
Type 2 Diabetes Mellitus and Liver Disease: Across the Gut–Liver Axis from Fibrosis to Cancer
 , and
, and 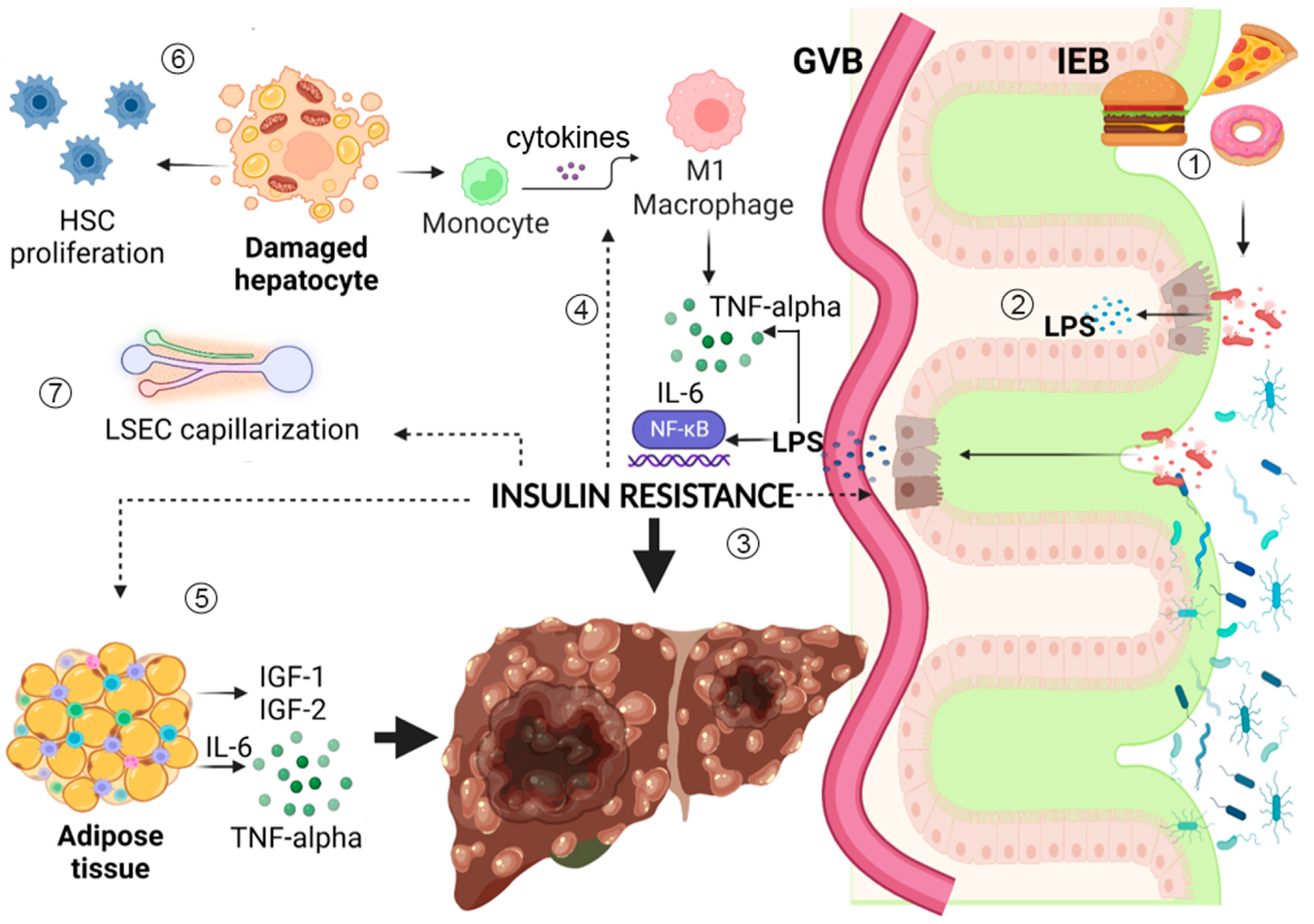{kind=link}
Abstract
:1. Introduction
2. Epidemiology
2.1. Diabetes and Insulin Resistance in Non-Alcoholic Fatty Liver Disease (NAFLD)
2.2. Prevalence of HCC in NAFLD
3. How Insulin Resistance and Diabetes Favor Liver Fibrosis Progression
3.1. Insulin Resistance Is Involved in Hepatocyte Damage in NAFLD and NASH
3.2. Insulin Resistance Promotes Endothelial Dysfunction Contributing to Liver Fibrosis Progression
4. Interplay between Insulin Resistance and the Gut–Liver Axis
4.1. From Insulin Resistance to Metabolic Endotoxemia
4.2. Dysbiosis and Metabolic Endotoxemia as a Trigger of Insulin Resistance and Diabetes: A Vicious Circle
5. Role of Insulin Resistance in the Development of Primary Liver Tumors
5.1. Hepatocellular Carcinoma
5.2. Cholangiocarcinoma
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HFD | High-fatty diet |
| AGEs | advanced glycation end products |
| HSC | hepatic stellate cells |
| LSEC | liver sinusoid endothelial cell |
| TNF-alpha | tumor necrosis factor-alpha |
| Il-6 | interleukin-6 |
| LPS | lypopolisaccharides |
| nf-kB | nuclear factor-kappa-B |
| IGF-1 | insulin growth factor-1 |
| IGF-2 | insulin growth factor-2 |
| GVB | gut vascular barrier |
| IEB | intestinal epithelial barrier |
References
- Ajmera, V.; Cepin, S.; Tesfai, K.; Hofflich, H.; Cadman, K.; Lopez, S.; Madamba, E.; Bettencourt, R.; Richards, L.; Behling, C.; et al. A prospective study on the prevalence of NAFLD, advanced fibrosis, cirrhosis and hepatocellular carcinoma in people with type 2 diabetes. J. Hepatol. 2023, 78, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R. Hepatogenous Diabetes: An Underestimated Problem of Liver Cirrhosis. Indian J. Endocrinol. Metab. 2018, 22, 552–559. [Google Scholar] [CrossRef] [PubMed]
- García-Compeán, D.; Jáquez-Quintana, J.O.; Lavalle-González, F.J.; Reyes-Cabello, E.; González-González, J.A.; Muñoz-Espinosa, L.E.; Vázquez-Elizondo, G.; Villarreal-Pérez, J.Z.; Maldonado-Garza, H.J. The prevalence and clinical characteristics of glucose metabolism disorders in patients with liver cirrhosis. A prospective study. Ann. Hepatol. 2012, 11, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-C.; Cheng, P.-N.; Kao, J.-H. Systematic review: Chronic viral hepatitis and metabolic derangement. Aliment. Pharmacol. Ther. 2020, 51, 216–230. [Google Scholar] [CrossRef]
