Shiga Toxin (Stx) Type 1a and Stx2a Translocate through a Three-Layer Intestinal Model

 , ,
, ,
Abstract
:1. Introduction
2. Results
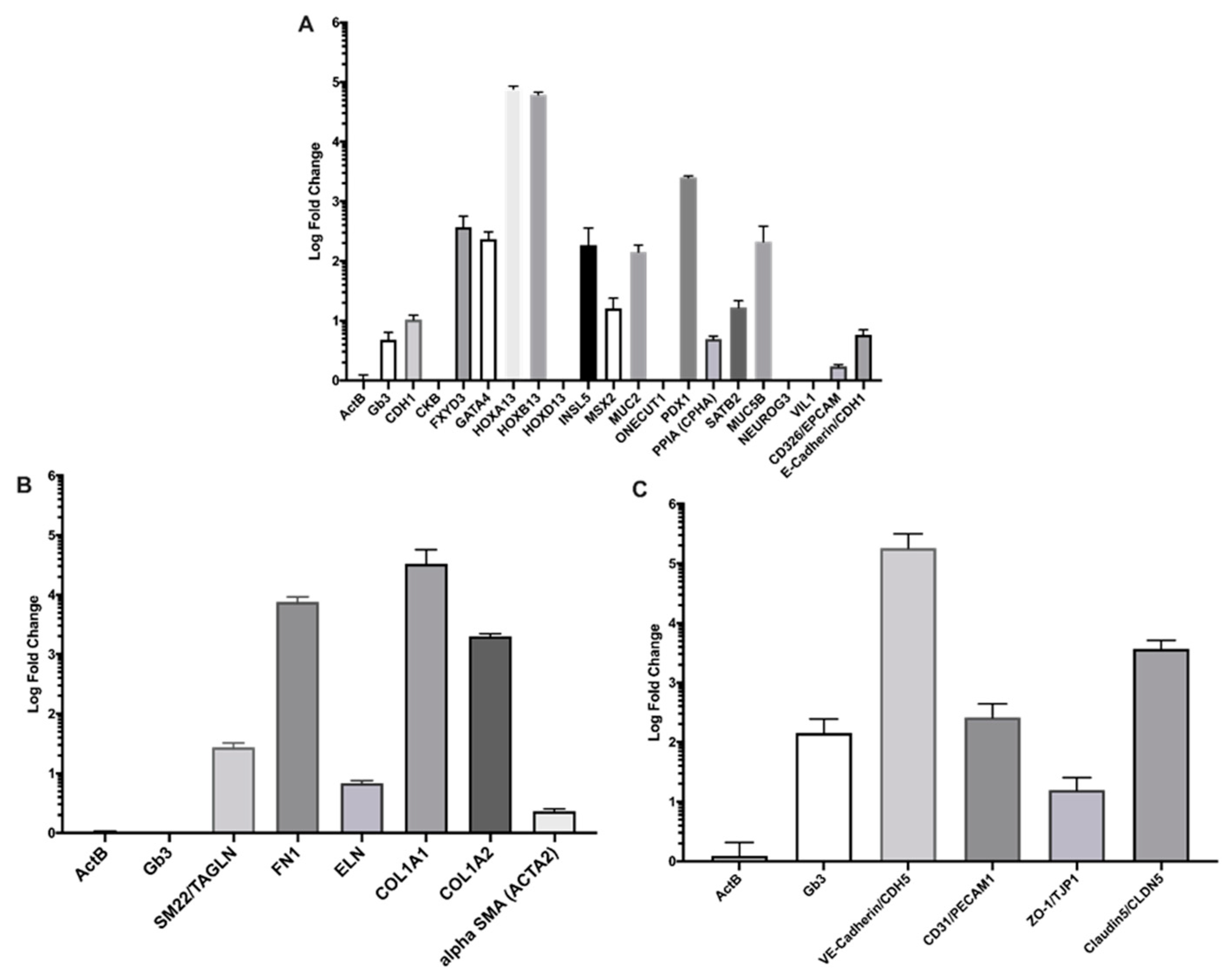
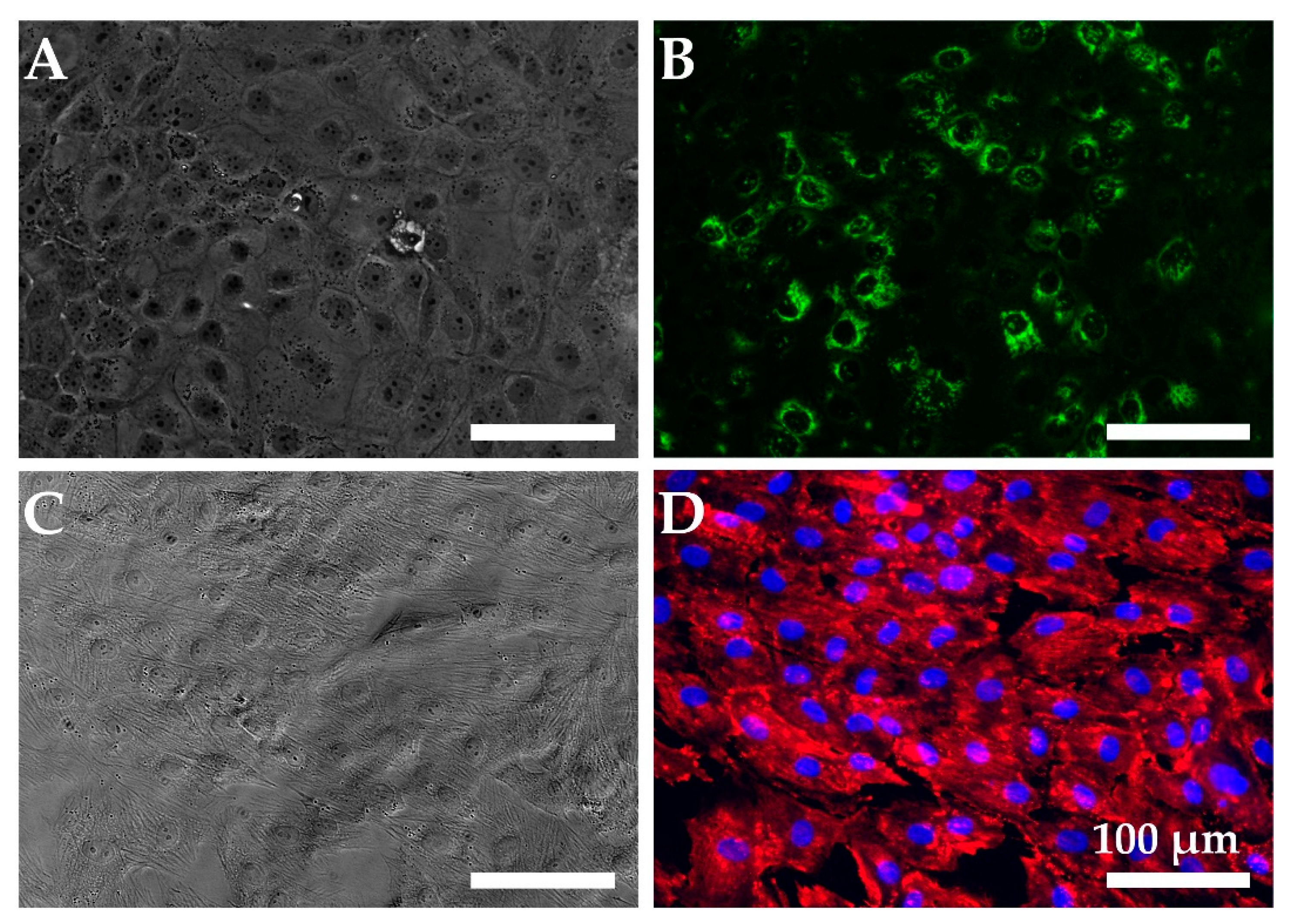
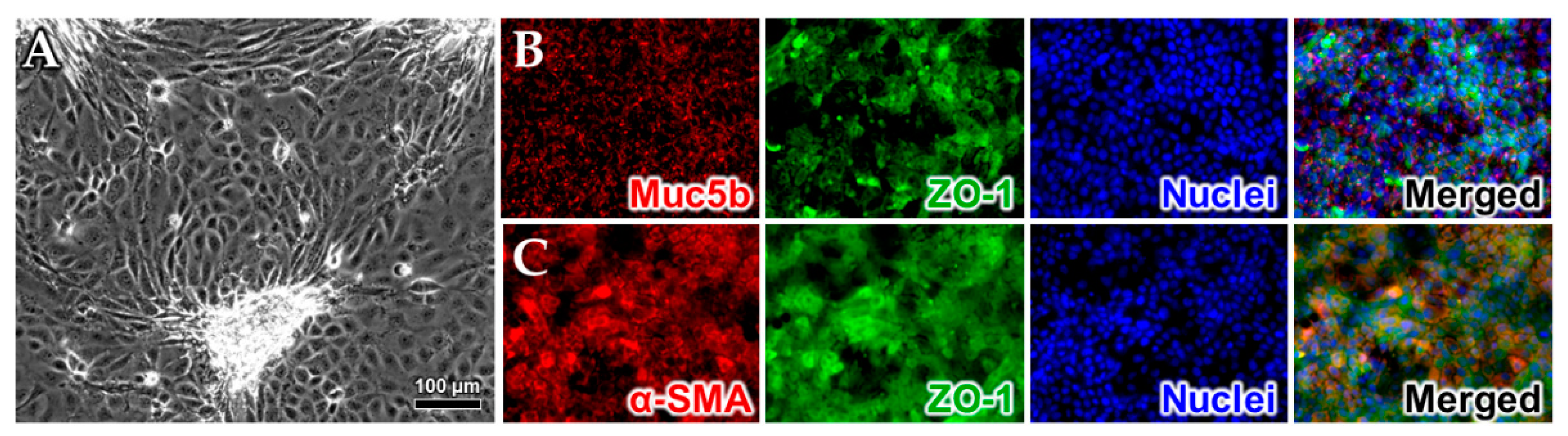
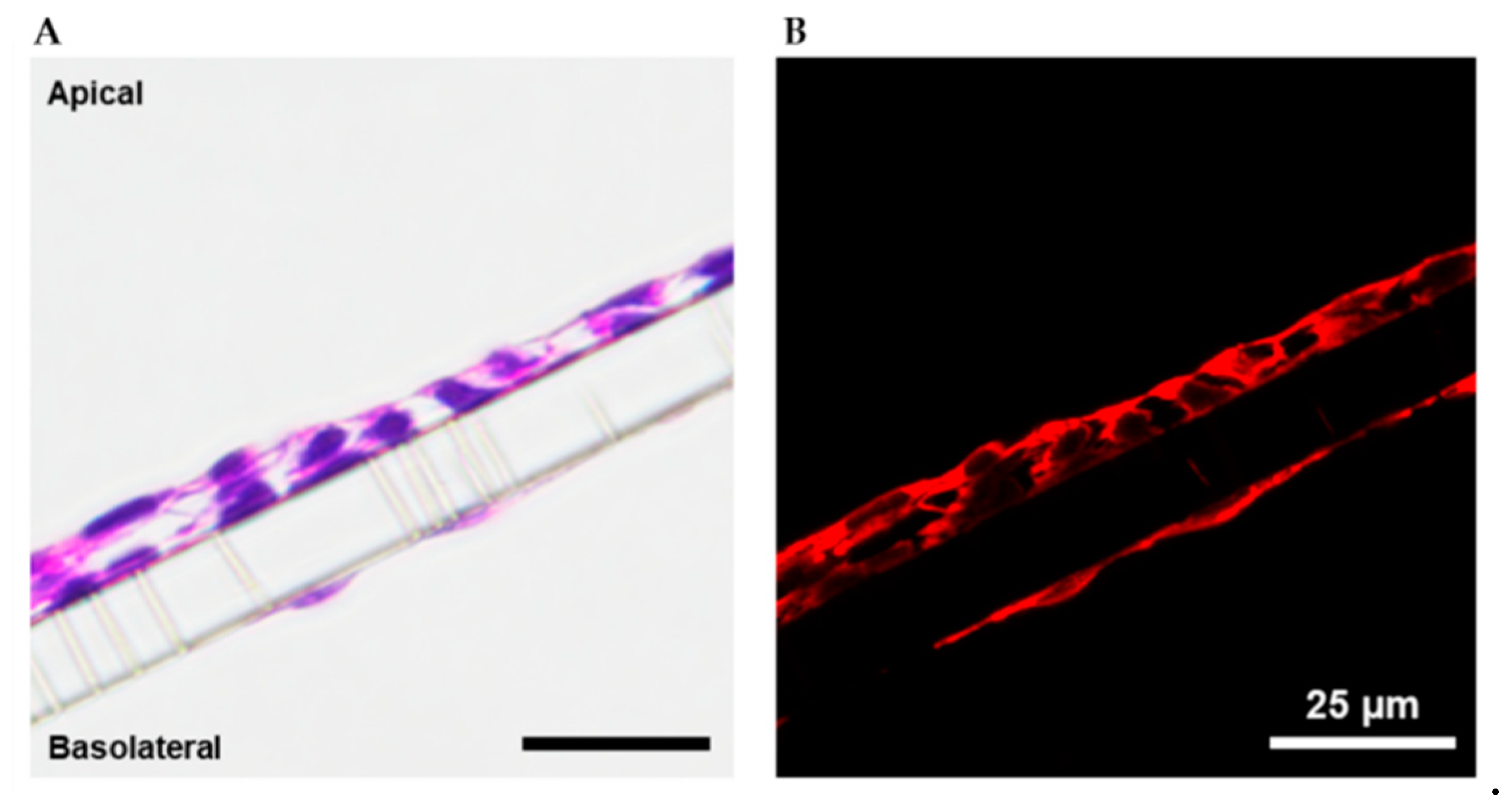
2.1. Cell Model Validation
Quantitative Reverse Transcription PCR (RT-qPCR)
2.2. Exposure of Cell Models to Purified Stxs
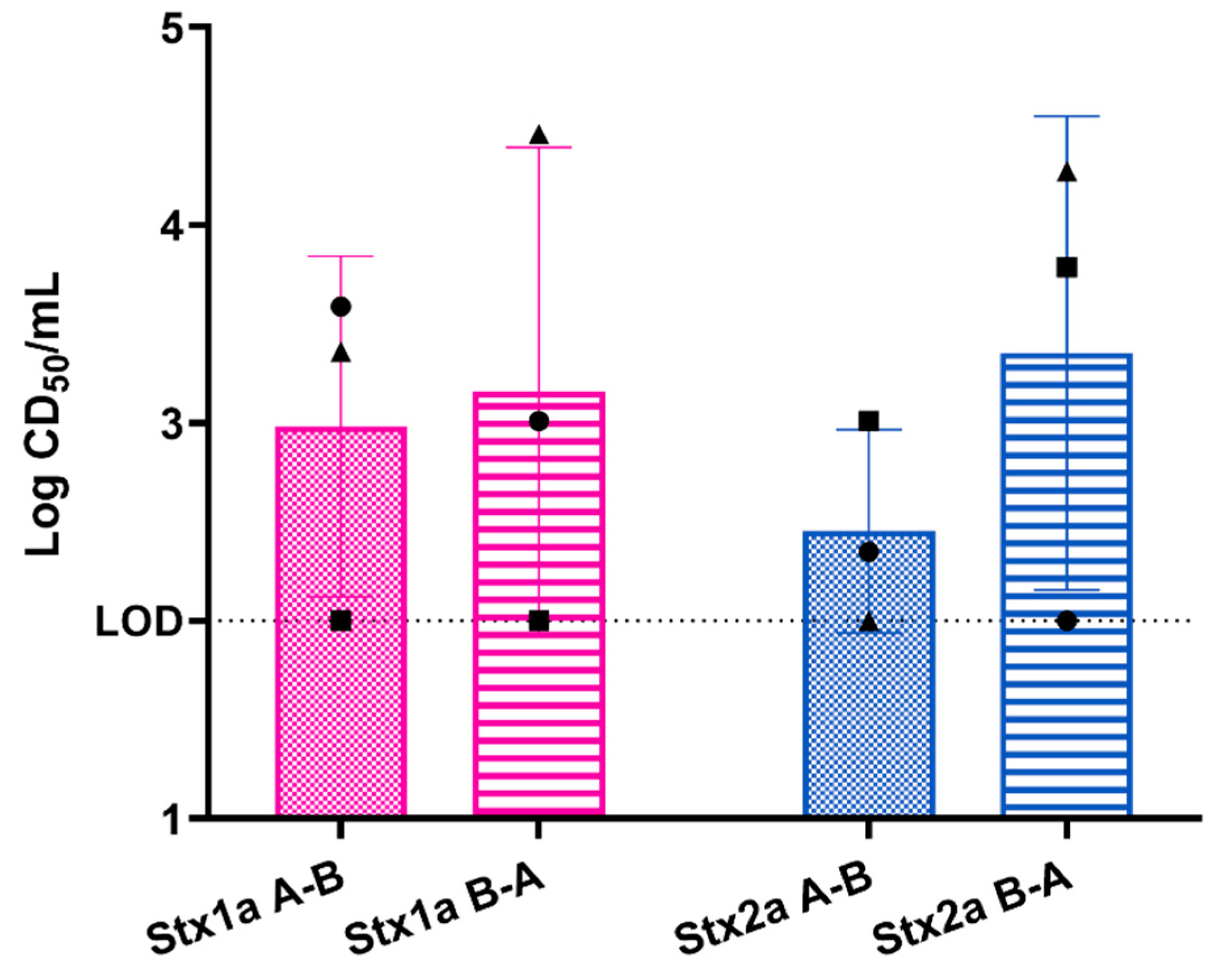
2.2.1. Response of Primary Colonic Epithelial Monolayer to Purified Stx
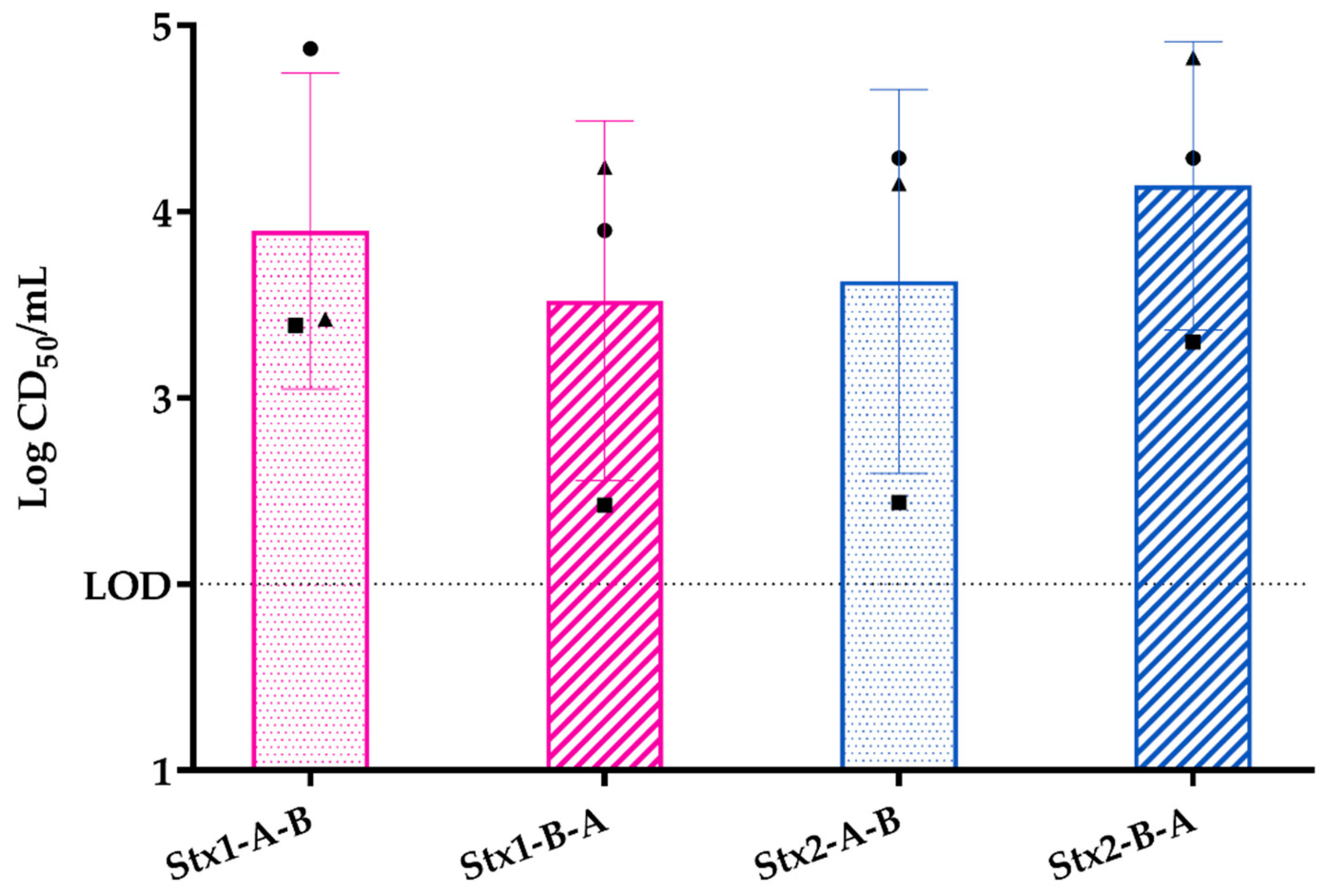
2.2.2. Response of Three-Layer Colonic Co-Culture Model to Purified Stx
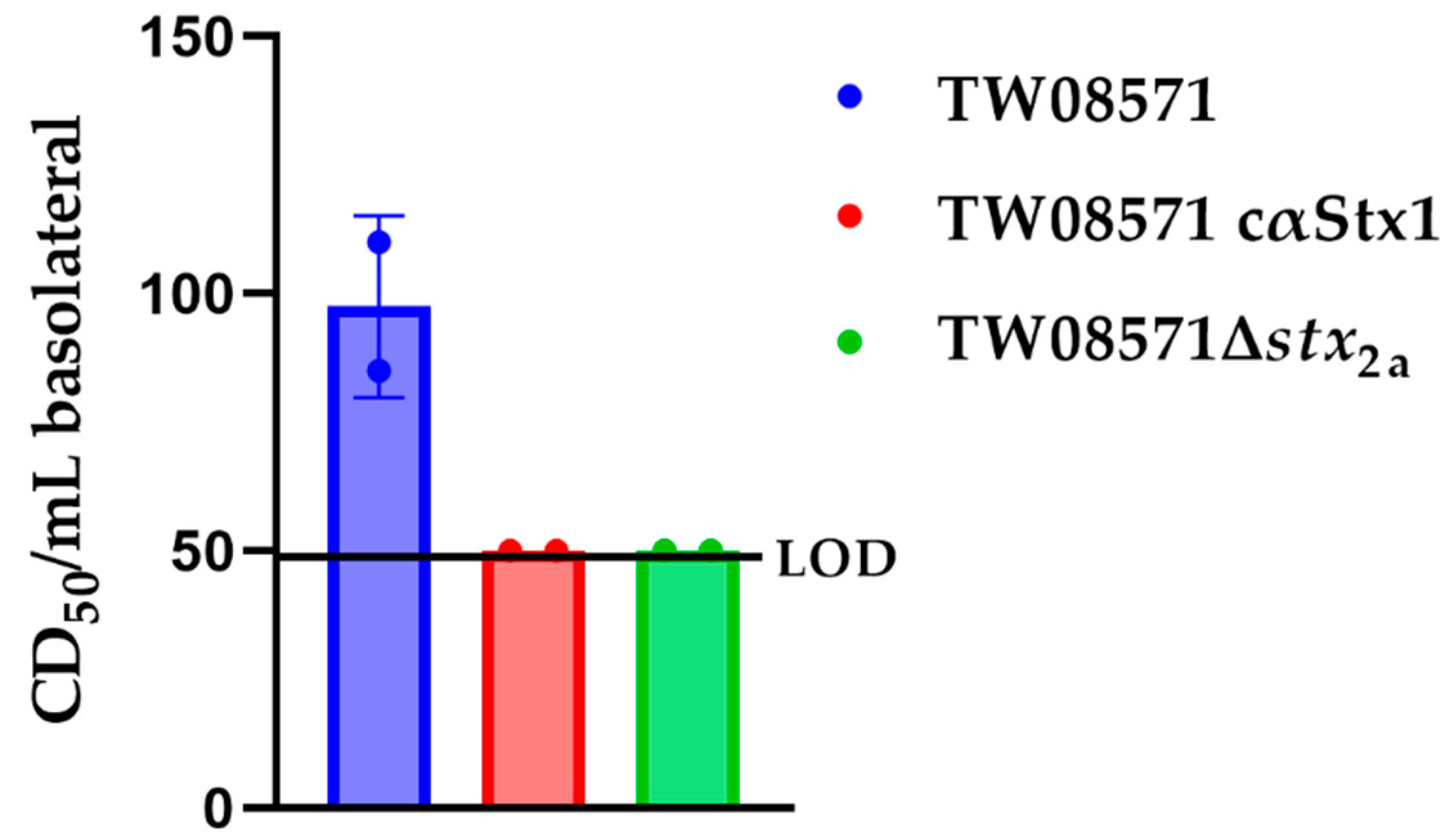
2.3. Exposure of the Three-Layer Model to STEC
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.1.1. Primary Cell Culture
4.1.2. Human Adipose-Derived Mesenchymal Stem/Stromal Cells
4.1.3. Myofibroblast Differentiation from hAD-MSCs
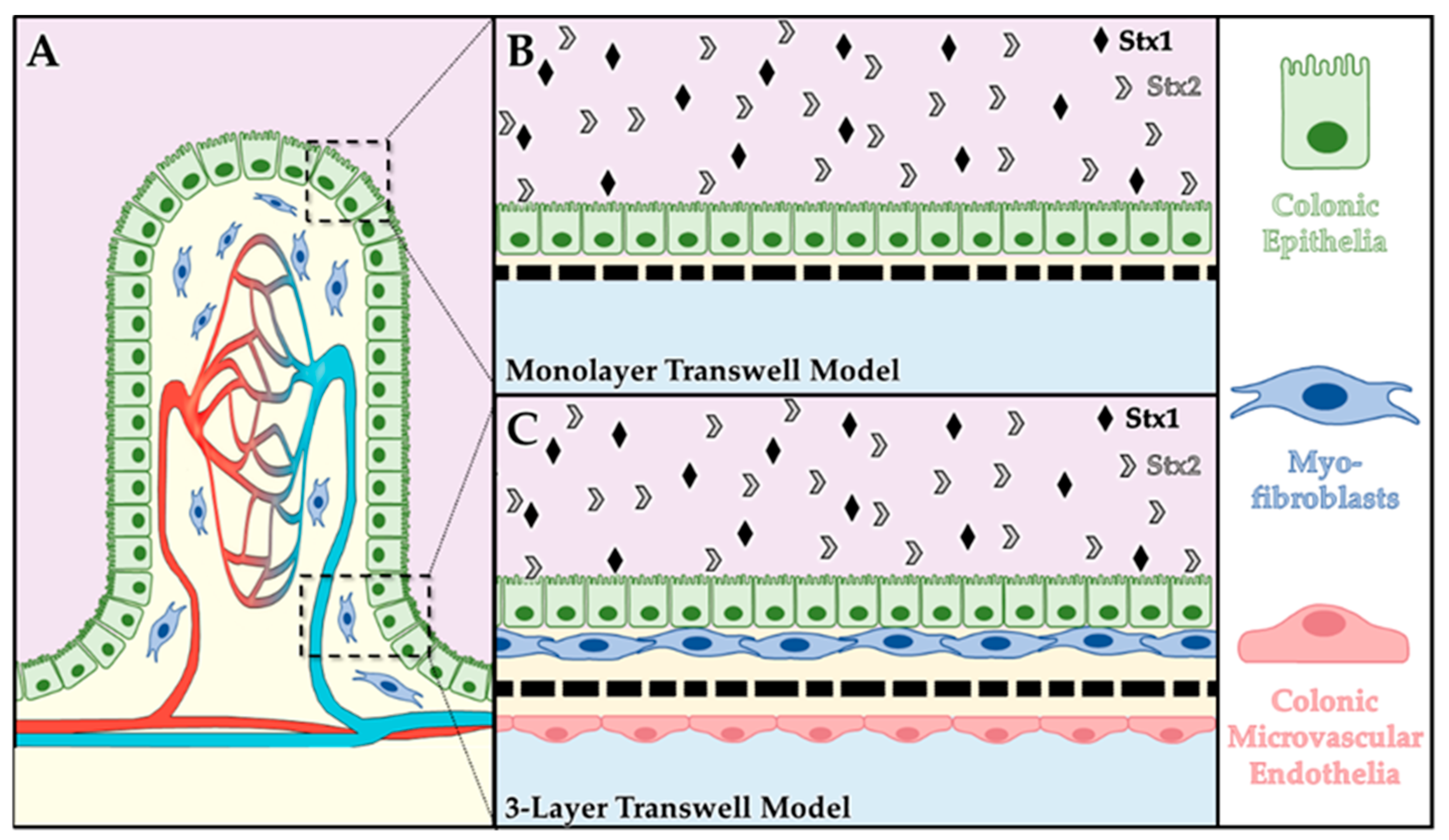
4.2. Transwell Model Systems
4.2.1. Human Primary Colonic Epithelial Cell Monolayer Model
4.2.2. Colonic 3-Layer Tissue Model
4.3. Immunohistochemistry, Histology, and Microscopy
4.4. RT-qPCR
4.5. Stx Purification and Cytotoxicity Assay
4.6. Exposure of Transwells to Purified Stxs or to STEC
4.7. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bruyand, M.; Mariani-Kurkdjian, P.; Gouali, M.; de Valk, H.; King, L.A.; Le Hello, S.; Bonacorsi, S.; Loirat, C. Hemolytic uremic syndrome due to Shiga toxin-producing Escherichia coli infection. Med. Mal. Infect. 2018, 48, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Biernbaum, E.N.; Kudva, I.T. AB(5) Enterotoxin-Mediated Pathogenesis: Perspectives Gleaned from Shiga Toxins. Toxins 2022, 14, 62. [Google Scholar] [CrossRef] [PubMed]
