, E.; Lisyanskaya, A.S.; Makhson, A.N.; Rolski, J.; et al. Trabectedin Plus Pegylated Liposomal Doxorubicin in Recurrent Ovarian Cancer. J. Clin. Oncol. 2010, 28, 3107–3114. [Google Scholar] [CrossRef]
- Monk, B.J.; Dalton, H.; Benjamin, I.; Tanovic, A. Trabectedin as a New Chemotherapy Option in the Treatment of Relapsed Platinum Sensitive Ovarian Cancer. Curr. Pharm. Des. 2012, 18, 3754–3769. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Yondelis. Available online: https://www.ema.europa.eu/en/medicines/human/referrals/yondelis#overview-section (accessed on 17 October 2022).
- Vodicka, P.; Urbanova, M.; Makovicky, P.; Tomasova, K.; Kroupa, M.; Stetina, R.; Opattova, A.; Kostovcikova, K.; Siskova, A.; Schneiderova, M.; et al. Oxidative Damage in Sporadic Colorectal Cancer: Molecular Mapping of Base Excision Repair Glycosylases in Colorectal Cancer Patients. Int. J. Mol. Sci. 2020, 21, 2473. [Google Scholar] [CrossRef] [Green Version]
- Stoffel, E.M.; Mangu, P.B.; Limburg, P.J. Hereditary Colorectal Cancer Syndromes: American Society of Clinical Oncology Clinical Practice Guideline Endorsement of the Familial Risk–Colorectal Cancer: European Society for Medical Oncology Clinical Practice Guidelines. J. Oncol. Pr. 2015, 11, e437–e441. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Lu, R.; Xie, S.; Zheng, H.; Wang, H.; Wang, Y.; Sun, J.; Gao, X.; Guo, L. APE1 overexpression promotes the progression of ovarian cancer and serves as a potential therapeutic target. Cancer Biomarkers 2016, 17, 313–322. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Xiang, D.; Wang, D.; Xin, X. Alterations in the expression of the apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE1/Ref-1) in human ovarian cancer and indentification of the therapeutic potential of APE1/Ref-1 inhibitor. Int. J. Oncol. 2009, 35, 1069–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, F.; Zhang, Y.; Chen, S.; Weng, X.; Rao, Y.; Fang, H. Mechanism and current progress of Poly ADP-ribose polymerase (PARP) inhibitors in the treatment of ovarian cancer. Biomed. Pharmacother. 2020, 123, 109661. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Nowsheen, S.; Maraboyina, S.; Xia, F. The role of poly(ADP-ribose) polymerase inhibitors in the treatment of cancer and methods to overcome resistance: A review. Cell Biosci. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 879. [Google Scholar] [CrossRef]
- Kyo, S.; Kanno, K.; Takakura, M.; Yamashita, H.; Ishikawa, M.; Ishibashi, T.; Sato, S.; Nakayama, K. Clinical Landscape of PARP Inhibitors in Ovarian Cancer: Molecular Mechanisms and Clues to Overcome Resistance. Cancers 2022, 14, 2504. [Google Scholar] [CrossRef]
- Miller, R.E.; El-Shakankery, K.H.; Lee, J.Y. PARP inhibitors in ovarian cancer: Overcoming resistance with combination strategies. J. Gynecol. Oncol. 2022, 33, e44. [Google Scholar] [CrossRef] [PubMed]
- Kakoti, S.; Sato, H.; Laskar, S.; Yasuhara, T.; Shibata, A. DNA Repair and Signaling in Immune-Related Cancer Therapy. Front. Mol. Biosci. 2020, 7, 205. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Plummer, E.R.; Elattar, A.; Soohoo, S.; Uzir, B.; Quinn, J.E.; McCluggage, W.G.; Maxwell, P.; Aneke, H.; Curtin, N.J.; et al. Clinicopathological Features of Homologous Recombination–Deficient Epithelial Ovarian Cancers: Sensitivity to PARP Inhibitors, Platinum, and Survival. Cancer Res. 2012, 72, 5675–5682. [Google Scholar] [CrossRef] [Green Version]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wise, H.C.; Iyer, G.V.; Moore, K.; Temkin, S.M.; Gordon, S.; Aghajanian, C.; Grisham, R.N. Activity of M3814, an Oral DNA-PK Inhibitor, In Combination with Topoisomerase II Inhibitors in Ovarian Cancer Models. Sci. Rep. 2019, 9, 18882. [Google Scholar] [CrossRef] [Green Version]
- Gordhandas, S.B.; Manning-Geist, B.; Henson, C.; Iyer, G.; Gardner, G.J.; Sonoda, Y.; Moore, K.N.; Aghajanian, C.; Chui, M.H.; Grisham, R.N. Pre-clinical activity of the oral DNA-PK inhibitor, peposertib (M3814), combined with radiation in xenograft models of cervical cancer. Sci. Rep. 2022, 12, 1–6. [Google Scholar] [CrossRef]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero, A.B.; San Miguel, J.; Gutierrez, N.C. Deregulation of DNA Double-Strand Break Repair in Multiple Myeloma: Implications for Genome Stability. PLoS ONE 2015, 10, e0121581. [Google Scholar] [CrossRef]
- Medova, M.; Medo, M.; Hovhannisyan, L.; Munoz-Maldonado, C.; Aebersold, D.M.; Zimmer, Y. DNA-PK in human malignant disorders: Mechanisms and implications for pharmacological interventions. Pharmacol Therapeut 2020, 215. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.Gov. A Study Combining the Peposertib (M3814) Pill with Standard Chemotherapy in Patients with Ovarian Cancer with an Expansion in High Grade Serous Ovarian Cancer and Low Grade Serous Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04092270 (accessed on 6 October 2022).
- Sekiguchi, M.; Matsushita, N. DNA Damage Response Regulation by Histone Ubiquitination. Int. J. Mol. Sci. 2022, 23, 8187. [Google Scholar] [CrossRef]
- Mayes, K.; Qiu, Z.; Alhazmi, A.; Landry, J.W. ATP-Dependent Chromatin Remodeling Complexes as Novel Targets for Cancer Therapy. Adv. Cancer Res. 2014, 121, 183–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klochendler-Yeivin, A.; Picarsky, E.; Yaniv, M. Increased DNA Damage Sensitivity and Apoptosis in Cells Lacking the Snf5/Ini1 Subunit of the SWI/SNF Chromatin Remodeling Complex. Mol. Cell. Biol. 2006, 26, 2661–2674. [Google Scholar] [CrossRef] [Green Version]
- Mandal, J.; Mandal, P.; Wang, T.-L.; Shih, I.-M. Treating ARID1A mutated cancers by harnessing synthetic lethality and DNA damage response. J. Biomed. Sci. 2022, 29, 1–15. [Google Scholar] [CrossRef]
- Gospodinov, A.; Vaissiere, T.; Krastev, D.B.; Legube, G.; Anachkova, B.; Herceg, Z. Mammalian Ino80 Mediates Double-Strand Break Repair through Its Role in DNA End Strand Resection. Mol. Cell. Biol. 2011, 31, 4735–4745. [Google Scholar] [CrossRef] [Green Version]
- Guillemette, S.; Serra, R.W.; Peng, M.; Hayes, J.A.; Konstantinopoulos, P.A.; Green, M.R.; Cantor, S.B. Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev. 2015, 29, 489–494. [Google Scholar] [CrossRef]
- Li, Y.; Gong, H.; Wang, P.; Zhu, Y.; Peng, H.; Cui, Y.; Li, H.; Liu, J.; Wang, Z. The emerging role of ISWI chromatin remodeling complexes in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 1–27. [Google Scholar] [CrossRef]
- Hall, M.J.; Bernhisel, R.; Hughes, E.; Larson, K.; Rosenthal, E.T.; Singh, N.A.; Lancaster, J.M.; Kurian, A.W. Germline Pathogenic Variants in the Ataxia Telangiectasia Mutated (ATM) Gene are Associated with High and Moderate Risks for Multiple Cancers. Cancer Prev. Res. 2021, 14, 433–440. [Google Scholar] [CrossRef]
- Sugino, K.; Tamura, R.; Nakaoka, H.; Yachida, N.; Yamaguchi, M.; Mori, Y.; Yamawaki, K.; Suda, K.; Ishiguro, T.; Adachi, S.; et al. Germline and somatic mutations of homologous recombination-associated genes in Japanese ovarian cancer patients. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Llorens-Agost, M.; Luessing, J.; van Beneden, A.; Eykelenboom, J.; O’Reilly, D.; Bicknell, L.S.; Reynolds, J.J.; van Koegelenberg, M.; Hurles, M.E.; Brady, A.F.; et al. Analysis of novel missense ATR mutations reveals new splicing defects underlying Seckel syndrome. Hum. Mutat. 2018, 39, 1847–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolarova, L.; Kleiblova, P.; Janatova, M.; Soukupova, J.; Zemankova, P.; Macurek, L.; Kleibl, Z. CHEK2 Germline Variants in Cancer Predisposition: Stalemate Rather than Checkmate. Cells 2020, 9, 2675. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, D.A. When more is less: Heritable gain-of-function chk1 mutations impair human fertility. FEBS J. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Martin, A.R.; Demange, L.; Benhida, R. ATM, ATR, CHK1, CHK2 and WEE1 inhibitors in cancer and cancer stem cells. MedChemComm 2016, 8, 295–319. [Google Scholar] [CrossRef]
- Chen, C.-W.; Buj, R.; Dahl, E.S.; Leon, K.E.; Aird, K.M. ATM inhibition synergizes with fenofibrate in high grade serous ovarian cancer cells. Heliyon 2020, 6, e05097. [Google Scholar] [CrossRef]
- Biegala, L.; Gajek, A.; Marczak, A.; Rogalska, A. Olaparib in Combination with Inhibitors of Atr/Chk1 Pathway Leads to Increased Cell Death in Ovarian Cancer Cells Sensitive and Resistant to Parpi. Int J Gynecol Cancer 2021, 31, A375–A376. [Google Scholar] [CrossRef]
- Barnieh, F.M.; Loadman, P.M.; Falconer, R.A. Progress towards a clinically-successful ATR inhibitor for cancer therapy. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100017. [Google Scholar] [CrossRef]