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.-A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The Role of Inflammation in Diabetes: Current Concepts and Future Perspectives. Eur. Cardiol. Rev. 2019, 14, 50–59. [Google Scholar] [CrossRef]
- El-Zayadi, A.-R. Hepatic steatosis: A benign disease or a silent killer. World J. Gastroenterol. 2008, 14, 4120–4126. [Google Scholar] [CrossRef]
- Li, Y.; Peng, Y.; Shen, Y.; Zhang, Y.; Liu, L.; Yang, X. Dietary polyphenols: Regulate the advanced glycation end products-RAGE axis and the microbiota-gut-brain axis to prevent neurodegenerative diseases. Crit. Rev. Food Sci. Nutr. 2022, 1–27. [Google Scholar] [CrossRef]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef]
- Clark, M.; Kroger, C.J.; Tisch, R.M. Type 1 Diabetes: A Chronic Anti-Self-Inflammatory Response. Front. Immunol. 2017, 8, 1898. [Google Scholar] [CrossRef]
- Budd, J.; Cusi, K. Nonalcoholic Fatty Liver Disease: What Does the Primary Care Physician Need to Know? Am. J. Med. 2020, 133, 536–543. [Google Scholar] [CrossRef]
- Younossi, Z.; Tacke, F.; Arrese, M.; Sharma, B.C.; Mostafa, I.; Bugianesi, E.; Wong, V.W.-S.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Trépo, E.; Valenti, L. Update on NAFLD genetics: From new variants to the clinic. J. Hepatol. 2020, 72, 1196–1209. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Leung, J.C.; Loong, T.C.; Wei, J.L.; Wong, G.L.; Chan, A.W.; Choi, P.C.; Shu, S.S.; Chim, A.M.; Chan, H.L.; Wong, V.W. Histological severity and clinical outcomes of nonalcoholic fatty liver disease in nonobese patients. Hepatology 2017, 65, 54–64. [Google Scholar] [CrossRef]
- Vos, B.; Moreno, C.; Nagy, N.; Féry, F.; Cnop, M.; Vereerstraeten, P.; Devière, J.; Adler, M. Lean non-alcoholic fatty liver disease (Lean-NAFLD): A major cause of cryptogenic liver dis-ease. Acta Gastroenterol. Belg. 2011, 74, 389–394. [Google Scholar]
- Younossi, Z.M.; Stepanova, M.; Negro, F.; Hallaji, S.; Younossi, Y.; Lam, B.; Srishord, M. Nonalcoholic Fatty Liver Disease in Lean Individuals in the United States. Medicine 2012, 91, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, H.J.; Lee, K.E.; Kim, D.J.; Kim, S.K.; Ahn, C.W.; Lim, S.-K.; Kim, K.R.; Lee, H.C.; Huh, K.B.; et al. Metabolic significance of nonalcoholic fatty liver disease in nonobese, nondiabetic adults. Arch. Intern. Med. 2004, 164, 2169–2175. [Google Scholar] [CrossRef]