- Scheutz, F.; Teel, L.D.; Beutin, L.; Pierard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S.; et al. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melton-Celsa, A.R. Shiga toxin (Stx) classification, structure, and function. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [Green Version]
- Detzner, J.; Pohlentz, G.; Müthing, J. Enterohemorrhagic Escherichia coli and a Fresh View on Shiga Toxin-Binding Glycosphingolipids of Primary Human Kidney and Colon Epithelial Cells and Their Toxin Susceptibility. Int. J. Mol. Sci. 2022, 23, 6884. [Google Scholar] [CrossRef]
- Zumbrun, S.D.; Hanson, L.; Sinclair, J.F.; Freedy, J.; Melton-Celsa, A.R.; Rodriguez-Canales, J.; Hanson, J.C.; O’Brien, A.D. Human intestinal tissue and cultured colonic cells contain globotriaosylceramide synthase mRNA and the alternate Shiga toxin receptor globotetraosylceramide. Infect. Immun. 2010, 78, 4488–4499. [Google Scholar] [CrossRef] [Green Version]
- Steil, D.; Schepers, C.L.; Pohlentz, G.; Legros, N.; Runde, J.; Humpf, H.U.; Karch, H.; Müthing, J. Shiga toxin glycosphingolipid receptors of Vero-B4 kidney epithelial cells and their membrane microdomain lipid environment. J. Lipid Res. 2015, 56, 2322–2336. [Google Scholar] [CrossRef] [Green Version]
