

Crosstalk between Gut Microbiota and Bile Acids in Cholestatic Liver Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. BA Homeostasis
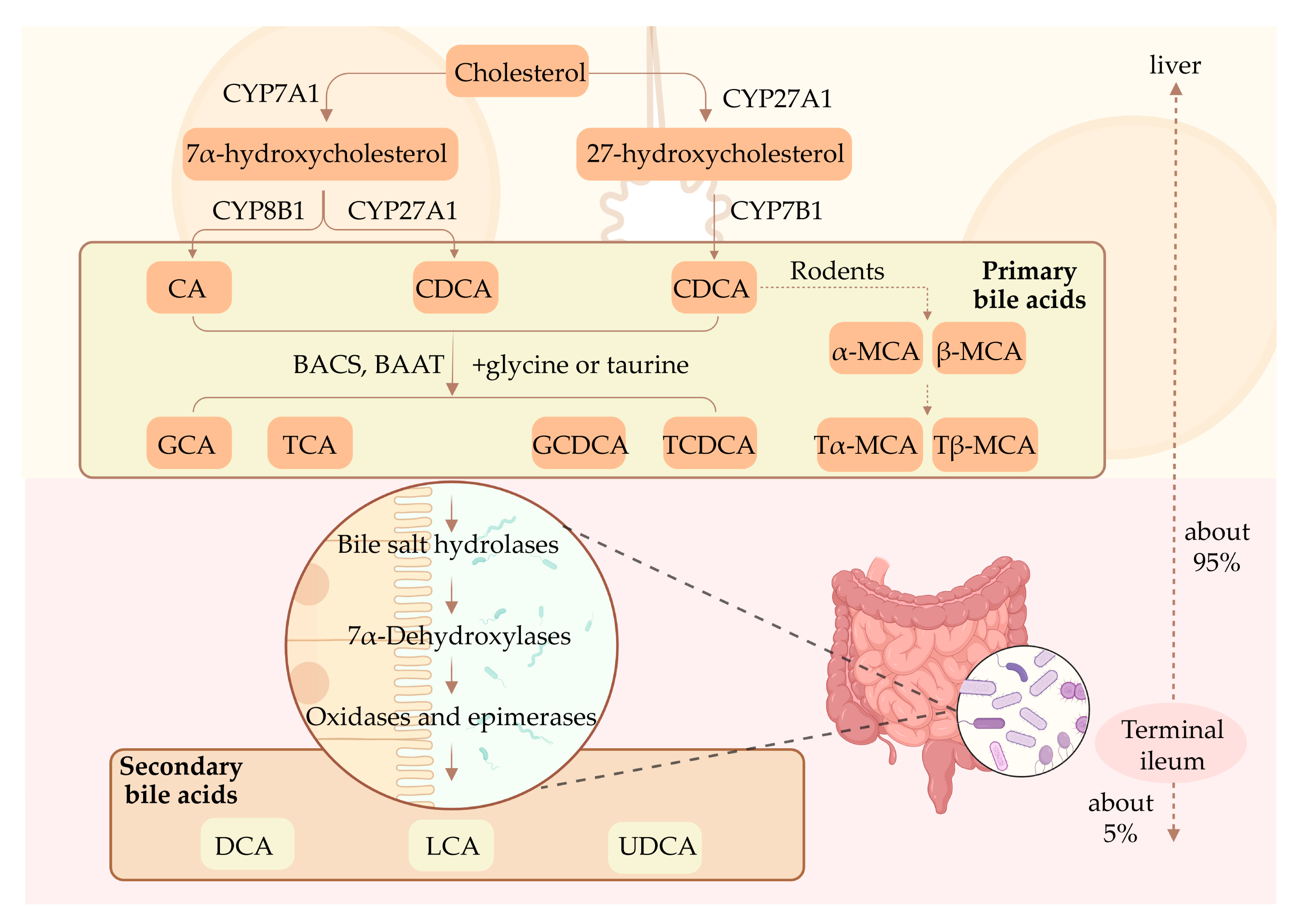
2.1. Synthesis of BAs
2.2. Enterohepatic Circulation of BAs
2.3. BA Signaling Receptors
3. Effect of Gut Microbiota on BA Metabolism
3.1. Gut Microbiota Can Alter the Composition of the BA Pool
3.1.1. Bile Salt Hydrolase (BSH)
3.1.2. Hydroxysteroid Dehydrogenase (HSDH)
3.1.3. 7α-Dehydroxylation Enzymes
3.1.4. Other Enzymes
3.2. Gut Microbiota Affects BA Metabolism through FXR Signaling Molecules
4. Effect of BAs on Gut Microbiota
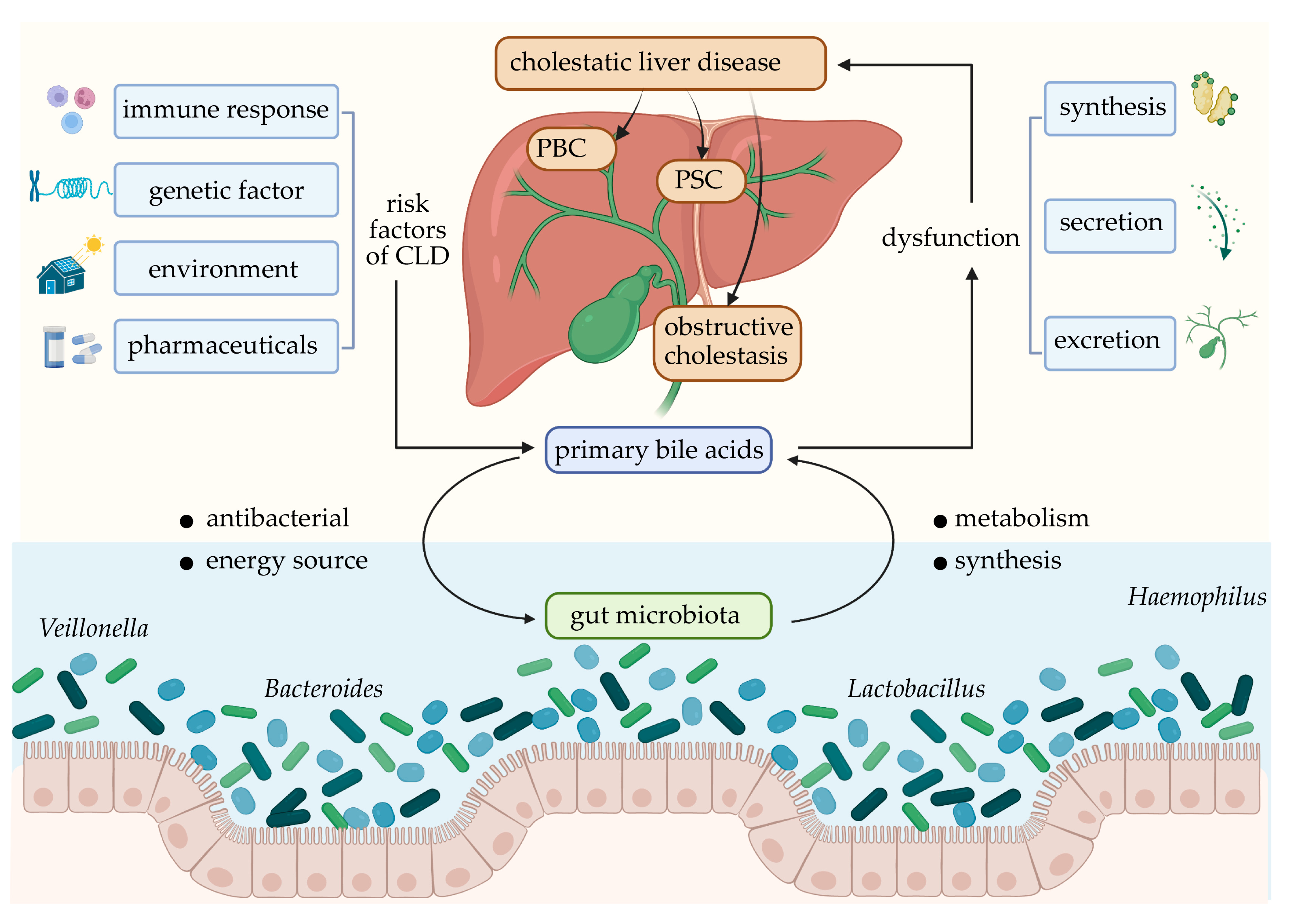
5. Effect and Mechanism of BA–Intestinal Microbiota Interaction in CLD
5.1. PBC
5.2. PSC
5.3. Obstructive Cholestasis
6. Potential Therapeutic Targets
6.1. FXR Agonist
6.2. Peroxisome Proliferator-Activated Receptor (PPAR) Agonists
6.3. FGF19 Analogs
6.4. Others
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Leviatan, S.; Segal, E. Identifying gut microbes that affect human health. Nature 2020, 587, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Liu, M.; Pu, J.; Zhu, Z.; Gao, Z.; Zhou, Q.; Gu, Q.; Li, P. Probiotics and Their Metabolites Ameliorate Inflammatory Bowel Disease: A Critical Review. Infect. Microbes Dis. 2021, 3, 4–13. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Pérez, O.; Cruz-Ramón, V.; Chinchilla-López, P.; Méndez-Sánchez, N. The Role of the Gut Microbiota in Bile Acid Metabolism. Ann. Hepatol. 2017, 16, s15–s20. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006, 124, 837–848. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef]
- Swann, J.R.; Want, E.J.; Geier, F.M.; Spagou, K.; Wilson, I.D.; Sidaway, J.E.; Nicholson, J.K.; Holmes, E. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc. Natl. Acad. Sci. USA 2011, 108, 4523–4530. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Bajaj, J.S. The human gut sterolbiome: Bile acid-microbiome endocrine aspects and therapeutics. Acta Pharm. Sin. B 2015, 5, 99–105. [Google Scholar] [CrossRef]
- Fuchs, C.D.; Trauner, M. Role of bile acids and their receptors in gastrointestinal and hepatic pathophysiology. Nat. Reviews. Gastroenterol. Hepatol. 2022, 19, 432–450. [Google Scholar] [CrossRef]
- Hofmann, A.F. Bile acid secretion, bile flow and biliary lipid secretion in humans. Hepatology 1990, 12, 17S–22S; discussion 22S–25S. [Google Scholar]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef]
- Cai, J.; Sun, L.; Gonzalez, F.J. Gut microbiota-derived bile acids in intestinal immunity, inflammation, and tumorigenesis. Cell Host Microbe 2022, 30, 289–300. [Google Scholar] [CrossRef]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef]
- de Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef]