- Sultana, R.; Abdel-Fatah, T.; Perry, C.; Moseley, P.; Albarakti, N.; Mohan, V.; Seedhouse, C.; Chan, S.; Madhusudan, S. Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Protein Kinase Inhibition Is Synthetically Lethal in XRCC1 Deficient Ovarian Cancer Cells. Plos One 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Gralewska, P.; Gajek, A.; Rybaczek, D.; Marczak, A.; Rogalska, A. The Influence of PARP, ATR, CHK1 Inhibitors on Premature Mitotic Entry and Genomic Instability in High-Grade Serous BRCA(MUT) and BRCA(WT) Ovarian Cancer Cells. Cells-Basel 2022, 11, 1889. [Google Scholar] [CrossRef]
- Yap, T.A.; Tan, D.S.; Stathis, A.; Shapiro, G.I.; Iwasa, S.; Joerger, M.; Zhang, J.; Plummer, R.; Sawyer, M.; Tan, A.C.; et al. Abstract CT006: Phase Ib expansion trial of the safety and efficacy of the oral ataxia telangiectasia and Rad3-related (ATR) inhibitor elimusertib in advanced solid tumors with DNA damage response (DDR) defects. Cancer Res 2022, 82, CT006. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; da Costa, A.A.B.A.; Gulhan, D.; Lee, E.K.; Cheng, S.-C.; Hendrickson, A.E.W.; Kochupurakkal, B.; Kolin, D.L.; Kohn, E.C.; Liu, J.F.; et al. A Replication stress biomarker is associated with response to gemcitabine versus combined gemcitabine and ATR inhibitor therapy in ovarian cancer. Nat. Commun. 2021, 12, 1–8. [Google Scholar] [CrossRef]
- Serda, M.; Becker, F.G.; Cleary, M.; Team, R.M.; Holtermann, H.; The, D.; Agenda, N.; Science, P.; Sk, S.K.; Hinnebusch, R.; et al. SRA737, a Novel Chk1 Inhibitor, Shows Efficacy in CCNE1-Amplified and MYCN-Overexpressing High-Grade Serous Ovarian Cancer Patient-Derived Xenograft Models. Eur. J. Cancer 2018, 103, e98. [Google Scholar]
- Park, S.J.; Chang, S.J.; Suh, D.H.; Kong, T.W.; Song, H.; Kim, T.H.; Kim, J.W.; Kim, H.S.; Lee, S.J. A Phase IA Dose-Escalation Study of PHI-101, a New Checkpoint Kinase 2 Inhibitor, for Platinum-Resistant Recurrent Ovarian Cancer. BMC Cancer 2022, 22, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.; Huang, T.T.; Murai, J.; Haynes, B.; Steeg, P.S.; Pommier, Y.; Lee, J.M. Resistance to the CHK1 inhibitor prexasertib involves functionally distinct CHK1 activities in BRCA wild-type ovarian cancer. Oncogene 2020, 39, 5520–5535. [Google Scholar] [CrossRef]
- Lampert, E.J.; An, D.; McCoy, A.; Kohn, E.C.; Annunziata, C.M.; Trewhitt, K.; Zimmer, A.D.S.; Lipkowitz, S.; Lee, J.-M. Prexasertib, a cell cycle checkpoint kinase 1 inhibitor, in BRCA mutant recurrent high-grade serous ovarian cancer (HGSOC): A proof-of-concept single arm phase II study. J. Clin. Oncol. 2020, 38, 6038. [Google Scholar] [CrossRef]
- Zhang, M.; Dominguez, D.; Chen, S.; Fan, J.; Qin, L.; Long, A.; Li, X.; Zhang, Y.; Shi, H.; Zhang, B. WEE1 inhibition by MK1775 as a single-agent therapy inhibits ovarian cancer viability. Oncol. Lett. 2017, 14, 3580–3586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Witte, C.J.; Valle-Inclan, J.E.; Hami, N.; Lõhmussaar, K.; Kopper, O.; Vreuls, C.P.H.; Jonges, G.N.; van Diest, P.; Nguyen, L.; Clevers, H.; et al. Patient-Derived Ovarian Cancer Organoids Mimic Clinical Response and Exhibit Heterogeneous Inter- and Intrapatient Drug Responses. Cell Rep. 2019, 31, 107762. [Google Scholar] [CrossRef]
- Zilfou, J.T.; Lowe, S.W. Tumor Suppressive Functions of p53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883. [Google Scholar] [CrossRef]
- Brown, C.J.; Cheok, C.F.; Verma, C.S.; Lane, D.P. Reactivation of p53: From peptides to small molecules. Trends Pharmacol. Sci. 2011, 32, 53–62. [Google Scholar] [CrossRef]
- Selivanova, G.; Wiman, K.G. Reactivation of mutant p53: Molecular mechanisms and therapeutic potential. Oncogene 2007, 26, 2243–2254. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, N.; Kajiyama, H.; Nakamura, K.; Utsumi, F.; Niimi, K.; Mitsui, H.; Sekiya, R.; Suzuki, S.; Shibata, K.; Callen, D.; et al. PRIMA-1MET induces apoptosis through accumulation of intracellular reactive oxygen species irrespective of p53 status and chemo-sensitivity in epithelial ovarian cancer cells. Oncol. Rep. 2016, 35, 2543–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Carrillo, A.M.; Hicks, M.; Khabele, D.; Eischen, C.M. Pharmacologically Increasing Mdm2 Inhibits DNA Repair and Cooperates with Genotoxic Agents to Kill p53-Inactivated Ovarian Cancer Cells. Mol. Cancer Res. 2015, 13, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Vanderstichele, A.; Loverix, L.; Busschaert, P.; van Nieuwenhuysen, E.; Han, S.N.; Concin, N.; Callewaert, T.; Olbrecht, S.; Salihi, R.; Berteloot, P.; et al. Randomized CLIO/BGOG-Ov10 Trial of Olaparib Monotherapy versus Physician’s Choice Chemotherapy in Relapsed Ovarian Cancer. Gynecol. Oncol. 2022, 165, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in Patients with Recurrent High-Grade Serous or Poorly Differentiated Ovarian Carcinoma or Triple-Negative Breast Cancer: A Phase 2, Multicentre, Open-Label, Non-Randomised Study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Domchek, S.M.; Aghajanian, C.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; et al. Efficacy and Safety of Olaparib Monotherapy in Germline BRCA1/2 Mutation Carriers with Advanced Ovarian Cancer and Three or More Lines of Prior Therapy. Gynecol. Oncol. 2016, 140, 199–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral Poly(ADP-Ribose) Polymerase Inhibitor Olaparib in Patients with BRCA1 or BRCA2 Mutations and Recurrent Ovarian Cancer: A Proof-of-Concept Trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Cadoo, K.A.; Simpkins, F.; Mathews, C.A.; Kabil, N.; Bennett, J.; Aghajanian, C. Olaparib Treatment in Patients (Pts) with Platinum-Sensitive Relapsed (PSR) Ovarian Cancer (OC) by BRCA Mutation (BRCAm) and Homologous Recombination Deficiency (HRD) Status: Phase II LIGHT Study. J. Clin. Oncol. 2020, 38, 6013. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Overall Survival in Patients with Platinum-Sensitive Recurrent Serous Ovarian Cancer Receiving Olaparib Maintenance Monotherapy: An Updated Analysis from a Randomised, Placebo-Controlled, Double-Blind, Phase 2 Trial. Lancet Oncol. 2016, 17, 1579–1589. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib Maintenance Therapy in Platinum-Sensitive Relapsed Ovarian Cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiSilvestro, P.; Banerjee, S.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; et al. Overall Survival With Maintenance Olaparib at a 7-Year Follow-Up in Patients With Newly Diagnosed Advanced Ovarian Cancer and a BRCA Mutation: The SOLO1/GOG 3004 Trial. J. Clin. Oncol. 2022. [Google Scholar] [CrossRef]
- Banerjee, S.; Moore, K.N.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; et al. Maintenance Olaparib for Patients with Newly Diagnosed Advanced Ovarian Cancer and a BRCA Mutation (SOLO1/GOG 3004): 5-Year Follow-up of a Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2021, 22, 1721–1731. [Google Scholar] [CrossRef]
- Friedlander, M.; Moore, K.N.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Lisyanskaya, A.; Sonke, G.S.; Gourley, C.; Banerjee, S.; et al. Patient-Centred Outcomes and Effect of Disease Progression on Health Status in Patients with Newly Diagnosed Advanced Ovarian Cancer and a BRCA Mutation Receiving Maintenance Olaparib or Placebo (SOLO1): A Randomised, Phase 3 Trial. Lancet Oncol. 2021, 22, 632–642. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Penson, R.T.; Valencia, R.V.; Cibula, D.; Colombo, N.; Leath, C.A.; Bidzinski, M.; Kim, J.W.; Nam, J.H.; Madry, R.; Hernández, C.; et al. Olaparib Versus Nonplatinum Chemotherapy in Patients With Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): A Randomized Phase III Trial. J. Clin. Oncol. 2020, 38, 1164–1174. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib Tablets as Maintenance Therapy in Patients with Platinum-Sensitive, Relapsed Ovarian Cancer and a BRCA1/2 Mutation (SOLO2/ENGOT-Ov21): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poveda, A.; Lheureux, S.; Colombo, N.; Cibula, D.; Lindemann, K.; Weberpals, J.; Bjurberg, M.; Oaknin, A.; Sikorska, M.; González-Martín, A.; et al. Olaparib Maintenance Monotherapy in Platinum-Sensitive Relapsed Ovarian Cancer Patients without a Germline BRCA1/BRCA2 Mutation: OPINION Primary Analysis. Gynecol. Oncol. 2022, 164, 498–504. [Google Scholar] [CrossRef]
- Gao, Q.; Zhu, J.; Zhao, W.; Huang, Y.; An, R.; Zheng, H.; Qu, P.; Wang, L.; Zhou, Q.; Wang, D.; et al. L-MOCA: An Open-Label Study of Olaparib Maintenance Monotherapy in Platinum-Sensitive Relapsed Ovarian Cancer. J. Clin. Oncol. 2021, 39, e17526. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Selle, F.; Scambia, G.; Asselain, B.; Marmé, F.; Lindemann, K.; Colombo, N.; Madry, R.; Glasspool, R.M.; Dubot, C.; et al. LBA33 Maintenance Olaparib Rechallenge in Patients (Pts) with Ovarian Carcinoma (OC) Previously Treated with a PARP Inhibitor (PARPi): Phase IIIb OReO/ENGOT Ov-38 Trial. Ann. Oncol. 2021, 32, S1308–S1309. [Google Scholar] [CrossRef]
- Prospective Multicentre Phase-IV Clinical Trial of Olaparib in Indian Patients With Ovarian and Metastatic Breast Cancer. Available online: https://clin.larvol.com/trial-detail/NCT04330040/261112 (accessed on 27 November 2022).