- Brigotti, M.; Carnicelli, D.; Arfilli, V.; Tamassia, N.; Borsetti, F.; Fabbri, E.; Tazzari, P.L.; Ricci, F.; Pagliaro, P.; Spisni, E.; et al. Identification of TLR4 as the receptor that recognizes Shiga toxins in human neutrophils. J. Immunol. 2013, 191, 4748–4758. [Google Scholar] [CrossRef] [Green Version]
- Mühlen, S.; Dersch, P. Treatment Strategies for Infections With Shiga Toxin-Producing Escherichia coli. Front. Cell. Infect. Microbiol. 2020, 10, 169. [Google Scholar] [CrossRef]
- Tarr, P.I.; Freedman, S.B. Why antibiotics should not be used to treat Shiga toxin-producing Escherichia coli infections. Curr. Opin. Gastroenterol. 2022, 38, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Acheson, D.W.; Moore, R.; De Breucker, S.; Lincicome, L.; Jacewicz, M.; Skutelsky, E.; Keusch, G.T. Translocation of Shiga toxin across polarized intestinal cells in tissue culture. Infect. Immun. 1996, 64, 3294–3300. [Google Scholar] [CrossRef] [Green Version]
- Hurley, B.P.; Jacewicz, M.; Thorpe, C.M.; Lincicome, L.L.; King, A.J.; Keusch, G.T.; Acheson, D.W. Shiga toxins 1 and 2 translocate differently across polarized intestinal epithelial cells. Infect. Immun. 1999, 67, 6670–6677. [Google Scholar] [CrossRef] [Green Version]
- Philpott, D.J.; Ackerley, C.A.; Kiliaan, A.J.; Karmali, M.A.; Perdue, M.H.; Sherman, P.M. Translocation of verotoxin-1 across T84 monolayers: Mechanism of bacterial toxin penetration of epithelium. Am. J. Physiol. 1997, 273, G1349–G1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, S.L.; Billoud, L.; Lewis, S.B.; Phillips, A.D.; Schüller, S. Shiga toxin production and translocation during microaerobic human colonic infection with Shiga toxin-producing E. coli O157:H7 and O104:H4. Cell. Microbiol. 2014, 16, 1255–1266. [Google Scholar] [CrossRef] [Green Version]