- Jia, W.; Wei, M.; Rajani, C.; Zheng, X. Targeting the alternative bile acid synthetic pathway for metabolic diseases. Protein Cell 2021, 12, 411–425. [Google Scholar] [CrossRef]
- Cortés, V.; Eckel, R.H. Insulin and Bile Acids in Cholesterol Homeostasis: New Players in Diabetes-Associated Atherosclerosis. Circulation 2022, 145, 983–986. [Google Scholar] [CrossRef]
- Collins, S.L.; Stine, J.G.; Bisanz, J.E.; Okafor, C.D.; Patterson, A.D. Bile acids and the gut microbiota: Metabolic interactions and impacts on disease. Nat. Rev. Microbiol. 2023, 21, 236–247. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef]
- Ding, L.; Yang, L.; Wang, Z.; Huang, W. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm. Sin. B 2015, 5, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Perino, A.; Yildiz, E.; El Alam, G.; Bou Sleiman, M.; Gioiello, A.; Pellicciari, R.; Schoonjans, K. Bile Acids Signal via TGR5 to Activate Intestinal Stem Cells and Epithelial Regeneration. Gastroenterology 2020, 159, 956–968.e8. [Google Scholar] [CrossRef] [PubMed]
- Reich, M.; Spomer, L.; Klindt, C.; Fuchs, K.; Stindt, J.; Deutschmann, K.; Höhne, J.; Liaskou, E.; Hov, J.R.; Karlsen, T.H.; et al. Downregulation of TGR5 (GPBAR1) in biliary epithelial cells contributes to the pathogenesis of sclerosing cholangitis. J. Hepatol. 2021, 75, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Holter, M.M.; Chirikjian, M.K.; Govani, V.N.; Cummings, B.P. TGR5 Signaling in Hepatic Metabolic Health. Nutrients 2020, 12, 2598. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of cholestatic liver diseases. J. Hepatol. 2009, 51, 237–267. [Google Scholar] [CrossRef]
- Heathcote, E.J. Diagnosis and management of cholestatic liver disease. Clin. Gastroenterol. Hepatol. 2007, 5, 776–782. [Google Scholar] [CrossRef]
- Jansen, P.L.; Ghallab, A.; Vartak, N.; Reif, R.; Schaap, F.G.; Hampe, J.; Hengstler, J.G. The ascending pathophysiology of cholestatic liver disease. Hepatology 2017, 65, 722–738. [Google Scholar] [CrossRef]
- Tsochatzis, E.A.; Gurusamy, K.S.; Gluud, C.; Burroughs, A.K. Ursodeoxycholic acid and primary biliary cirrhosis: EASL and AASLD guidelines. J. Hepatol. 2009, 51, 1084–1085; author reply 1085–1086. [Google Scholar] [CrossRef]
- Tint, G.S.; Salen, G.; Shefer, S. Effect of ursodeoxycholic acid and chenodeoxycholic acid on cholesterol and bile acid metabolism. Gastroenterology 1986, 91, 1007–1018. [Google Scholar] [CrossRef]
- Sabino, J.; Vieira-Silva, S.; Machiels, K.; Joossens, M.; Falony, G.; Ballet, V.; Ferrante, M.; Van Assche, G.; Van der Merwe, S.; Vermeire, S.; et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 2016, 65, 1681–1689. [Google Scholar] [CrossRef]