- Pignata, S.; Oza, A.; Hall, G.; Pardo, B.; Madry, R.; Cibula, D.; Klat, J.; Montes, A.; Glasspool, R.; Colombo, N.; et al. ORZORA: Maintenance Olaparib in Patients with Platinum-Sensitive Relapsed Ovarian Cancer: Outcomes by Somatic and Germline BRCA and Other Homologous Recombination Repair Gene Mutation Status. Gynecol. Oncol. 2021, 162, S29. [Google Scholar] [CrossRef]
- Marme, F.; Hilpert, F.; Welslau, M.; El-Balat, A.; Grischke, E.-M.; Schinkothe, T.; Glowik, R.; Sehouli, J. Olaparib in German Routine Clinical Practice: Updated Interim Results of the Non-Interventional Study C-PATROL. J. Clin. Oncol. 2018, 36, e17549. [Google Scholar] [CrossRef]
- de La Motte Rouge, T.; Bengrine Lefevre, L.; Mouret-Reynier, M.-A.; Asselain, B.; Lucas, B.; Gavoille, C.; Cornila, C.; Spaeth, D.; Colomba, E.; Patsouris, A.; et al. 823P Extended Follow-up of a Real-World Cohort of Patients (Pts) with BRCA Mutation (BRCAm) Relapsed Epithelial Ovarian Cancer (EOC) Receiving Olaparib Maintenance Therapy: The GINECO RETROLA Study. Ann. Oncol. 2020, 31, S621–S622. [Google Scholar] [CrossRef]
- Lorusso, D.; Gourley, C.; Garbay, D.; Jean, B.; Asselain, R.; Raspagliesi, F.; Zamagni, C.; Leary, A.; Bellier, C.; Glasspool, R.; et al. 2022-RA-944-ESGO Real-World Safety, Baseline Characteristics and First-Year Therapy Management in Patients with BRCA1/BRCA2-Mutated Advanced Ovarian Cancer Treated with Olaparib Tablets in the First-Line Maintenance Setting: First Analysis of the Pan-European OVAL-1 Study. Int J Gynecol Cancer 2022, 32. [Google Scholar] [CrossRef]
- Konner, J.A.; Boucicaut, N.N.; O’Cearbhaill, R.E.; Zamarin, D.; Makker, V.; Sabbatini, P.; Tew, W.P.; Cadoo, K.A.; Grisham, R.N.; Aghajanian, C. A Phase I Dose-Escalation Study of Intraperitoneal (IP) Cisplatin, IV/IP Paclitaxel, IV Bevacizumab, and Oral Olaparib for Newly Diagnosed Adenxal Carcinoma. J. Clin. Oncol. 2017, 35, 5572. [Google Scholar] [CrossRef]
- Rivkin, S.E.; Moon, J.; Iriarte, D.S.; Bailey, E.; Sloan, H.L.; Goodman, G.E.; Bondurant, A.E.; Velijovich, D.; Wahl, T.; Jiang, P.; et al. Phase Ib with Expansion Study of Olaparib plus Weekly (Metronomic) Carboplatin and Paclitaxel in Relapsed Ovarian Cancer Patients. Int J Gynecol Cancer 2019, 29, 325–333. [Google Scholar] [CrossRef]
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.J.; Sonke, G.S.; Colombo, N.; Špaček, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib Combined with Chemotherapy for Recurrent Platinum-Sensitive Ovarian Cancer: A Randomised Phase 2 Trial. Lancet Oncol. 2015, 16, 87–97. [Google Scholar] [CrossRef]
- Kaye, S.B.; Lubinski, J.; Matulonis, U.; Ang, J.E.; Gourley, C.; Karlan, B.Y.; Amnon, A.; Bell-McGuinn, K.M.; Chen, L.M.; Friedlander, M.; et al. Phase II, Open-Label, Randomized, Multicenter Study Comparing the Efficacy and Safety of Olaparib, a Poly (ADP-Ribose) Polymerase Inhibitor, and Pegylated Liposomal Doxorubicin in Patients with BRCA1 or BRCA2 Mutations and Recurrent Ovarian Cancer. J. Clin. Oncol. 2012, 30, 372–379. [Google Scholar] [CrossRef]
- Harter, P.; Mouret-Reynier, M.A.; Pignata, S.; Cropet, C.; González-Martín, A.; Bogner, G.; Fujiwara, K.; Vergote, I.; Colombo, N.; Nøttrup, T.J.; et al. Efficacy of Maintenance Olaparib plus Bevacizumab According to Clinical Risk in Patients with Newly Diagnosed, Advanced Ovarian Cancer in the Phase III PAOLA-1/ENGOT-Ov25 Trial. Gynecol. Oncol. 2022, 164, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- González-Martín, A.; Desauw, C.; Heitz, F.; Cropet, C.; Gargiulo, P.; Berger, R.; Ochi, H.; Vergote, I.; Colombo, N.; Mirza, M.R.; et al. Maintenance Olaparib plus Bevacizumab in Patients with Newly Diagnosed Advanced High-Grade Ovarian Cancer: Main Analysis of Second Progression-Free Survival in the Phase III PAOLA-1/ENGOT-Ov25 Trial. Eur J Cancer 2022, 174, 221–231. [Google Scholar] [CrossRef]
- Ray-Coquard, I.L.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Sevelda, P.; Fujiwara, K.; Vergote, I.B.; Colombo, N.; Mäenpää, J.; et al. Phase III PAOLA-1/ENGOT-Ov25 Trial: Olaparib plus Bevacizumab (Bev) as Maintenance Therapy in Patients (Pts) with Newly Diagnosed, Advanced Ovarian Cancer (OC) Treated with Platinum-Based Chemotherapy (PCh) plus Bev. Ann. Oncol. 2019, 30, v894–v895. [Google Scholar] [CrossRef]
- LEE, J.-Y.; Kim, J.-W.; Kim, B.-G.; Kim, S.W.; Kim, H.S.; Kim, S.I.; Lim, M.C.; Choi, C.H.; Ngoi, N.; Tan, D.S.P.; et al. Interim Analysis from a Phase II Study of Olaparib Maintenance with Pembrolizumab and Bevacizumab in BRCA Non-Mutated Patients with Platinum-Sensitive Recurrent Ovarian Cancer: APGOT-Ov4/ OPEB-01. J. Clin. Oncol. 2022, 40, e17579. [Google Scholar] [CrossRef]
- Peer, C.J.; Lee, J.M.; Roth, J.; Rodgers, L.; Nguyen, J.; Annunziata, C.M.; Minasian, L.; Kohn, E.C.; Figg, W.D. Population Pharmacokinetic Analyses of the Effect of Carboplatin Pretreatment on Olaparib in Recurrent or Refractory Women’s Cancers. Cancer Chemother. Pharmacol. 2017, 80, 165. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Hays, J.L.; Annunziata, C.M.; Noonan, A.M.; Minasian, L.; Zujewski, J.A.; Yu, M.; Gordon, N.; Ji, J.; Sissung, T.M.; et al. Phase I/Ib Study of Olaparib and Carboplatin in BRCA1 or BRCA2 Mutation-Associated Breast or Ovarian Cancer With Biomarker Analyses. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Geenen, J.J.J.; Dackus, G.M.H.E.; Schouten, P.C.; Pluim, D.; Marchetti, S.; Sonke, G.S.; Jóźwiak, K.; Huitema, A.D.R.; Beijnen, J.H.; Schellens, J.H.M.; et al. A Phase I Dose-escalation Study of Two Cycles Carboplatin-olaparib Followed by Olaparib Monotherapy in Patients with Advanced Cancer. Int. J. Cancer 2021, 148, 3041. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.J.; Buss, M.K.; Nattam, S.R.; Hurteau, J.; et al. Overall Survival and Updated Progression-Free Survival Outcomes in a Randomized Phase II Study of Combination Cediranib and Olaparib versus Olaparib in Relapsed Platinum-Sensitive Ovarian Cancer. Ann. Oncol. 2019, 30, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.J.; Buss, M.K.; Nattam, S.; Hurteau, J.; et al. A Randomized Phase 2 Study of Combination Cediranib and Olaparib versus Olaparib Alone as Recurrence Therapy in Platinum-Sensitive Ovarian Cancer. Lancet Oncol. 2014, 15, 1207. [Google Scholar] [CrossRef] [Green Version]
- Colombo, N.; Tomao, F.; Benedetti Panici, P.; Nicoletto, M.O.; Tognon, G.; Bologna, A.; Lissoni, A.A.; DeCensi, A.; Lapresa, M.; Mancari, R.; et al. Randomized Phase II Trial of Weekly Paclitaxel vs. Cediranib-Olaparib (Continuous or Intermittent Schedule) in Platinum-Resistant High-Grade Epithelial Ovarian Cancer. Gynecol. Oncol. 2022, 164, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Oaknin, A.; Garg, S.; Bruce, J.P.; Madariaga, A.; Dhani, N.C.; Bowering, V.; White, J.; Accardi, S.; Tan, Q.; et al. EVOLVE: A Multicenter Open-Label Single-Arm Clinical and Translational Phase II Trial of Cediranib Plus Olaparib for Ovarian Cancer after PARP Inhibition Progression. Clin. Cancer Res. 2020, 26, 4206–4215. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Moore, R.G.; Ghamande, S.; Park, M.S.; Diaz, J.P.; Chapman, J.; Kendrick, J.; Slomovitz, B.M.; Tewari, K.S.; Lowe, E.S.; et al. Cediranib in Combination with Olaparib in Patients without a Germline BRCA1/2 Mutation and with Recurrent Platinum-Resistant Ovarian Cancer: Phase IIb CONCERTO Trial. Clin. Cancer Res. 2022, 28, 4186–4193. [Google Scholar] [CrossRef]
- Nicum, S.; Holmes, J.; McGregor, N.; Dunn, R.; Collins, L.; Kaye, S.; McNeish, I.; Glasspool, R.M.; Hall, M.; Roux, R.; et al. 722O Randomised Phase II Trial of Olaparib Compared to Weekly Paclitaxel or Olaparib plus Cediranib in Patients with Platinum-Resistant Ovarian Cancer (OCTOVA). Ann. Oncol. 2021, 32, S725–S726. [Google Scholar] [CrossRef]
- Liu, J.F.; Brady, M.F.; Matulonis, U.A.; Miller, A.; Kohn, E.C.; Swisher, E.M.; Tew, W.P.; Cloven, N.G.; Muller, C.; Bender, D.; et al. A Phase III Study Comparing Single-Agent Olaparib or the Combination of Cediranib and Olaparib to Standard Platinum-Based Chemotherapy in Recurrent Platinum-Sensitive Ovarian Cancer. J. Clin. Oncol. 2020, 38, 6003. [Google Scholar] [CrossRef]
- Liu, J.F.; Brady, M.F.; Matulonis, U.A.; Miller, A.; Kohn, E.C.; Swisher, E.M.; Cella, D.; Tew, W.P.; Cloven, N.G.; Muller, C.Y.; et al. Olaparib With or Without Cediranib Versus Platinum-Based Chemotherapy in Recurrent Platinum-Sensitive Ovarian Cancer (NRG-GY004): A Randomized, Open-Label, Phase III Trial. J. Clin. Oncol. 2022, 40, 2138–2147. [Google Scholar] [CrossRef]
- Kurnit, K.C.; Meric-Bernstam, F.; Hess, K.; Coleman, R.L.; Bhosale, P.; Savelieva, K.; Janku, F.; Hong, D.; Naing, A.; Pant, S.; et al. Abstract CT020: Phase I Dose Escalation of Olaparib (PARP Inhibitor) and Selumetinib (MEK Inhibitor) Combination in Solid Tumors with Ras Pathway Alterations. Cancer Res 2019, 79, CT020. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Wulf, G.M.; Barry, W.T.; Birrer, M.; Westin, S.N.; Farooq, S.; Bell-McGuinn, K.M.; Obermayer, E.; Whalen, C.; Spagnoletti, T.; et al. Phase I Dose Escalation Study of the PI3kinase Pathway Inhibitor BKM120 and the Oral Poly (ADP Ribose) Polymerase (PARP) Inhibitor Olaparib for the Treatment of High-Grade Serous Ovarian and Breast Cancer. Ann. Oncol. 2017, 28, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Phase II Study of Olaparib plus Durvalumab with or without Bevacizumab (MEDIOLA): Final Analysis of Overall Survival in Patients with Non-Germline... | OncologyPRO. Available online: https://oncologypro.esmo.org/meeting-resources/esmo-congress/phase-ii-study-of-olaparib-plus-durvalumab-with-or-without-bevacizumab-mediola-final-analysis-of-overall-survival-in-patients-with-non-germline (accessed on 28 November 2022).