- Russo, L.M.; Melton-Celsa, A.R.; Smith, M.J.; O’Brien, A.D. Comparisons of native Shiga toxins (Stxs) type 1 and 2 with chimeric toxins indicate that the source of the binding subunit dictates degree of toxicity. PLoS ONE 2014, 9, e93463. [Google Scholar] [CrossRef] [Green Version]
- Lukyanenko, V.; Malyukova, I.; Hubbard, A.; Delannoy, M.; Boedeker, E.; Zhu, C.; Cebotaru, L.; Kovbasnjuk, O. Enterohemorrhagic Escherichia coli infection stimulates Shiga toxin 1 macropinocytosis and transcytosis across intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 2011, 301, C1140–C1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malyukova, I.; Murray, K.F.; Zhu, C.; Boedeker, E.; Kane, A.; Patterson, K.; Peterson, J.R.; Donowitz, M.; Kovbasnjuk, O. Macropinocytosis in Shiga toxin 1 uptake by human intestinal epithelial cells and transcellular transcytosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G78–G92. [Google Scholar] [CrossRef] [Green Version]
- Philpott, D.J.; McKay, D.M.; Mak, W.; Perdue, M.H.; Sherman, P.M. Signal transduction pathways involved in enterohemorrhagic Escherichia coli-induced alterations in T84 epithelial permeability. Infect. Immun. 1998, 66, 1680–1687. [Google Scholar] [CrossRef] [Green Version]