- Tang, R.; Wei, Y.; Li, Y.; Chen, W.; Chen, H.; Wang, Q.; Yang, F.; Miao, Q.; Xiao, X.; Zhang, H.; et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut 2018, 67, 534–541. [Google Scholar] [CrossRef]
- Mattner, J. Impact of Microbes on the Pathogenesis of Primary Biliary Cirrhosis (PBC) and Primary Sclerosing Cholangitis (PSC). Int. J. Mol. Sci. 2016, 17, 1864. [Google Scholar] [CrossRef]
- Li, B.; Zhang, J.; Chen, Y.; Wang, Q.; Yan, L.; Wang, R.; Wei, Y.; You, Z.; Li, Y.; Miao, Q.; et al. Alterations in microbiota and their metabolites are associated with beneficial effects of bile acid sequestrant on icteric primary biliary Cholangitis. Gut Microbes 2021, 13, 1946366. [Google Scholar] [CrossRef]
- Liao, L.; Schneider, K.M.; Galvez, E.J.C.; Frissen, M.; Marschall, H.-U.; Su, H.; Hatting, M.; Wahlström, A.; Haybaeck, J.; Puchas, P.; et al. Intestinal dysbiosis augments liver disease progression via NLRP3 in a murine model of primary sclerosing cholangitis. Gut 2019, 68, 1477–1492. [Google Scholar] [CrossRef]
- Blesl, A.; Stadlbauer, V. The Gut-Liver Axis in Cholestatic Liver Diseases. Nutrients 2021, 13, 1018. [Google Scholar] [CrossRef]
- Li, Y.; Tang, R.; Leung, P.S.C.; Gershwin, M.E.; Ma, X. Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmun. Rev. 2017, 16, 885–896. [Google Scholar] [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef]
- Liwinski, T.; Zenouzi, R.; John, C.; Ehlken, H.; Rühlemann, M.C.; Bang, C.; Groth, S.; Lieb, W.; Kantowski, M.; Andersen, N.; et al. Alterations of the bile microbiome in primary sclerosing cholangitis. Gut 2020, 69, 665–672. [Google Scholar] [CrossRef]
- Axelson, M.; Ellis, E.; Mörk, B.; Garmark, K.; Abrahamsson, A.; Björkhem, I.; Ericzon, B.G.; Einarsson, C. Bile acid synthesis in cultured human hepatocytes: Support for an alternative biosynthetic pathway to cholic acid. Hepatology 2000, 31, 1305–1312. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef]
- Takahashi, S.; Fukami, T.; Masuo, Y.; Brocker, C.N.; Xie, C.; Krausz, K.W.; Wolf, C.R.; Henderson, C.J.; Gonzalez, F.J. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J. Lipid Res. 2016, 57, 2130–2137. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chiang, J.Y. Regulation of human sterol 27-hydroxylase gene (CYP27A1) by bile acids and hepatocyte nuclear factor 4alpha (HNF4alpha). Gene 2003, 313, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Boyer, J.L. Bile salt transporters: Molecular characterization, function, and regulation. Physiol. Rev. 2003, 83, 633–671. [Google Scholar] [CrossRef] [PubMed]
- Stieger, B. The role of the sodium-taurocholate cotransporting polypeptide (NTCP) and of the bile salt export pump (BSEP) in physiology and pathophysiology of bile formation. Handb. Exp. Pharmacol. 2011, 201, 205–259. [Google Scholar] [CrossRef]