- Fracanzani, A.L.; Valenti, L.; Bugianesi, E.; Vanni, E.; Grieco, A.; Miele, L.; Consonni, D.; Fatta, E.; Lombardi, R.; Marchesini, G.; et al. Risk of nonalcoholic steatohepatitis and fibrosis in patients with nonalcoholic fatty liver disease and low visceral adiposity. J. Hepatol. 2011, 54, 1244–1249. [Google Scholar] [CrossRef]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef]
- Fracanzani, A.L.; Valenti, L.; Bugianesi, E.; Andreoletti, M.; Colli, A.; Vanni, E.; Bertelli, C.; Fatta, E.; Bignamini, D.; Marchesini, G.; et al. Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: A role for insulin resistance and diabetes. Hepatology 2008, 48, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Ciardullo, S.; Monti, T.; Perseghin, G. High Prevalence of Advanced Liver Fibrosis Assessed by Transient Elastography among U.S. Adults with Type 2 Diabetes. Diabetes Care 2021, 44, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Bugianesi, E.; Pajvani, U.; Targher, G. Nonalcoholic fatty liver disease: Cause or consequence of type 2 diabetes? Liver Int. 2016, 36, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Mohamed, H.A.; Cvinar, J.L.; Gores, G.J.; Roberts, L.R.; Kim, R.W. Diabetes Mellitus Heightens the Risk of Hepatocellular Carcinoma Except in Patients with Hepatitis C Cirrhosis. Am. J. Gastroenterol. 2016, 111, 1573–1580. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Li, L.; Dai, J.; Natarajan, Y.; Yu, X.; Asch, S.M.; El-Serag, H.B. Effect of Metabolic Traits on the Risk of Cirrhosis and Hepatocellular Cancer in Nonalcoholic Fatty Liver Disease. Hepatology 2020, 71, 808–819. [Google Scholar] [CrossRef]
- Alexander, M.; Loomis, A.K.; van der Lei, J.; Duarte-Salles, T.; Prieto-Alhambra, D.; Ansell, D.; Pasqua, A.; Lapi, F.; Rijnbeek, P.; Mosseveld, M.; et al. Risks and clinical predictors of cirrhosis and hepatocellular carcinoma diagnoses in adults with diagnosed NAFLD: Real-world study of 18 million patients in four European cohorts. BMC Med. 2019, 17, 95. [Google Scholar] [CrossRef]
- Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 1 April 2023).