- Adams, S.F.; Rixe, O.; Lee, J.-H.; McCance, D.J.; Westgate, S.; Eberhardt, S.C.; Rutledge, T.; Muller, C. Phase I Study Combining Olaparib and Tremelimumab for the Treatment of Women with BRCA-Deficient Recurrent Ovarian Cancer. J. Clin. Oncol. 2017, 35, e17052. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, H.; Gao, Y.; Lou, G.; Yin, R.; Ji, D.; Li, W.; Wang, W.; Xia, B.; Wang, D.; et al. Phase I Pharmacokinetic Study of Niraparib in Chinese Patients with Epithelial Ovarian Cancer. Oncologist 2020, 25, 19. [Google Scholar] [CrossRef] [Green Version]
- Akce, M.; El-Khoueiry, A.; Piha-Paul, S.A.; Bacque, E.; Pan, P.; Zhang, Z.Y.; Ewesuedo, R.; Gupta, D.; Tang, Y.; Milton, A.; et al. Pharmacokinetics and Safety of Niraparib in Patients with Moderate Hepatic Impairment. Cancer Chemother. Pharmacol. 2021, 88, 825–836. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib Monotherapy for Late-Line Treatment of Ovarian Cancer (QUADRA): A Multicentre, Open-Label, Single-Arm, Phase 2 Trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, W.; Liu, R.; Shan, W.; Li, H.; Liu, J.; Xia, B.; He, S.; Xia, Y.; Wang, S.; et al. Effectiveness and Safety of Niraparib as Neoadjuvant Therapy in Advanced Ovarian Cancer with Homologous Recombination Deficiency: NANT Study Protocol for a Prospective, Multicenter, Exploratory, Phase 2, Single-Arm Study (041). Gynecol. Oncol. 2022, 166, S28–S29. [Google Scholar] [CrossRef]
- Takehara, K.; Matsumoto, T.; Hamanishi, J.; Hasegawa, K.; Matsuura, M.; Miura, K.; Nagao, S.; Nakai, H.; Tanaka, N.; Tokunaga, H.; et al. Phase 2 Single-Arm Study on the Safety of Maintenance Niraparib in Japanese Patients with Platinum-Sensitive Relapsed Ovarian Cancer. J. Gynecol. Oncol. 2021, 32, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, A.; Kondo, E.; Nakamura, T.; Yanagida, S.; Hamanishi, J.; Harano, K.; Hasegawa, K.; Hirasawa, T.; Hori, K.; Komiyama, S.; et al. Phase 2 Single-Arm Study on the Efficacy and Safety of Niraparib in Japanese Patients with Heavily Pretreated, Homologous Recombination-Deficient Ovarian Cancer. J. Gynecol. Oncol. 2021, 32, 1–12. [Google Scholar] [CrossRef]
- Yin, R.; Li, N.; Wu, L.; Wang, J.; Zhu, J.; Pan, L.; Kong, B.; Zheng, H.; Liu, J.; Wu, X.; et al. Efficacy of Niraparib Maintenance Therapy in Patients with Newly Diagnosed Advanced Ovarian Cancer in Phase 3 PRIME Study: A Subgroup Analysis by Response to First-Line Platinum-Based Chemotherapy. J. Clin. Oncol. 2022, 40, 5551. [Google Scholar] [CrossRef]
- O’Cearbhaill, R.E.; Pérez-Fidalgo, J.A.; Monk, B.J.; Tusquets, I.; McCormick, C.; Fuentes, J.; Moore, R.G.; Vulsteke, C.; Shahin, M.S.; Forget, F.; et al. Efficacy of Niraparib by Time of Surgery and Postoperative Residual Disease Status: A Post Hoc Analysis of Patients in the PRIMA/ENGOT-OV26/GOG-3012 Study. Gynecol. Oncol. 2022, 166, 36–43. [Google Scholar] [CrossRef]
- del Campo, J.M.; Matulonis, U.A.; Malander, S.; Provencher, D.; Mahner, S.; Follana, P.; Waters, J.; Berek, J.S.; Woie, K.; Oza, A.M.; et al. Niraparib Maintenance Therapy in Patients With Recurrent Ovarian Cancer After a Partial Response to the Last Platinum-Based Chemotherapy in the ENGOT-OV16/NOVA Trial. J. Clin. Oncol. 2019, 37, 2968–2973. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Walder, L.; Nøttrup, T.J.; Bessette, P.; Mahner, S.; Gil-Martin, M.; Kalbacher, E.; Ledermann, J.A.; Wenham, R.M.; Woie, K.; et al. Niraparib Maintenance Treatment Improves Time Without Symptoms or Toxicity (TWiST) Versus Routine Surveillance in Recurrent Ovarian Cancer: A TWiST Analysis of the ENGOT-OV16/NOVA Trial. J. Clin. Oncol. 2019, 37, 3183–3191. [Google Scholar] [CrossRef]
- Vilming, B.; Dahl, J.F.; Aune, G.; Zucknick, M.; Lindemann, K. Real-Life Use of Niraparib in a Patient Access Program in Norway. 2021. Available online: https://hdl.handle.net/11250/2775965 (accessed on 31 October 2022).
- Liu, G.; Feng, Y.; Li, J.; Deng, T.; Yin, A.; Yan, L.; Zheng, M.; Xiong, Y.; Li, J.; Huang, Y.; et al. A Novel Combination of Niraparib and Anlotinib in Platinum-Resistant Ovarian Cancer: Efficacy and Safety Results from the Phase II, Multi-Center ANNIE Study. EClinicalMedicine 2022, 0, 101767. [Google Scholar] [CrossRef]
- Mirza, M.R.; Nyvang, G.-B.; Lund, B.; Christensen, R.d.; Werner, T.L.; Malander, S.; Bjørge, L.; Birrer, M.J.; Anttila, M.; Lindahl, G.; et al. Final Survival Analysis of NSGO-AVANOVA2/ENGOT-OV24: Combination of Niraparib and Bevacizumab versus Niraparib Alone as Treatment of Recurrent Platinum-Sensitive Ovarian Cancer—A Randomized Controlled Chemotherapy-Free Study. J. Clin. Oncol. 2020, 38, 6012. [Google Scholar] [CrossRef]
- Mirza, M.R.; Åvall Lundqvist, E.; Birrer, M.J.; dePont Christensen, R.; Nyvang, G.B.; Malander, S.; Anttila, M.; Werner, T.L.; Lund, B.; Lindahl, G.; et al. Niraparib plus Bevacizumab versus Niraparib Alone for Platinum-Sensitive Recurrent Ovarian Cancer (NSGO-AVANOVA2/ENGOT-Ov24): A Randomised, Phase 2, Superiority Trial. Lancet Oncol. 2019, 20, 1409–1419. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Bergmann, T.K.; Mau-Sørensen, M.; Christensen, R. de P.; Åvall-Lundqvist, E.; Birrer, M.J.; Jørgensen, M.; Roed, H.; Malander, S.; Nielsen, F.; et al. A Phase I Study of the PARP Inhibitor Niraparib in Combination with Bevacizumab in Platinum-Sensitive Epithelial Ovarian Cancer: NSGO AVANOVA1/ENGOT-OV24. Cancer Chemother. Pharmacol. 2019, 84, 791–798. [Google Scholar] [CrossRef]
- Hardesty, M.M.; Krivak, T.C.; Wright, G.S.; Hamilton, E.; Fleming, E.L.; Belotte, J.; Keeton, E.K.; Wang, P.; Gupta, D.; Clements, A.; et al. OVARIO Phase II Trial of Combination Niraparib plus Bevacizumab Maintenance Therapy in Advanced Ovarian Cancer Following First-Line Platinum-Based Chemotherapy with Bevacizumab. Gynecol. Oncol. 2022, 166, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gaillard, S.; Hendrickson, A.W.; Moroney, J.; Yeku, O.; Diver, E.; Gunderson, C.; Arend, R.; Ratner, E.; Samnotra, V.; et al. An Open-Label Phase II Study of Dostarlimab (TSR-042), Bevacizumab (Bev), and Niraparib Combination in Patients (Pts) with Platinum-Resistant Ovarian Cancer (PROC): Cohort A of the OPAL Trial. Gynecol. Oncol. 2021, 162, S17–S18. [Google Scholar] [CrossRef]
- Randall, L.M.; O’Malley, D.M.; Monk, B.J.; Coleman, R.L.; Gaillard, S.; Adams, S.F.; Duska, L.R.; Cappuccini, F.; Dalton, H.; Holloway, R.W.; et al. MOONSTONE/GOG-3032: Interim Analysis of a Phase 2 Study of Niraparib + Dostarlimab in Patients (Pts) with Platinum-Resistant Ovarian Cancer (PROC). J. Clin. Oncol. 2022, 40, 5573. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- de Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov 2017, 7, 620. [Google Scholar] [CrossRef] [Green Version]