- Borst, P.; Zelcer, N.; van de Wetering, K. MRP2 and 3 in health and disease. Cancer Lett. 2006, 234, 51–61. [Google Scholar] [CrossRef]
- Chothe, P.P.; Czuba, L.C.; Ayewoh, E.N.; Swaan, P.W. Tyrosine Phosphorylation Regulates Plasma Membrane Expression and Stability of the Human Bile Acid Transporter ASBT (SLC10A2). Mol. Pharm. 2019, 16, 3569–3576. [Google Scholar] [CrossRef]
- Dawson, P.A.; Lan, T.; Rao, A. Bile acid transporters. J. Lipid Res. 2009, 50, 2340–2357. [Google Scholar] [CrossRef]
- Kullak-Ublick, G.A.; Stieger, B.; Meier, P.J. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology 2004, 126, 322–342. [Google Scholar] [CrossRef]
- Thibaut, M.M.; Bindels, L.B. Crosstalk between bile acid-activated receptors and microbiome in entero-hepatic inflammation. Trends Mol. Med. 2022, 28, 223–236. [Google Scholar] [CrossRef]
- Fiorucci, S.; Distrutti, E.; Carino, A.; Zampella, A.; Biagioli, M. Bile acids and their receptors in metabolic disorders. Prog. Lipid Res. 2021, 82, 101094. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Bonfrate, L.; Baj, J.; Khalil, M.; Garruti, G.; Stellaard, F.; Wang, H.H.; Wang, D.Q.; Portincasa, P. Recent Advances in the Digestive, Metabolic and Therapeutic Effects of Farnesoid X Receptor and Fibroblast Growth Factor 19: From Cholesterol to Bile Acid Signaling. Nutrients 2022, 14, 4950. [Google Scholar] [CrossRef]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef]
- Holt, J.A.; Luo, G.; Billin, A.N.; Bisi, J.; McNeill, Y.Y.; Kozarsky, K.F.; Donahee, M.; Wang, D.Y.; Mansfield, T.A.; Kliewer, S.A.; et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003, 17, 1581–1591. [Google Scholar] [CrossRef]
- Kir, S.; Zhang, Y.; Gerard, R.D.; Kliewer, S.A.; Mangelsdorf, D.J. Nuclear receptors HNF4α and LRH-1 cooperate in regulating Cyp7a1 in vivo. J. Biol. Chem. 2012, 287, 41334–41341. [Google Scholar] [CrossRef]
- Ananthanarayanan, M.; Balasubramanian, N.; Makishima, M.; Mangelsdorf, D.J.; Suchy, F.J. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J. Biol. Chem. 2001, 276, 28857–28865. [Google Scholar] [CrossRef]
- Boyer, J.L.; Trauner, M.; Mennone, A.; Soroka, C.J.; Cai, S.Y.; Moustafa, T.; Zollner, G.; Lee, J.Y.; Ballatori, N. Upregulation of a basolateral FXR-dependent bile acid efflux transporter OSTalpha-OSTbeta in cholestasis in humans and rodents. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1124–G1130. [Google Scholar] [CrossRef]
- Kast, H.R.; Goodwin, B.; Tarr, P.T.; Jones, S.A.; Anisfeld, A.M.; Stoltz, C.M.; Tontonoz, P.; Kliewer, S.; Willson, T.M.; Edwards, P.A. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J. Biol. Chem. 2002, 277, 2908–2915. [Google Scholar] [CrossRef]