- Dyson, J.; Jaques, B.; Chattopadyhay, D.; Lochan, R.; Graham, J.; Das, D.; Aslam, T.; Patanwala, I.; Gaggar, S.; Cole, M.; et al. Hepatocellular cancer: The impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 2014, 60, 110–117. [Google Scholar] [CrossRef]
- Lomonaco, R.; Leiva, E.G.; Bril, F.; Shrestha, S.; Mansour, L.; Budd, J.; Romero, J.P.; Schmidt, S.; Chang, K.-L.; Samraj, G.; et al. Advanced Liver Fibrosis Is Common in Patients With Type 2 Diabetes Followed in the Outpatient Setting: The Need for Systematic Screening. Diabetes Care 2021, 44, 399–406. [Google Scholar] [CrossRef]
- Leite, N.C.; Villela-Nogueira, C.A.; Pannain, V.L.N.; Bottino, A.C.; Rezende, G.F.M.; Cardoso, C.R.L.; Salles, G.F. Histopathological stages of nonalcoholic fatty liver disease in type 2 diabetes: Prevalences and correlated factors. Liver Int. 2011, 31, 700–706. [Google Scholar] [CrossRef]
- Stine, J.G.; Wentworth, B.J.; Zimmet, A.; Rinella, M.E.; Loomba, R.; Caldwell, S.H.; Argo, C.K. Systematic review with meta-analysis: Risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment. Pharmacol. Ther. 2018, 48, 696–703. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans Is Associated with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131. [Google Scholar] [CrossRef]
- Asrih, M.; Jornayvaz, F.R. Metabolic syndrome and nonalcoholic fatty liver disease: Is insulin resistance the link? Mol. Cell. Endocrinol. 2015, 418 Pt 1, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Lomonaco, R.; Ortiz-Lopez, C.; Orsak, B.; Webb, A.; Hardies, J.; Darland, C.; Finch, J.; Gastaldelli, A.; Harrison, S.; Tio, F.; et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Léveillé, M.; Estall, J.L. Mitochondrial Dysfunction in the Transition from NASH to HCC. Metabolites 2019, 9, 233. [Google Scholar] [CrossRef]
- Longo, M.; Meroni, M.; Paolini, E.; Erconi, V.; Carli, F.; Fortunato, F.; Ronchi, D.; Piciotti, R.; Sabatini, S.; Macchi, C.; et al. TM6SF2/PNPLA3/MBOAT7 Loss-of-Function Genetic Variants Impact on NAFLD Development and Progression Both in Patients and in In Vitro Models. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 759–788. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, C.; Lee, S.; Kim, W.; Klevstig, M.; Harzandi, A.M.; Sikanic, N.; Arif, M.; Ståhlman, M.; Nielsen, J.; et al. Pyruvate kinase L/R is a regulator of lipid metabolism and mitochondrial function. Metab. Eng. 2019, 52, 263–272. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R.M.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol. 2013, 19, 6969–6978. [Google Scholar] [CrossRef]
- Moschen, A.; Wieser, V.; Tilg, H. Adiponectin: Key player in the adipose tissue-liver crosstalk. Curr. Med. Chem. 2012, 19, 5467–5473. [Google Scholar] [CrossRef]
- Daemen, S.; Schilling, J.D. The Interplay Between Tissue Niche and Macrophage Cellular Metabolism in Obesity. Front. Immunol. 2020, 10, 3133. [Google Scholar] [CrossRef]
- Ehses, J.A.; Zeman-Meier, D.; Wueest, S.; Rytka, J.; Boller, S.; Wielinga, P.Y.; Schraenen, A.; Lemaire, K.; Debray, S.; Van Lommel, L.; et al. Toll-like receptor 2-deficient mice are protected from insulin resistance and beta cell dysfunction induced by a high-fat diet. Diabetologia 2010, 53, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Peng, Y.; Wu, J.; Wang, Y.; Yao, L. Toll-like receptor 2/4 links to free fatty acid-induced inflammation and β-cell dysfunction. J. Leukoc. Biol. 2014, 95, 47–52. [Google Scholar] [CrossRef]
- Zhang, L.; Xie, Z.; Yu, H.; Du, H.; Wang, X.; Cai, J.; Qiu, Y.; Chen, R.; Jiang, X.; Liu, Z.; et al. TLR2 inhibition ameliorates the amplification effect of LPS on lipid accumulation and lipotoxicity in hepatic cells. Ann. Transl. Med. 2021, 9, 1429. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Yang, L.; van Rooijen, N.; Brenner, D.A.; Ohnishi, H.; Seki, E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology 2013, 57, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver—Linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef]