- Te Loo, D.M.; Monnens, L.A.; van Der Velden, T.J.; Vermeer, M.A.; Preyers, F.; Demacker, P.N.; van Den Heuvel, L.P.; van Hinsbergh, V.W. Binding and transfer of verocytotoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood 2000, 95, 3396–3402. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, A.L.; Arvidsson, I.; Johansson, K.E.; Chromek, M.; Rebetz, J.; Loos, S.; Kristoffersson, A.C.; Békássy, Z.D.; Mörgelin, M.; Karpman, D. A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles. PLoS Pathog. 2015, 11, e1004619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, L.M.; Melton-Celsa, A.R.; Smith, M.A.; Smith, M.J.; O’Brien, A.D. Oral intoxication of mice with Shiga toxin type 2a (Stx2a) and protection by anti-Stx2a monoclonal antibody 11E10. Infect. Immun. 2014, 82, 1213–1221. [Google Scholar] [CrossRef] [Green Version]
- Russo, L.M.; Melton-Celsa, A.R.; O’Brien, A.D. Shiga Toxin (Stx) Type 1a Reduces the Oral Toxicity of Stx Type 2a. J. Infect. Dis. 2016, 213, 1271–1279. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, S.; Karve, S.S.; Weiss, A.A.; Hawkins, J.; Poling, H.M.; Helmrath, M.A.; Wells, J.M.; McCauley, H.A. Tissue Responses to Shiga Toxin in Human Intestinal Organoids. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 171–190. [Google Scholar] [CrossRef] [Green Version]
- Karve, S.S.; Pradhan, S.; Ward, D.V.; Weiss, A.A. Intestinal organoids model human responses to infection by commensal and Shiga toxin producing Escherichia coli. PLoS ONE 2017, 12, e0178966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, P.; Acheson, D.; Chitrakar, R.; Cnaan, A.; Gibbs, K.; Hirschman, G.H.; Christen, E.; Trachtman, H. Basic fibroblast growth factor among children with diarrhea-associated hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2002, 13, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Desai, V.D.; Hsia, H.C.; Schwarzbauer, J.E. Reversible modulation of myofibroblast differentiation in adipose-derived mesenchymal stem cells. PLoS ONE 2014, 9, e86865. [Google Scholar] [CrossRef] [Green Version]
- Forbester, J.L.; Goulding, D.; Vallier, L.; Hannan, N.; Hale, C.; Pickard, D.; Mukhopadhyay, S.; Dougan, G. Interaction of Salmonella enterica Serovar Typhimurium with Intestinal Organoids Derived from Human Induced Pluripotent Stem Cells. Infect. Immun. 2015, 83, 2926–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- In, J.; Foulke-Abel, J.; Zachos, N.C.; Hansen, A.M.; Kaper, J.B.; Bernstein, H.D.; Halushka, M.; Blutt, S.; Estes, M.K.; Donowitz, M.; et al. Enterohemorrhagic Escherichia coli reduce mucus and intermicrovillar bridges in human stem cell-derived colonoids. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 48–62.e3. [Google Scholar] [CrossRef] [Green Version]
- Tesh, V.L.; Burris, J.A.; Owens, J.W.; Gordon, V.M.; Wadolkowski, E.A.; O’Brien, A.D.; Samuel, J.E. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect. Immun. 1993, 61, 3392–3402. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Donohue-Rolfe, A.; Krautz-Peterson, G.; Sevo, M.; Parry, N.; Abeijon, C.; Tzipori, S. Gnotobiotic piglet infection model for evaluating the safe use of antibiotics against Escherichia coli O157:H7 infection. J. Infect. Dis. 2009, 199, 486–493. [Google Scholar] [CrossRef] [Green Version]