- Denson, L.A.; Sturm, E.; Echevarria, W.; Zimmerman, T.L.; Makishima, M.; Mangelsdorf, D.J.; Karpen, S.J. The orphan nuclear receptor, shp, mediates bile acid-induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology 2001, 121, 140–147. [Google Scholar] [CrossRef]
- Keitel, V.; Reinehr, R.; Gatsios, P.; Rupprecht, C.; Görg, B.; Selbach, O.; Häussinger, D.; Kubitz, R. The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology 2007, 45, 695–704. [Google Scholar] [CrossRef]
- Pols, T.W.; Noriega, L.G.; Nomura, M.; Auwerx, J.; Schoonjans, K. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J. Hepatol. 2011, 54, 1263–1272. [Google Scholar] [CrossRef]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Keitel, V.; Häussinger, D. Role of TGR5 (GPBAR1) in Liver Disease. Semin. Liver Dis. 2018, 38, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.; Chow, E.C.Y.; Quach, H.P.; Groothuis, G.M.M.; Tirona, R.G.; Pang, K.S. Significance of the Vitamin D Receptor on Crosstalk with Nuclear Receptors and Regulation of Enzymes and Transporters. AAPS J. 2022, 24, 71. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Sphingosine-1-phosphate receptor 2: A novel bile acid receptor and regulator of hepatic lipid metabolism? Hepatology 2015, 61, 1118–1120. [Google Scholar] [CrossRef]
- Nagahashi, M.; Takabe, K.; Liu, R.; Peng, K.; Wang, X.; Wang, Y.; Hait, N.C.; Wang, X.; Allegood, J.C.; Yamada, A.; et al. Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatology 2015, 61, 1216–1226. [Google Scholar] [CrossRef]
- Dussurget, O.; Cabanes, D.; Dehoux, P.; Lecuit, M.; Buchrieser, C.; Glaser, P.; Cossart, P. Listeria monocytogenes bile salt hydrolase is a PrfA-regulated virulence factor involved in the intestinal and hepatic phases of listeriosis. Mol. Microbiol. 2002, 45, 1095–1106. [Google Scholar] [CrossRef]
- Islam, K.B.M.S.; Fukiya, S.; Hagio, M.; Fujii, N.; Ishizuka, S.; Ooka, T.; Ogura, Y.; Hayashi, T.; Yokota, A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 2011, 141, 1773–1781. [Google Scholar] [CrossRef]
- Bourgin, M.; Kriaa, A.; Mkaouar, H.; Mariaule, V.; Jablaoui, A.; Maguin, E.; Rhimi, M. Bile Salt Hydrolases: At the Crossroads of Microbiota and Human Health. Microorganisms 2021, 9, 1122. [Google Scholar] [CrossRef]
- Begley, M.; Sleator, R.D.; Gahan, C.G.; Hill, C. Contribution of three bile-associated loci, bsh, pva, and btlB, to gastrointestinal persistence and bile tolerance of Listeria monocytogenes. Infect. Immun. 2005, 73, 894–904. [Google Scholar] [CrossRef]
- Bustos, A.Y.; Font de Valdez, G.; Fadda, S.; Taranto, M.P. New insights into bacterial bile resistance mechanisms: The role of bile salt hydrolase and its impact on human health. Food Res. Int. 2018, 112, 250–262. [Google Scholar] [CrossRef]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.M.; Marchesi, J.R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar] [CrossRef]