- Gora, I.M.; Ciechanowska, A.; Ladyzynski, P. NLRP3 Inflammasome at the Interface of Inflammation, Endothelial Dysfunction, and Type 2 Diabetes. Cells 2021, 10, 314. [Google Scholar] [CrossRef]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 2021, 74, 156–167. [Google Scholar] [CrossRef]
- King, R.J.; Harrison, L.; Gilbey, S.G.; Santhakumar, A.; Wyatt, J.; Jones, R.; Bodansky, H.J. Diabetic hepatosclerosis: Another diabetes microvascular complication? Diabet. Med. 2016, 33, e5–e7. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Kazazis, C.; Ioannidis, G. Diabetic hepatosclerosis: True clinical entity or ghost disease? Diabetes Metab. Syndr. 2017, 11 (Suppl. 2), S775–S776. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Caparrós, E.; Fernández-Iglesias, A.; Francés, R. Role of liver sinusoidal endothelial cells in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 411–431. [Google Scholar] [CrossRef]
- Du, W.; Wang, L. The Crosstalk Between Liver Sinusoidal Endothelial Cells and Hepatic Microenvironment in NASH Related Liver Fibrosis. Front. Immunol. 2022, 13, 936196. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Marrone, G.; Fernández-Iglesias, A. Hepatic microcirculation and mechanisms of portal hypertension. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Marrone, G.; Shah, V.H.; Gracia-Sancho, J. Sinusoidal communication in liver fibrosis and regeneration. J. Hepatol. 2016, 65, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Pasarín, M.; Abraldes, J.G.; Rodríguez-Vilarrupla, A.; La Mura, V.; García-Pagán, J.C.; Bosch, J. Insulin resistance and liver microcirculation in a rat model of early NAFLD. J. Hepatol. 2011, 55, 1095–1102. [Google Scholar] [CrossRef]
- Khalid, M.; Petroianu, G.; Adem, A. Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives. Biomolecules 2022, 12, 542. [Google Scholar] [CrossRef]
- Pereira, E.N.G.D.S.; Silvares, R.R.; Flores, E.E.I.; Rodrigues, K.L.; Ramos, I.P.; da Silva, I.J.; Machado, M.P.; Miranda, R.A.; Pazos-Moura, C.C.; Gonçalves-De-Albuquerque, C.F.; et al. Hepatic microvascular dysfunction and increased advanced glycation end products are components of non-alcoholic fatty liver disease. PLoS ONE 2017, 12, e0179654. [Google Scholar] [CrossRef]
- Francque, S.; Wamutu, S.; Chatterjee, S.; Van Marck, E.; Herman, A.; Ramon, A.; Jung, A.; Vermeulen, W.; De Winter, B.; Pelckmans, P.; et al. Non-alcoholic steatohepatitis induces non-fibrosis-related portal hypertension associated with splanchnic vasodilation and signs of a hyperdynamic circulation in vitro and in vivo in a rat model. Liver Int. 2010, 30, 365–375. [Google Scholar] [CrossRef]
- Nababan, S.H.H.; Lesmana, C.R.A. Portal Hypertension in Nonalcoholic Fatty Liver Disease: From Pathogenesis to Clinical Practice. J. Clin. Transl. Hepatol. 2022, 10, 979–985. [Google Scholar] [CrossRef]
- Westley, R.L.; May, F.E.B. A Twenty-first century cancer epidemic caused by obesity: The involvement of insulin, diabetes, and insulin-like growth factors. Int. J. Endocrinol. 2013, 2013, 632461. [Google Scholar] [CrossRef]
- Adamek, A.; Kasprzak, A. Insulin-Like Growth Factor (IGF) System in Liver Diseases. Int. J. Mol. Sci. 2018, 19, 1308. [Google Scholar] [CrossRef]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol.-Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chu, J.; Hao, W.; Zhang, J.; Li, H.; Yang, C.; Yang, J.; Chen, X.; Wang, H. Gut Microbiota and Type 2 Diabetes Mellitus: Association, Mechanism, and Translational Applications. Mediat. Inflamm. 2021, 2021, 5110276. [Google Scholar] [CrossRef]
- Manilla, V.; Di Tommaso, N.; Santopaolo, F.; Gasbarrini, A.; Ponziani, F.R. Endotoxemia and Gastrointestinal Cancers: Insight into the Mechanisms Underlying a Dangerous Relationship. Microorganisms 2023, 11, 267. [Google Scholar] [CrossRef]
- Di Tommaso, N.; Santopaolo, F.; Gasbarrini, A.; Ponziani, F.R. The Gut–Vascular Barrier as a New Protagonist in Intestinal and Extraintestinal Diseases. Int. J. Mol. Sci. 2023, 24, 1470. [Google Scholar] [CrossRef]