- Piha-Paul, S.A.; Xiong, W.W.; Moss, T.; Mostorino, R.M.; Sedelmeier, S.; Hess, K.; Fu, S.; Hong, D.; Janku, F.; Karp, D.; et al. Abstract A096: Phase II Study of the PARP Inhibitor Talazoparib in Advanced Cancer Patients with Somatic Alterations in BRCA1/2, Mutations/Deletions in PTEN or PTEN Loss, Aberrations in Other BRCA Pathway Genes, and Germline Mutations in BRCA1/2 (Not Breast or Ovarian Cancer). Mol Cancer Ther 2018, 17, A096. [Google Scholar] [CrossRef]
- Schram, A.M.; Colombo, N.; Arrowsmith, E.; Narayan, V.; Yonemori, K.; Scambia, G.; Zelnak, A.; Bauer, T.M.; Jin, N.; Ulahannan, S.v.; et al. Avelumab Plus Talazoparib in Patients With BRCA1/2- or ATM-Altered Advanced Solid Tumors: Results From JAVELIN BRCA/ATM, an Open-Label, Multicenter, Phase 2b, Tumor-Agnostic Trial. JAMA Oncol. 2022. [Google Scholar] [CrossRef]
- Yap, T.A.; Bardia, A.; Dvorkin, M.; Galsky, M.D.; Thaddeus Beck, J.; Wise, D.R.; Karyakin, O.; Rubovszky, G.; Kislov, N.; Rohrberg, K.; et al. Avelumab Plus Talazoparib in Patients With Advanced Solid Tumors: The JAVELIN PARP Medley Nonrandomized Controlled Trial. JAMA Oncol. 2022. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, J.; Wang, J.; Lin, Z.; Yin, R.; Sun, W.; Zhou, Q.; Zhang, S.; Wang, D.; Shi, H.; et al. Pamiparib Monotherapy for Patients with Germline BRCA1/2-Mutated Ovarian Cancer Previously Treated with at Least Two Lines of Chemotherapy: A Multicenter, Open-Label, Phase II Study. Clin. Cancer Res. 2022, 28, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Galsky, M.; Barve, M.; Goel, S.; Park, H.; Du, B.; Mu, S.; Ramakrishnan, V.; Wood, K.; Wang, V.; et al. Preliminary Results of Pamiparib (BGB-290), a PARP1/2 Inhibitor, in Combination with Temozolomide (TMZ) in Patients (Pts) with Locally Advanced or Metastatic Solid Tumors. Ann. Oncol. 2018, 29, viii138. [Google Scholar] [CrossRef]
- Kristeleit, R.; Shapiro, G.I.; Burris, H.A.; Oza, A.M.; LoRusso, P.; Patel, M.R.; Domchek, S.M.; Balmaña, J.; Drew, Y.; Chen, L.M.; et al. A Phase I-II Study of the Oral PARP Inhibitor Rucaparib in Patients with Germline BRCA1/2-Mutated Ovarian Carcinoma or Other Solid Tumors. Clin. Cancer Res. 2017, 23, 4095–4106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, G.I.; Kristeleit, R.S.; Burris, H.A.; LoRusso, P.; Patel, M.R.; Drew, Y.; Giordano, H.; Maloney, L.; Watkins, S.; Goble, S.; et al. Pharmacokinetic Study of Rucaparib in Patients With Advanced Solid Tumors. Clin Pharmacol Drug Dev 2019, 8, 107. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.v.; O’Malley, D.M.; et al. Rucaparib in Relapsed, Platinum-Sensitive High-Grade Ovarian Carcinoma (ARIEL2 Part 1): An International, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Swisher, E.M.; Kwan, T.T.; Oza, A.M.; Tinker, A.v.; Ray-Coquard, I.; Oaknin, A.; Coleman, R.L.; Aghajanian, C.; Konecny, G.E.; O’Malley, D.M.; et al. Molecular and Clinical Determinants of Response and Resistance to Rucaparib for Recurrent Ovarian Cancer Treatment in ARIEL2 (Parts 1 and 2). Nat. Commun. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib Maintenance Treatment for Recurrent Ovarian Carcinoma after Response to Platinum Therapy (ARIEL3): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 390, 1949. [Google Scholar] [CrossRef] [Green Version]
- Ledermann, J.A.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.R.; Scambia, G.; et al. Rucaparib for Patients with Platinum-Sensitive, Recurrent Ovarian Carcinoma (ARIEL3): Post-Progression Outcomes and Updated Safety Results from a Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2020, 21, 710–722. [Google Scholar] [CrossRef]
- Colombo, N.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Weberpals, J.I.; Clamp, A.R.; Scambia, G.; Leary, A.; et al. The Effect of Age on Efficacy, Safety and Patient-Centered Outcomes with Rucaparib: A Post Hoc Exploratory Analysis of ARIEL3, a Phase 3, Randomized, Maintenance Study in Patients with Recurrent Ovarian Carcinoma. Gynecol. Oncol. 2020, 159, 101. [Google Scholar] [CrossRef]
- Kristeleit, R.; Lisyanskaya, A.; Fedenko, A.; Dvorkin, M.; de Melo, A.C.; Shparyk, Y.; Rakhmatullina, I.; Bondarenko, I.; Colombo, N.; Svintsitskiy, V.; et al. Rucaparib versus Standard-of-Care Chemotherapy in Patients with Relapsed Ovarian Cancer and a Deleterious BRCA1 or BRCA2 Mutation (ARIEL4): An International, Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2022, 23, 465–478. [Google Scholar] [CrossRef]
- Yubero-Esteban, A.; Barquin, A.; Estévez, P.; Pajares Hachero, B.; Sánchez, L.; Reche, P.; Alarcón, J.; Calzas, J.; Gaba, L.; Fuentes-Pradera, J.; et al. 403 Analysis of the Clinical Experience within Rucaparib’s Early Access Program in Spain – a GEICO Study. Int. J. Gynecol. Cancer 2021, 31, A232.2–A233. [Google Scholar] [CrossRef]
- Lorusso, D.; Maltese, G.; Sabatucci, I.; Cresta, S.; Matteo, C.; Ceruti, T.; D’Incalci, M.; Zucchetti, M.; Raspagliesi, F.; Sonetto, C.; et al. Phase I Study of Rucaparib in Combination with Bevacizumab in Ovarian Cancer Patients: Maximum Tolerated Dose and Pharmacokinetic Profile. Target. Oncol. 2021, 16, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Dumbrava, E.E.; Shapiro, G.; Bendell, J.C.; Yap, T.A.; Jeselsohn, R.; Lepley, D.M.; Hurley, S.; Lin, K.K.; Liao, M.; Habeck, J.; et al. Phase 1b/2 SEASTAR Trial: Safety, Pharmacokinetics, and Preliminary Efficacy of the Poly(ADP)-Ribose Polymerase (PARP) Inhibitor Rucaparib and Angiogenesis Inhibitor Lucitanib in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2021, 39, 3102. [Google Scholar] [CrossRef]
- Yap, T.A.; Hamilton, E.; Bauer, T.; Dumbrava, E.E.; Jeselsohn, R.; Enke, A.; Hurley, S.; Lin, K.K.; Habeck, J.; Giordano, H.; et al. Phase Ib SEASTAR Study: Combining Rucaparib and Sacituzumab Govitecan in Patients With Cancer With or Without Mutations in Homologous Recombination Repair Genes. JCO Precis. Oncol. 2022, 6. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Parkinson, C.; Lim, M.C.; O’malley, D.M.; Oaknin, A.; Wilson, M.K.; Coleman, R.L.; Lorusso, D.; Bessette, P.; Ghamande, S.; et al. A Randomized, Phase III Trial to Evaluate Rucaparib Monotherapy as Maintenance Treatment in Patients With Newly Diagnosed Ovarian Cancer (ATHENA-MONO/GOG-3020/ENGOT-Ov45). J. Clin. Oncol. 2022, 40. [Google Scholar] [CrossRef]



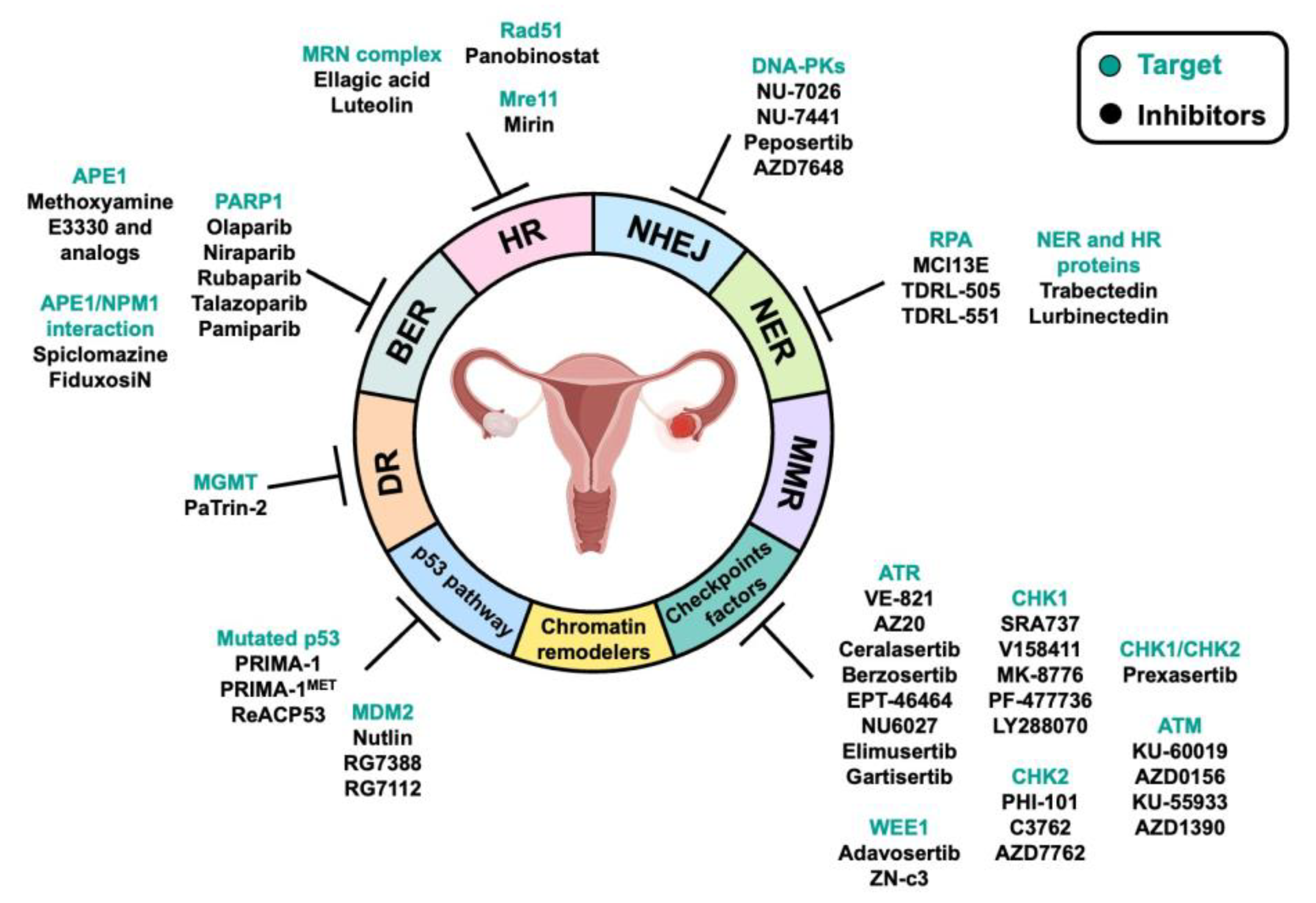{kind=link}
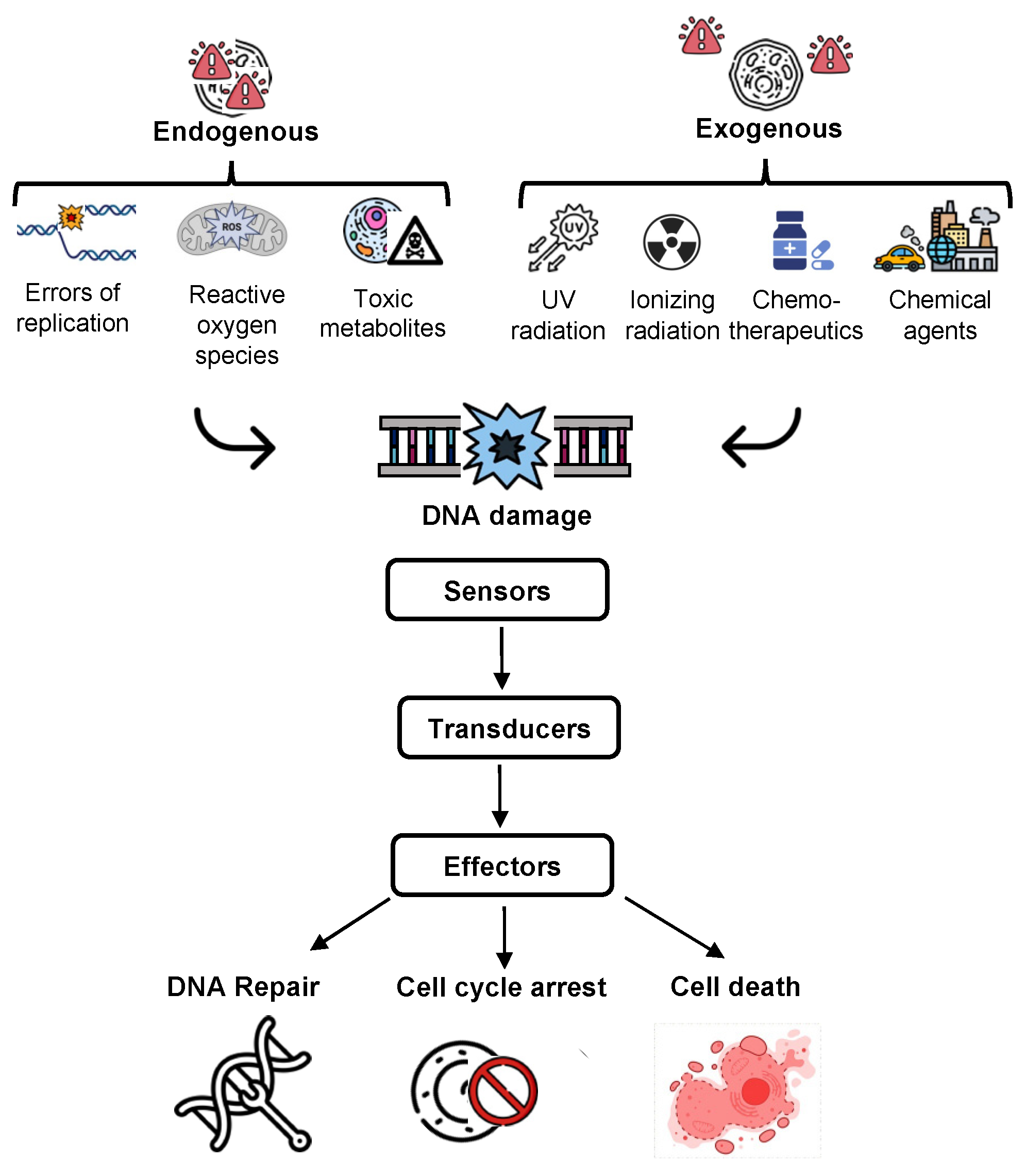{kind=link}
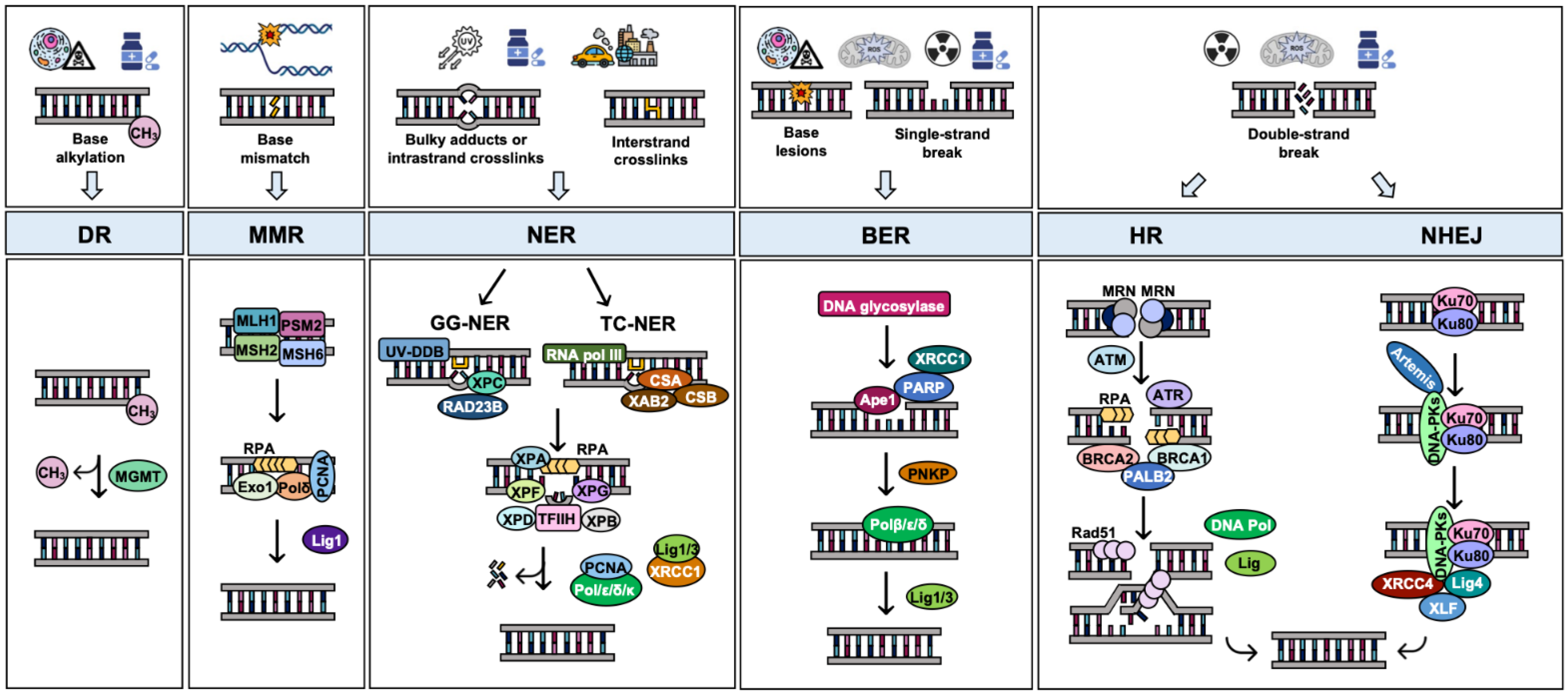{kind=link}
| Subtype | High-Grade Serous | Endometrioid | Clear cell | Mucinous | Low-Grade Serous |
|---|---|---|---|---|---|
| Prevalence | 70% | 10% | 10% | 5% | <5% |
| Stage at diagnosis | Advanced | Early | Early | Early | Early or advanced |