- Hauser, J.R.; Atitkar, R.R.; Petro, C.D.; Lindsey, R.L.; Strockbine, N.; O’Brien, A.D.; Melton-Celsa, A.R. The Virulence of Escherichia coli O157:H7 Isolates in Mice Depends on Shiga Toxin Type 2a (Stx2a)-Induction and High Levels of Stx2a in Stool. Front. Cell. Infect. Microbiol. 2020, 10, 62. [Google Scholar] [CrossRef] [Green Version]
- Matussek, A.; Jernberg, C.; Einemo, I.M.; Monecke, S.; Ehricht, R.; Engelmann, I.; Löfgren, S.; Mernelius, S. Genetic makeup of Shiga toxin-producing Escherichia coli in relation to clinical symptoms and duration of shedding: A microarray analysis of isolates from Swedish children. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 1433–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melton-Celsa, A.R.; O’Brien, A.D. Shiga toxins of Shigella dysenteriae and Escherichia coli. In Handbook of Experimental Pharmacology; Aktories, K., Just, I., Eds.; Springer: Berlin/Heidelberg, Germany, 2000; Volume 145, pp. 385–406. [Google Scholar]
- Petro, C.D.; Trojnar, E.; Sinclair, J.; Liu, Z.M.; Smith, M.; O’Brien, A.D.; Melton-Celsa, A. Shiga toxin (Stx) type 1a reduces the toxicity of the more potent Stx2a in vivo and in vitro. Infect. Immun. 2019, 87, e00787-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnenberg, M.S.; Kaper, J.B. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun. 1991, 59, 4310–4317. [Google Scholar] [CrossRef] [Green Version]