- Fang, J.; Wang, H.; Zhou, Y.; Zhang, H.; Zhou, H.; Zhang, X. Slimy partners: The mucus barrier and gut microbiome in ulcerative colitis. Exp. Mol. Med. 2021, 53, 772–787. [Google Scholar] [CrossRef] [PubMed]
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834. [Google Scholar] [CrossRef]
- Shemtov, S.J.; Emani, R.; Bielska, O.; Covarrubias, A.J.; Verdin, E.; Andersen, J.K.; Winer, D.A. The intestinal immune system and gut barrier function in obesity and ageing. FEBS J. 2022. [Google Scholar] [CrossRef]
- Burcelin, R. Gut microbiota and immune crosstalk in metabolic disease. Mol. Metab. 2016, 5, 771–781. [Google Scholar] [CrossRef]
- Pitocco, D.; DI Leo, M.; Tartaglione, L.; De Leva, F.; Petruzziello, C.; Saviano, A.; Pontecorvi, A.; Ojetti, V. The role of gut microbiota in mediating obesity and diabetes mellitus. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1548–1562. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.M.G.; Costa, J.A.; Alfenas, R.C.G. Metabolic endotoxemia and diabetes mellitus: A systematic review. Metabolism 2017, 68, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Zawada, A.; Moszak, M.; Skrzypczak, D.; Grzymisławski, M. Gastrointestinal complications in patients with diabetes mellitus. Adv. Clin. Exp. Med. 2018, 27, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Creely, S.J.; McTernan, P.G.; Kusminski, C.M.; Fisher, F.M.; Da Silva, N.F.; Khanolkar, M.; Evans, M.; Harte, A.L.; Kumar, S. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E740–E747. [Google Scholar] [CrossRef]
- Martin, A.; Devkota, S. Hold the Door: Role of the Gut Barrier in Diabetes. Cell Metab. 2018, 27, 949–951. [Google Scholar] [CrossRef]
- Nagai, Y.; Takatsu, K. Role of the Immune System in Obesity-Associated Inflammation and Insulin Resistance. In Nutrition in the Prevention and Treatment of Abdominal Obesity; Academic Press: Cambridge, MA, USA, 2014; pp. 281–293. [Google Scholar] [CrossRef]
- Nawrot, M.; Peschard, S.; Lestavel, S.; Staels, B. Intestine-liver crosstalk in Type 2 Diabetes and non-alcoholic fatty liver disease. Metabolism 2021, 123, 154844. [Google Scholar] [CrossRef]
- Mantovani, A.; Targher, G. Type 2 diabetes mellitus and risk of hepatocellular carcinoma: Spotlight on nonalcoholic fatty liver disease. Ann. Transl. Med. 2017, 5, 270. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef]
- Dong, L.; Li, Y.; Chen, Q.; Liu, Y.; Qiao, Z.; Sang, S.; Zhang, J.; Zhan, S.; Wu, Z.; Liu, L. Research advances of advanced glycation end products in milk and dairy products: Formation, determination, control strategy and immunometabolism via gut microbiota. Food Chem. 2023, 417, 135861. [Google Scholar] [CrossRef]
- Delzenne, N.M.; Knudsen, C.; Beaumont, M.; Rodriguez, J.; Neyrinck, A.M.; Bindels, L.B. Contribution of the gut microbiota to the regulation of host metabolism and energy balance: A focus on the gut–liver axis. Proc. Nutr. Soc. 2019, 78, 319–328. [Google Scholar] [CrossRef]
- Sanna, S.; Van Zuydam, N.R.; Mahajan, A.; Kurilshikov, A.; Vila, A.V.; Võsa, U.; Mujagic, Z.; Masclee, A.A.M.; Jonkers, D.M.A.E.; Oosting, M.; et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat. Genet. 2019, 51, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Couto, M.R.; Gonçalves, P.; Magro, F.; Martel, F. Microbiota-derived butyrate regulates intestinal inflammation: Focus on inflammatory bowel disease. Pharmacol. Res. 2020, 159, 104947. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Silva, C.; Kashani, A.; Hansen, T.H.; Pinna, N.K.; Anjana, R.M.; Dutta, A.; Saxena, S.; Støy, J.; Kampmann, U.; Nielsen, T.; et al. Trans-ethnic gut microbiota signatures of type 2 diabetes in Denmark and India. Genome Med. 2021, 13, 37. [Google Scholar] [CrossRef]
- Chen, Z.; Radjabzadeh, D.; Chen, L.; Kurilshikov, A.; Kavousi, M.; Ahmadizar, F.; Ikram, M.A.; Uitterlinden, A.G.; Zhernakova, A.; Fu, J.; et al. Association of Insulin Resistance and Type 2 Diabetes with Gut Microbial Diversity: A Microbiome-Wide Analysis From Popula-tion Studies. JAMA Netw. Open 2021, 4, e2118811. [Google Scholar] [CrossRef] [PubMed]