| Progression speed | High | Low (90%) High (10%) | Low | Low (50%) High (50%) | Low |
| Genetic alterations | TP53 BRCA1 BRCA2 | CTNNB1 PIK3CA ARID1A KRAS PPP2R1 PTEN | ARID1A PTEN PIK3CA KRAS MET | KRAS TP53 HER2/Neu | BRAF KRAS HER2/Neu |
| Chemotherapeutic response | High (at early stages) | Low | Low | Low | Low |
| DDR Pathway | Proteins Involved (References) | |||
|---|---|---|---|---|
| DR | MGMT [28] | |||
| MMR | Sensors | MSH2, MSH6, MLH1, PMS1, PMS2, and MSH3 [29,30,31] | ||
| Transducers | RPA, PCNA, and RFC [32] | |||
| Effectors | Exo1, Pol δ, and DNA ligase I [32,33] | |||
| NER | Sensors | TC-NER | RNA polymerase, CSA, CSB, and XAB2 [26,34] | |
| GG-NER | XPC-RAD23B and UV-DDB [26,34] | |||
| Transducers | XPA, XPG, XPF/ERCC1, RPA, TFIIH complex [34,35] | |||
| Effectors | PCNA, RFC, DNA polymerases δ, ε, and κ, and DNA ligase I and LIG3 [34,35] | |||
| BER | Sensors | DNA glycosylase (OGG1, UNG, or MUTYH), APE1, and PARP1 [36] | ||
| Transducers | PNKP [37,38] | |||
| Effectors | XRCC1, LIG3, DNA polymerase β [37,38] | |||
| HR | Sensors | MRN complex (MRE11, RAD50, NSB1) [39,40] | ||
| Transducers | ATM, BRCA1, ATR, RPA, PALB2, BRCA2 [41,42] | |||
| Effectors | RAD51, DNA ligase, DNA polymerase [39,40] | |||
| NHEJ | Sensors | C-NHEJ | Ku70, Ku80 [43,44] | |
| Alt-NHEJ | PARP1 [45,46] | |||
| Transducers | DNA-PKcs, ATM, ATR, and Artemis [43,44] | |||
| Effectors | C-NHEJ | XRCC4, XLF, and DNA ligase IV [43,44] | ||
| Alt-NHEJ | XRCC1, DNA polymerase Θ, and LIG3 [45,46] | |||
| Associated pathways | Chromatin remodelers | SWI/SNF, INO80, CHD, ISWI [47] | ||
| Checkpoints factors | Transducers | ATM, ATR, CHK1, CHK2 [48] | ||
| Effectors | p53, p21, CDC25C, WEE1, CDK1, CDK2 [48,49,50] | |||
| DDR Pathway | Genomic/Epigenomic Alterations in OC (References) |
|---|---|
| DR | MGMT promoter hypermethylation [51,52] |
| MMR | Mutations in MLH1, MSH2, MSH6, and PMS2 [53,54,55] MLH1 promoter hypermethylation [56,57,58] |
| NER | SNPs in NER genes [59] Homozygous deletions, missense, or splice site mutations in NER genes [60,61] |
| BER | SNPs in OGG1, APE1, and XRCC1 [62,63,64,65,66,67,68] APE1 overexpression [69] |
| HR | Genetic and epigenetic modifications of genes encoding HR proteins [70,71,72] Mutations in BRCA1, BRCA2, RAD51C, RAD51D and MRN complex genes [70,71,72,73,74,75,76] Downregulation of RAD50 [75,77] RAD51 promoter hypermethylation [60] |
| NHEJ | Mutations or overexpression of genes that encoded for NHEJ proteins (DNA-PKs, DNA polymerase Θ, or XRCC4) [60,78,79,80] SNPs in NHEJ genes (DNA ligase IV, XRCC1) [60] |
| Chromatin remodelers | Mutations in ARID1A [81,82,83,84] Mutations in CHD4 [85,86,87], CHD5 [88], and CHD8 [88] subunits Amplification of CHD4 [85,89] and ACT6LA [88,90] subunits |
| Checkpoints factors | Mutations in ATM [91,92] |
| Somatic mutations in CHK2 [93] | |
| p53 pathway | Mutations in TP53 [94,95] Loss of heterozygosity in the chromosome that contains TP53 [96] |
| DDR Pathway | Drug | Target(s) | Preclinical Evidence(s) in OC Cells or Xenograft Models |
|---|---|---|---|
| DR | PaTrin-2 | MGMT | Sensitization to temozolomide [97] |
| NER | MCI13E | RPA | Antitumor activity [98] |
| TDRL-505 | RPA | Antitumor activity [98] | |
| TDRL-551 | RPA | Antitumor activity and synergistic cytotoxic effect in combination with etoposide and platinum [98] | |
| Trabectedin/ Lurbinectedin | NER and HR proteins | Antitumor activity and synergistic cytotoxic effect in combination with cisplatin, doxorubicin, and irinotecan [99,100,101,102] | |
| BER | Methoxyamine | APE1 | Enhancement of temozolomide cytotoxicity [103] |
| E3330 and analogs | APE1 | Inhibition of cell proliferation [104,105] | |
| Spiclomazine/ fiduxosin | APE1/NPM1 interaction | Inhibition of cell proliferation and sensitization to bleomycin [106] | |
| PARPi | PARP | Specific killing of BRCA-deficient tumors [107,108] | |
| HR | Mirin | Mre11 | Increase sensitivity to DNA-damaging agents (cisplatin, carboplatin, chloroquine) [109,110,111] |
| Panobinostat | Rad51 | Inhibition of OC cell proliferation, induction of apoptosis, and inhibition of DNA repair by altering the correct repair of Rad51 [109,112,113]. Synergistic cytotoxic effect in combination with chloroquine or cisplatin [109,114] | |
| Ellagic acid/ luteolin | MRN complex | Decrease cellular proliferation and migration [115] | |
| NHEJ | NU-7026/NU-7441 | DNA-PKs | Enhancement of DNA-damaging agents’ cytotoxicity (irradiation, chloroquine, cisplatin) [116,117,118] |
| Peposertib | DNA-PKs | Increase radiosensitivity and chemosensitivity to etoposide or doxorubicin [119,120] | |
| AZD7648 | DNA-PKs | Increase the cytotoxicity to irradiation, doxorubicin, or PARP inhibitors [121,122] | |
| Checkpoints factors | KU-60019 | ATM | Inhibition of cell migration and induction of apoptosis [123]. Synergistic cytotoxic effect in combination with fenofibrate (PPARA inhibitor) [123]. Sensitization to ionizing radiation, trabectedin, and lurbinectedin [124,125] |
| AZD0156 | ATM | Synergistic cytotoxic effect in combination with fenofibrate (PPARA inhibitor) [123] | |
| KU-55933 | ATM | Sensitization to ionizing radiation, trabectedin and lurbinectedin [124,125] | |
| AZD1390 | ATM | Synergistic cytotoxic effect in combination with an aldehyde dehydrogenase 1 inhibitor (67A) [126]. | |
| VE-821 | ATR | Inhibit cell proliferation and enhancement of cisplatin, topotecan, gemcitabine, and veliparib cytotoxicity [127,128,129,130,131]. Enhancement of lurbinectedin and trabectedin cytotoxicity in combination with KU-60019 [125] | |
| AZ20 | ATR | Sensitize PARPi-resistant cells to PARPi [131] | |
| Ceralasertib | ATR | Synergistic cytotoxic effect in combination with belotecan, an aldehyde dehydrogenase 1 inhibitor (67A), and PARPi [126,132,133,134,135] | |
| Berzosertib | ATR | Reduction of cell proliferation and cell survival [127,129,136,137] | |
| EPT-46464 | ATR | Synergistic cytotoxic effect in combination with cisplatin, carboplatin, and radiation [124] | |
| NU6027 | ATR/ CDK2 | Enhancement of the cytotoxic effect of cisplatin and temozolomide [138] | |
| Elimusertib | ATR | Inhibition of cell proliferation [139,140] and enhancement of the cytotoxic effect of carboplatin and therapeutic radiopharmaceuticals [141,140] | |
| Gartisertib | ATR | Enhancement of topotecan, irinotecan, gemcitabine, cisplatin, and talazoparib cytotoxicity [142] | |
| SRA737 | CHK1 | Synergistic cytotoxic effect in combination with PARPi [143] | |
| V158411 | CHK1 | Inhibition of cell proliferation and enhancement of carboplatin and cisplatin cytotoxicity [144] | |