- Fang, F.; Li, Y.; Bumann, M.; Raftis, E.J.; Casey, P.G.; Cooney, J.C.; Walsh, M.A.; O’Toole, P.W. Allelic variation of bile salt hydrolase genes in Lactobacillus salivarius does not determine bile resistance levels. J. Bacteriol. 2009, 191, 5743–5757. [Google Scholar] [CrossRef]
- Doden, H.L.; Wolf, P.G.; Gaskins, H.R.; Anantharaman, K.; Alves, J.M.P.; Ridlon, J.M. Completion of the gut microbial epi-bile acid pathway. Gut Microbes 2021, 13, 1907271. [Google Scholar] [CrossRef]
- Macdonald, I.A.; Sutherland, J.D.; Cohen, B.I.; Mosbach, E.H. Effect of bile acid oxazoline derivatives on microorganisms participating in 7 alpha-hydroxyl epimerization of primary bile acids. J. Lipid Res. 1983, 24, 1550–1559. [Google Scholar] [CrossRef]
- Devendran, S.; Méndez-García, C.; Ridlon, J.M. Identification and characterization of a 20β-HSDH from the anaerobic gut bacterium Butyricicoccus desmolans ATCC 43058. J. Lipid Res. 2017, 58, 916–925. [Google Scholar] [CrossRef]
- Hirano, S.; Masuda, N. Epimerization of the 7-hydroxy group of bile acids by the combination of two kinds of microorganisms with 7 alpha- and 7 beta-hydroxysteroid dehydrogenase activity, respectively. J. Lipid Res. 1981, 22, 1060–1068. [Google Scholar] [CrossRef]
- Doden, H.; Sallam, L.A.; Devendran, S.; Ly, L.; Doden, G.; Daniel, S.L.; Alves, J.M.P.; Ridlon, J.M. Metabolism of Oxo-Bile Acids and Characterization of Recombinant 12α-Hydroxysteroid Dehydrogenases from Bile Acid 7α-Dehydroxylating Human Gut Bacteria. Appl. Environ. Microbiol. 2018, 84, e00235-18. [Google Scholar] [CrossRef]
- Mythen, S.M.; Devendran, S.; Méndez-García, C.; Cann, I.; Ridlon, J.M. Targeted Synthesis and Characterization of a Gene Cluster Encoding NAD(P)H-Dependent 3α-, 3β-, and 12α-Hydroxysteroid Dehydrogenases from Eggerthella CAG:298, a Gut Metagenomic Sequence. Appl. Environ. Microbiol. 2018, 84, e02475-17. [Google Scholar] [CrossRef]
- Bhowmik, S.; Chiu, H.-P.; Jones, D.H.; Chiu, H.-J.; Miller, M.D.; Xu, Q.; Farr, C.L.; Ridlon, J.M.; Wells, J.E.; Elsliger, M.-A.; et al. Structure and functional characterization of a bile acid 7α dehydratase BaiE in secondary bile acid synthesis. Proteins 2016, 84, 316–331. [Google Scholar] [CrossRef]
- Kriaa, A.; Bourgin, M.; Potiron, A.; Mkaouar, H.; Jablaoui, A.; Gérard, P.; Maguin, E.; Rhimi, M. Microbial impact on cholesterol and bile acid metabolism: Current status and future prospects. J. Lipid Res. 2019, 60, 323–332. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L.; Ferrell, J.M. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G554–G573. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Park, D.; Jia, B.; Baek, J.H.; Hahn, Y.; Jeon, C.O. Identification and Characterization of Major Bile Acid 7α-Dehydroxylating Bacteria in the Human Gut. MSystems 2022, 7, e0045522. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.A.; Mallonee, D.H.; Björkhem, I.; Hylemon, P.B. Expression and characterization of a C24 bile acid 7 alpha-dehydratase from Eubacterium sp. strain VPI 12708 in Escherichia coli. J. Lipid Res. 1996, 37, 1258–1267. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.-J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef]
- Qiu, Y.; Yu, J.; Li, Y.; Yang, F.; Yu, H.; Xue, M.; Zhang, F.; Jiang, X.; Ji, X.; Bao, Z. Depletion of gut microbiota induces skeletal muscle atrophy by FXR-FGF15/19 signalling. Ann. Med. 2021, 53, 508–522. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.-U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018, 24, 1919–1929. [Google Scholar] [CrossRef]
- Sun, L.; Pang, Y.; Wang, X.; Wu, Q.; Liu, H.; Liu, B.; Liu, G.; Ye, M.; Kong, W.; Jiang, C. Ablation of gut microbiota alleviates obesity-induced hepatic steatosis and glucose intolerance by modulating bile acid metabolism in hamsters. Acta Pharm. Sin. B 2019, 9, 702–710. [Google Scholar] [CrossRef]
- Xiong, F.; Wu, S.-G.; Zhang, J.; Jakovlić, I.; Li, W.-X.; Zou, H.; Li, M.; Wang, G.-T. Dietary Bile Salt Types Influence the Composition of Biliary Bile Acids and Gut Microbiota in Grass Carp. Front. Microbiol. 2018, 9, 2209. [Google Scholar] [CrossRef]
- van Best, N.; Rolle-Kampczyk, U.; Schaap, F.G.; Basic, M.; Olde Damink, S.W.M.; Bleich, A.; Savelkoul, P.H.M.; von Bergen, M.; Penders, J.; Hornef, M.W. Bile acids drive the newborn’s gut microbiota maturation. Nat. Commun. 2020, 11, 3692. [Google Scholar] [CrossRef]
- Begley, M.; Gahan, C.G.M.; Hill, C. The interaction between bacteria and bile. FEMS Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef]
- Inagaki, T.; Moschetta, A.; Lee, Y.-K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef]
- Parséus, A.; Sommer, N.; Sommer, F.; Caesar, R.; Molinaro, A.; Ståhlman, M.; Greiner, T.U.; Perkins, R.; Bäckhed, F. Microbiota-induced obesity requires farnesoid X receptor. Gut 2017, 66, 429–437. [Google Scholar] [CrossRef]
- Friedman, E.S.; Li, Y.; Shen, T.-C.D.; Jiang, J.; Chau, L.; Adorini, L.; Babakhani, F.; Edwards, J.; Shapiro, D.; Zhao, C.; et al. FXR-Dependent Modulation of the Human Small Intestinal Microbiome by the Bile Acid Derivative Obeticholic Acid. Gastroenterology 2018, 155, 1741–1752.e5. [Google Scholar] [CrossRef]
- Gossard, A.A.; Talwalkar, J.A. Cholestatic liver disease. Med. Clin. North Am. 2014, 98, 73–85. [Google Scholar] [CrossRef]
- Terziroli Beretta-Piccoli, B.; Mieli-Vergani, G.; Vergani, D.; Vierling, J.M.; Adams, D.; Alpini, G.; Banales, J.M.; Beuers, U.; Björnsson, E.; Bowlus, C.; et al. The challenges of primary biliary cholangitis: What is new and what needs to be done. J. Autoimmun. 2019, 105, 102328. [Google Scholar] [CrossRef]