- Parada Venegas, D.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.V.; Duca, F.A.; Waise, T.Z.; Dranse, H.J.; Rasmussen, B.A.; Puri, A.; Rasti, M.; O'Brien, C.A.; Lam, T.K. Lactobacillus gasseri in the Upper Small Intestine Impacts an ACSL3-Dependent Fatty Acid-Sensing Pathway Regulating Whole-Body Glucose Homeostasis. Cell Metab. 2018, 27, 572–587. [Google Scholar] [CrossRef]
- Kang, J.-H.; Yun, S.-I.; Park, M.-H.; Park, J.-H.; Jeong, S.-Y.; Park, H.-O. Anti-obesity effect of Lactobacillus gasseri BNR17 in high-sucrose diet-induced obese mice. PLoS ONE 2013, 8, e54617. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Saad, M.J.A.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology (Bethesda) 2016, 31, 283–293. [Google Scholar] [CrossRef]
- Amalia, R.; Pramono, A.; Afifah, D.N.; Noer, E.R.; Muniroh, M.; Kumoro, A.C. Mangrove fruit (Bruguiera gymnorhiza) increases circulating GLP-1 and PYY, modulates lipid profiles, and reduces systemic inflammation by improving SCFA levels in obese wistar rats. Heliyon 2022, 8, e10887. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, L.E.; Bäckhed, F. The Metabolic Role and Therapeutic Potential of the Microbiome. Endocr. Rev. 2022, 43, 907–926. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.H.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef]
- Tajiri, K.; Shimizu, Y. Branched-chain amino acids in liver diseases. World J. Gastroenterol. 2013, 19, 7620–7629. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Karpen, S.J.; Dawson, P.A.; Arrese, M.; Trauner, M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 2016, 65, 350–362. [Google Scholar] [CrossRef]
- Leoni, S.; Tovoli, F.; Napoli, L.; Serio, I.; Ferri, S.; Bolondi, L. Current guidelines for the management of non-alcoholic fatty liver disease: A systematic review with comparative analysis. World J. Gastroenterol. 2018, 24, 3361–3373. [Google Scholar] [CrossRef]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.-S.; Harrison, S.A. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef]
- He, Q.; Sha, S.; Sun, L.; Zhang, J.; Dong, M. GLP-1 analogue improves hepatic lipid accumulation by inducing autophagy via AMPK/mTOR pathway. Biochem. Biophys. Res. Commun. 2016, 476, 196–203. [Google Scholar] [CrossRef]
- Yamada, N.; Matsushima-Nishiwaki, R.; Kobayashi, K.; Tachi, J.; Kozawa, O. GLP-1 reduces the migration of hepatocellular carcinoma cells via suppression of the stress-activated protein kinase/c-Jun N-terminal kinase pathway. Arch. Biochem. Biophys. 2021, 703, 108851. [Google Scholar] [CrossRef]
- Kojima, M.; Takahashi, H.; Kuwashiro, T.; Tanaka, K.; Mori, H.; Ozaki, I.; Kitajima, Y.; Matsuda, Y.; Ashida, K.; Eguchi, Y.; et al. Glucagon-Like Peptide-1 Receptor Agonist Prevented the Progression of Hepatocellular Carcinoma in a Mouse Model of Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 5722. [Google Scholar] [CrossRef]
- Ahmad, T.R.; Haeusler, R.A. Bile acids in glucose metabolism and insulin signaling—Mechanisms and research needs. Nat. Rev. Endocrinol. 2019, 15, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Hameed, B.; Terrault, N.A.; Gill, R.M.; Loomba, R.; Chalasani, N.; Hoofnagle, J.H.; Van Natta, M.L.; Crn, F.T.N. Clinical and metabolic effects associated with weight changes and obeticholic acid in non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2018, 47, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Alvarez, M.I.; Zorzano, A. Mitochondrial Dynamics and Liver Cancer. Cancers 2021, 13, 2571. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, Y.; Liu, D.; Liu, J. LPS promotes epithelial-mesenchymal transition and activation of TLR4/JNK signaling. Tumor Biol. 2014, 35, 10429–10435. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Quetglas, I.; Pinyol, R.; Dauch, D.; Torrecilla, S.; Tovar, V.; Moeini, A.; Alsinet, C.; Portela, A.; Rodriguez-Carunchio, L.; Solé, M.; et al. IGF2 Is Up-regulated by Epigenetic Mechanisms in Hepatocellular Carcinomas and Is an Actionable Oncogene Product in Experimental Models. Gastroenterology 2016, 151, 1192–1205. [Google Scholar] [CrossRef]