| MK-8776 | CHK1 | Inhibition of cell proliferation and enhancement of gemcitabine and olaparib efficacy [128,133,145] | |
| PF-477736 | CHK1 | Synergistic antiproliferative effect in combination with topotecan [146] | |
| PV1019 | CHK2 | Inhibition of cell proliferation and synergistic cytotoxic effect in combination with topotecan or camptothecins [147] | |
| PHI-101 | CHK2 | Antitumor activity [148] | |
| C3742 | CHK2 | Synergistic cytotoxic effect in combination with cisplatin [149] | |
| AZD7762 | CHK2 | Synergistic cytotoxic effect in combination with cisplatin [150]. Sensitization to PARG inhibition [151] | |
| Prexasertib | CHK1/ CHK2 | Antitumor activity and synergistic cytotoxic effect in combination with olaparib or gemcitabine [128,152,153] | |
| Adavosertib | WEE1 | Antitumor activity, and inhibition of cell proliferation and migration. Induction of DNA damage, apoptosis, and G2/M cell cycle arrest [154]. Synergistic cytotoxic effect in combination with ATR inhibitor (AZD6738) [155], CHK1inhibitor (PF-00477736) [156], and radioimmunotherapy [157] | |
| p53 pathway | PRIMA-1 | Mutated p53 | Induction of cell death and re-sensitization of chemoresistant-cells to cisplatin [158,159] |
| PRIMA-1MET | Mutated p53 | Re-sensitization of cisplatin/doxorubicin-cells to cisplatin/doxorubicin [160]. Synergistic cytotoxic effect in combination with cisplatin, carboplatin, or doxorubicin [160,161] | |
| ReACP53 | Mutated p53 | Cell proliferation decrease and synergistic cytotoxic effect in combination with carboplatin [162,163] | |
| Nutlin | MDM2 | Reduction of cell viability, induction of apoptosis, and synergistic cytotoxic effect in combination with cisplatin, rucaparib, or etoposide [164,165,166,167,168] | |
| RG7388 | MDM2 | Reduction of cell viability, induction of apoptosis, and synergistic cytotoxic effect in combination with cisplatin, rucaparib, or etoposide [164,167,169] | |
| RG7112 | MDM2 | Cell growth reduction [170] |
| DDR Pathway | Target(s) | Drug | Combined with | Condition | Phase | Clinical ID | Results |
|---|---|---|---|---|---|---|---|
| DR | MGMT | PaTrin-2 | Temozolomide | OC | I | [171] | One patient with OC has a decrease of around 50% of tumor markers and stable radiology over 5–6 cycles of treatment [171] |
| NER | NER and HR proteins | Trabectedin | - | Advanced OC | II | NCT00050414 | Trabectedin was active and well-tolerated in advanced OC platinum-sensitive patients. The optimal regimen was established [172] |
| Advanced tumor malignancies | II | NCT00786838 | Trabectedin efficacy was confirmed [173] | ||||
| Advanced soft tissue sarcomas | II | NCT00003939 | Trabectedin has proven to control tumor progression in highly pretreated, progression, advanced, metastatic resistant, or refractory sarcoma patients [174] | ||||
| Ovarian carcinosarcoma | II | NCT02993705 | Trabectedin conferred a modest benefit to patients with advanced OC and it was well-tolerated [175] | ||||
| BRCA mutated OC | II III | NCT01772979 NCT02903004 | OC patients with BRCAness phenotype could benefit from trabectedin treatment. However, this treatment did not improve survival compared to standard chemotherapy in BRCA-mutated and BRCAness phenotype OC patients [176,177] | ||||
| PLD | Relapsed OC | II | NCT04887961 | No results posted | |||
| Partially platinum-sensitive OC | III | NCT01379989 | Trabectedin/PLD combination showed a similar overall survival that carboplatin/PLD combination and could be considered for treating patients who need a longer recovery time from platinum toxicities [178] | ||||
| Advanced relapsed OC | III | NCT00113607 NCT01846611 | Trabectedin/PLD combination did not show a favorable overall survival benefit or safety. Specifically, patients with BRCA-mutation or a platinum-free interval of 6–12 months seemed to present a survival benefit from this combination [179,180,181,182] | ||||
| Recurrent OC | III | NCT03690739 | No results posted | ||||
| Platinum-sensitive recurrent OC | IV | NCT03164980 | OC patients treated with trabectedin/PLD did not show inferiority signals compared to standard platinum-based chemotherapy. The study is ongoing and recruiting new patients [183] | ||||
| Platinum-sensitive recurrent OC | Observational | NCT02394015 NCT05512676 | The intercalation with a nonplatinum regimen, such as trabectedin/PLD, could improve the response to a platinum-base therapy platinum-sensitive OC patients [184] | ||||
| Partially platinum-sensitive recurrent OC | Observational | NCT03446495 | No results posted | ||||
| Relapsed OC | Observational | NCT02825420 | The combination of trabectedin and PLD represented a therapeutical safe option for platinum-sensitive recurrent OC regardless of prior anti-angiogenic treatment [185] | ||||
| Platinum-sensitive relapsed OC | Observational | NCT02163720 NCT01869400 | PLD/trabectedin supposed a therapeutic option for partially or fully platinum-sensitive recurrent OC patients [186,187] | ||||
| PLD +/− olaparib | Recurrent OC | II | NCT03470805 | No results posted | |||
| Durvalumab | OC | I | NCT03085225 | The combination of trabectedin and durvalumab presented a manageable toxicity and a promising antitumor activity in platinum-refractory OC patients [188] | |||
| Docetaxel + pegfilgrastim/filgastrim | Recurrent or persistent OC | II | NCT00569673 | Docetaxel/trabectedin was well-tolerated and seemed to be more active than docetaxel treatment in recurrent OC patients [189] | |||
| Bevacizumab +/− carboplatin | Recurrent OC | II | NCT01735071 | Bevacizumab/trabectedin combination had clinical activity and could be a therapeutic option for partially platinum-sensitive OC patients. Bevacizumab/trabectedin/carboplatin demonstrated a positive activity and should be further studied [190] | |||
| Lurbinectedin | Paclitaxel +/− bevacizumab | Advanced solid tumors | I | NCT01831089 | Lubinectedin combined with paclitaxel and/or bevacizumab presented a manageable toxicity and promising antitumor activity in patients with advanced solid tumors, including OC [191] | ||
| Olaparib | Advanced solid tumors | I/II | NCT02684318 | Lurbinectedin/olaparib combination was feasible and recommended doses were obtained for each drug [192] | |||
| Irinotecan | Solid tumors | I/II | NCT02611024 | One OC BRCA1-mutated patient presented an extraordinary response with a time to further progression of 8 months. No more results have been posted [193] | |||
| BER | APE1 | Methoxyamine | - | Platinum-resistant OC | III | NCT02421588 | Lurbinectedin showed antitumor activity similar to PLD and it was better tolerated [194] |
| Temozolomide | Granulosa cell OC | I/II | NCT01851369 | Two patients with granulosa cell OC experienced a partial response [195] | |||