- Edwards, A.C.; Melton-Celsa, A.R.; Arbuthnott, K.; Stinson, J.R.; Schmitt, C.K.; Wong, H.C.; O’Brien, A.D. Vero cell neutralization and mouse protective efficacy of humanized monoclonal antibodies against Escherichia coli toxins Stx1 and Stx2. In Escherichia coli O157:H7 and Other Shiga Toxin-Producing E. coli Strains; Kaper, J.B., O’Brien, A.D., Eds.; ASM Press: Washington, DC, USA, 1998; pp. 388–392. [Google Scholar]
- Ostroff, S.M.; Griffin, P.M.; Tauxe, R.V.; Shipman, L.D.; Greene, K.D.; Wells, J.G.; Lewis, J.H.; Blake, P.A.; Kobayashi, J.M. A statewide outbreak of Escherichia coli O157:H7 infections in Washington State. Am. J. Epidemiol. 1990, 132, 239–247. [Google Scholar] [CrossRef]
- McKee, M.L.; Melton-Celsa, A.R.; Moxley, R.A.; Francis, D.H.; O’Brien, A.D. Enterohemorrhagic Escherichia coli O157:H7 requires intimin to colonize the gnotobiotic pig intestine and to adhere to HEp-2 cells. Infect. Immun. 1995, 63, 3739–3744. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bova, R.A.; Lamont, A.C.; Picou, T.J.; Ho, V.B.; Gilchrist, K.H.; Melton-Celsa, A.R. Shiga Toxin (Stx) Type 1a and Stx2a Translocate through a Three-Layer Intestinal Model. Toxins 2023, 15, 207. https://doi.org/10.3390/toxins15030207
Bova RA, Lamont AC, Picou TJ, Ho VB, Gilchrist KH, Melton-Celsa AR. Shiga Toxin (Stx) Type 1a and Stx2a Translocate through a Three-Layer Intestinal Model. Toxins. 2023; 15(3):207. https://doi.org/10.3390/toxins15030207
Chicago/Turabian StyleBova, Rebecca A., Andrew C. Lamont, Theodore J. Picou, Vincent B. Ho, Kristin H. Gilchrist, and Angela R. Melton-Celsa. 2023. "Shiga Toxin (Stx) Type 1a and Stx2a Translocate through a Three-Layer Intestinal Model" Toxins 15, no. 3: 207. https://doi.org/10.3390/toxins15030207