- Gibert-Ramos, A.; Sanfeliu-Redondo, D.; Aristu-Zabalza, P.; Martínez-Alcocer, A.; Gracia-Sancho, J.; Guixé-Muntet, S.; Fernández-Iglesias, A. The Hepatic Sinusoid in Chronic Liver Disease: The Optimal Milieu for Cancer. Cancers 2021, 13, 5719. [Google Scholar] [CrossRef]
- Kim, H.; Lee, D.S.; An, T.H.; Park, H.-J.; Kim, W.K.; Bae, K.-H.; Oh, K.-J. Metabolic Spectrum of Liver Failure in Type 2 Diabetes and Obesity: From NAFLD to NASH to HCC. Int. J. Mol. Sci. 2021, 22, 4495. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Sterbini, F.P.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated with Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef]
- Yang, Q.; Ouyang, J.; Sun, F.; Yang, J. Short-Chain Fatty Acids: A Soldier Fighting Against Inflammation and Protecting From Tumorigenesis in People With Diabetes. Front. Immunol. 2020, 11, 590685. [Google Scholar] [CrossRef]
- Clements, O.; Eliahoo, J.; Kim, J.U.; Taylor-Robinson, S.D.; Khan, S.A. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: A systematic review and meta-analysis. J. Hepatol. 2020, 72, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Yang, X.; Wu, H.; Gong, J.; Li, X. Diabetes and PKM2 affect prognosis in patients with intrahepatic cholangiocarcinoma. Oncol. Lett. 2020, 20, 265. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Kubo, S.; Tanaka, S.; Wakasa, K.; Takemura, S.; Kinoshita, M.; Hamano, G.; Kuwae, Y.; Shibata, T.; Suehiro, S. Outcomes of Hepatic Resection in Intrahepatic Cholangiocarcinoma Patients with Diabetes, Hypertension, and Dyslipidemia: Significance of Routine Follow-Up. Liver Cancer 2016, 5, 107–120. [Google Scholar] [CrossRef]
- Cadamuro, M.; Lasagni, A.; Sarcognato, S.; Guido, M.; Fabris, R.; Strazzabosco, M.; Strain, A.J.; Simioni, P.; Villa, E.; Fabris, L. The Neglected Role of Bile Duct Epithelial Cells in NASH. Semin. Liver Dis. 2022, 42, 034–047. [Google Scholar] [CrossRef]
- Vernia, S.; Cavanagh-Kyros, J.; Garcia-Haro, L.; Sabio, G.; Barrett, T.; Jung, D.Y.; Kim, J.K.; Xu, J.; Shulha, H.P.; Garber, M.; et al. The PPARα-FGF21 hormone axis contributes to metabolic regulation by the hepatic jnk signaling pathway. Cell Metab. 2014, 20, 512–525. [Google Scholar] [CrossRef]
- Manieri, E.; Folgueira, C.; Rodríguez, M.E.; Leiva-Vega, L.; Esteban-Lafuente, L.; Chen, C.; Cubero, F.J.; Barrett, T.; Cavanagh-Kyros, J.; Seruggia, D.; et al. JNK-mediated disruption of bile acid homeostasis promotes intrahepatic cholangiocarcinoma. Proc. Natl. Acad. Sci. USA 2020, 117, 16492–16499. [Google Scholar] [CrossRef]
- Saengboonmee, C.; Seubwai, W.; Lert-Itthiporn, W.; Sanlung, T.; Wongkham, S. Association of Diabetes Mellitus and Cholangiocarcinoma: Update of Evidence and the Effects of Antidiabetic Medication. Can. J. Diabetes 2021, 45, 282–290. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, Y.; Graham, S.; Wang, X.; Cai, D.; Huang, M.; Pique-Regi, R.; Dong, X.C.; Chen, Y.E.; Willer, C.; et al. Causal relationships between NAFLD, T2D and obesity have implications for disease sub-phenotyping. J. Hepatol. 2020, 73, 263–276. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manilla, V.; Santopaolo, F.; Gasbarrini, A.; Ponziani, F.R. Type 2 Diabetes Mellitus and Liver Disease: Across the Gut–Liver Axis from Fibrosis to Cancer. Nutrients 2023, 15, 2521. https://doi.org/10.3390/nu15112521
Manilla V, Santopaolo F, Gasbarrini A, Ponziani FR. Type 2 Diabetes Mellitus and Liver Disease: Across the Gut–Liver Axis from Fibrosis to Cancer. Nutrients. 2023; 15(11):2521. https://doi.org/10.3390/nu15112521
Chicago/Turabian StyleManilla, Vittoria, Francesco Santopaolo, Antonio Gasbarrini, and Francesca Romana Ponziani. 2023. "Type 2 Diabetes Mellitus and Liver Disease: Across the Gut–Liver Axis from Fibrosis to Cancer" Nutrients 15, no. 11: 2521. https://doi.org/10.3390/nu15112521