| Permetrexed + cisplatin | Advanced solid tumors | I/II | NCT02535312 | No results posted | |||
| PARP | Olaparib | Described in Table S1 | |||||
| Niraparib | |||||||
| Talazoparib | |||||||
| Pamiparib | |||||||
| Rucaparib | |||||||
| HR | Rad51 | Panobinostat | - | Advanced solid tumors | I | NCT00739414 | Panobinostat treatment was safe and potentially effective against advanced solid tumors like OC [196] |
| Gemcitabine | Solid tumors | I | NCT00550199 | Recommended doses for panobinostat/gemcitabine were established [197] | |||
| NHEJ | DNA-PKs | Peposertib | PLD | Recurrent OC | I | NCT04092270 | No results posted |
| Checkpoints factors | ATR | Ceralasertib | Olaparib | Recurrent OC | II | NCT03462342 | The combination of ceralasertib and olaparib was well-tolerated. No objective response was observed; however, a signal of activity was observed and depended on BRCA1 status [198] |
| Recurrent OC | II | NCT03579316 | No results posted about ceralasertib/olaparib combination | ||||
| Gynecological cancers | II | NCT04065269 | No results posted | ||||
| Advanced solid tumors | II | NCT02576444 | Ceralasertib/olaparib combination has demonstrated preliminary activity in ATM-mutated tumors and in BRCA-mutated PARPi-resistant HGSC OC patients [199] | ||||
| Carboplatin/Olaparib/Durvalumab | Advanced tumors | I | NCT02264678 | No results posted | |||
| Berzosertib | Carboplatin + Gemcitabine | Recurrent and metastatic OC | I | NCT02627443 | No results posted | ||
| Carboplatin + Avelumab | PARPi resistant, recurrent, and platinum sensitive OC | I | NCT03704467 | Berzosertib/carboplatin/carboplatin safe doses were established; however, phase II was not started [200] | |||
| Gemcitabine/Cisplatin/Etoposide/Carboplatin/Irinotecan | Advanced solid tumors | I | NCT02157792 | Berzosertib/cisplatin and berzosertib/carboplatin combinations were well-tolerated and presented preliminary preclinical activity in patients with advanced solid tumors, including OC patients. [201,202] | |||
| Topotecan | Ovarian neoplasms | I | NCT02487095 | Only one patient with OC was recruited, this patient presented a short duration of the response and a progressive disease [203] | |||
| Gemcitabine | Recurrent OC | II | NCT02595892 | The addition of berzosertib to gemcitabine in platinum-resistant HGSC increased PFS rate compared to gemcitabine in monotherapy [204] | |||
| Gartisertib | Niraparib | PARPi resistant and recurrent OC | I | NCT04149145 | Not yet recruiting patients | ||
| Elimusertib | Niraparib | Advanced OC | I | NCT04267939 | No results posted | ||
| Gemcitabine | Advanced OC | I | NCT04616534 | No results posted | |||
| Gemcitabine +/− Cisplatin | Advanced OC | I | NCT04491942 | No results posted | |||
| CHK1 | SRA737 | - | HGSC OC with/without CCNE1 gene amplification | I/II | NCT02797964 | SRA737-maximum tolerated dose was established. Based on tolerability and pharmacokinetics, phase II was recommended [205] | |
| Gemcitabine +/− Cisplatin | HGSC OC | I/II | NCT02797977 | Low-dose gemcitabine combined with SRA737 has been well-tolerated in HGSC OC patients [206] | |||
| LY2880070 | +/− Gemcitabine | Advanced or metastatic OC | I/II | NCT02632448 | LY2880070 together with low-dose gemcitabine was well-tolerated [207] | ||
| CHK2 | PHI-101 | - | Platinum-resistant or refractory OC | I | NCT04678102 | No results posted | |
| CHK1/ CHK2 | Prexasertib | - | Platinum-resistant or refractory OC | II | NCT03414047 | Prexasertib has demonstrated durable single agent activity in a subset of OC patients regardless their clinical characteristics, BRCA status of prior therapies [208] | |
| - | HGSC OC with/without BRCA mutations | II | NCT02203513 | Prexasertib presented clinical activity and was tolerable in HGSC OC patients with BRCA-wild type [209] | |||
| Olaparib | Advanced solid tumors | I | NCT03057145 | The combination of prexasertib and olaparib had preclinical activity in BRCA-mutant HGSC OC patients who had previously progressed on a PARPi [210] | |||
| WEE1 | Adavosertib | - | Advanced OC | I | NCT02659241 | No results posted | |
| Carboplatin/Paclitaxel/Gemcitabine/PLD | Platinum-resistant OC | II | NCT02272790 | 3% of platinum-resistant OC patients presented a completed response and 29% a partial response. The highest response rate was obtained with adavosertib/carboplatin combination [211] | |||
| Carboplatin | TP53-mutated refractory and resistant OC | II | NCT01164995 | Adavosertib/carboplatin combination demonstrated a manageable toxicity. The overall response rate was 43% and one patient (5%) presented prolonged complete response [212] | |||
| Gemcitabine | Recurrent OC | II | NCT02101775 | Recurrent OC patients-treated with adavosertib/gemcitabine presented a longer PFS [213] | |||
| Paclitaxel + carboplatin | TP53-mutated platinum- sensitive OC | II | NCT01357161 | The addition of adavosertib to chemotherapy treatment (paclitaxel/carboplatin) improved PFS [214] | |||
| Olaparib | Recurrent OC Refractory solid tumors Advanced solid tumors | II I II | NCT03579316 NCT02511795 NCT02576444 | Adavosertib in monotherapy or combined with olaparib demonstrated efficacy in patients with resistance to PARPi. Adavosertib/olaparib combination presented manageable toxicities [215,216,217] | |||
| ZN-c3 | Niraparib | Platinum-resistant OC | I/II | NCT05198804 | No results posted, recruiting patients | ||
| p53 pathway | Mutated p53 | PRIMA-1MET | PLD | Platinum-resistant HGSC OC | II | NCT03268382 | 36 patients were enrolled in this study which have been treated with several doses of PLD and APR-246. That combination was feasible and adverse effects were manageable |
| +/− Carboplatin/PLD | Recurrent HGSC OC | I/II | NCT02098343 | APR-246/carboplatin or APR-246/PLD combinations were effective in HGSC OC patients with TP53-mutated and recommended phase II dose has been established [218] | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ovejero-Sánchez, M.; González-Sarmiento, R.; Herrero, A.B. DNA Damage Response Alterations in Ovarian Cancer: From Molecular Mechanisms to Therapeutic Opportunities. Cancers 2023, 15, 448. https://doi.org/10.3390/cancers15020448
Ovejero-Sánchez M, González-Sarmiento R, Herrero AB. DNA Damage Response Alterations in Ovarian Cancer: From Molecular Mechanisms to Therapeutic Opportunities. Cancers. 2023; 15(2):448. https://doi.org/10.3390/cancers15020448
Chicago/Turabian StyleOvejero-Sánchez, María, Rogelio González-Sarmiento, and Ana Belén Herrero. 2023. "DNA Damage Response Alterations in Ovarian Cancer: From Molecular Mechanisms to Therapeutic Opportunities" Cancers 15, no. 2: 448. https://doi.org/10.3390/cancers15020448