- Furukawa, M.; Moriya, K.; Nakayama, J.; Inoue, T.; Momoda, R.; Kawaratani, H.; Namisaki, T.; Sato, S.; Douhara, A.; Kaji, K.; et al. Gut dysbiosis associated with clinical prognosis of patients with primary biliary cholangitis. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2020, 50, 840–852. [Google Scholar] [CrossRef]
- Han, W.; Huang, C.; Zhang, Q.; Tao, S.; Hu, X.; Xu, J.; Jiang, R.; Xu, B.; Liu, Y.; Hou, J. Alterations in gut microbiota and elevated serum bilirubin in primary biliary cholangitis patients treated with ursodeoxycholic acid. Eur. J. Clin. Investig. 2022, 52, e13714. [Google Scholar] [CrossRef]
- Chen, W.; Wei, Y.; Xiong, A.; Li, Y.; Guan, H.; Wang, Q.; Miao, Q.; Bian, Z.; Xiao, X.; Lian, M.; et al. Comprehensive Analysis of Serum and Fecal Bile Acid Profiles and Interaction with Gut Microbiota in Primary Biliary Cholangitis. Clin. Rev. Allergy Immunol. 2020, 58, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Sawitza, I.; Kordes, C.; Götze, S.; Herebian, D.; Häussinger, D. Bile acids induce hepatic differentiation of mesenchymal stem cells. Sci. Rep. 2015, 5, 13320. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Zhao, T.; Gao, X.; Wu, Y. Liver X receptor α is essential for the capillarization of liver sinusoidal endothelial cells in liver injury. Sci. Rep. 2016, 6, 21309. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Duan, Z. Bile Acids and the Potential Role in Primary Biliary Cirrhosis. Digestion 2016, 94, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Samant, H. Primary Sclerosing Cholangitis. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2023. [Google Scholar]
- Torres, J.; Palmela, C.; Brito, H.; Bao, X.; Ruiqi, H.; Moura-Santos, P.; Pereira da Silva, J.; Oliveira, A.; Vieira, C.; Perez, K.; et al. The gut microbiota, bile acids and their correlation in primary sclerosing cholangitis associated with inflammatory bowel disease. United Eur. Gastroenterol. J. 2018, 6, 112–122. [Google Scholar] [CrossRef]
- Tabibian, J.H.; O’Hara, S.P.; Trussoni, C.E.; Tietz, P.S.; Splinter, P.L.; Mounajjed, T.; Hagey, L.R.; LaRusso, N.F. Absence of the intestinal microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology 2016, 63, 185–196. [Google Scholar] [CrossRef]
- Awoniyi, M.; Wang, J.; Ngo, B.; Meadows, V.; Tam, J.; Viswanathan, A.; Lai, Y.; Montgomery, S.; Farmer, M.; Kummen, M.; et al. Protective and aggressive bacterial subsets and metabolites modify hepatobiliary inflammation and fibrosis in a murine model of PSC. Gut 2023, 72, 671–685. [Google Scholar] [CrossRef]
- Caforio, A.; Argenziano Cutolo, A.; Skoff, G. The presence of mercury in the ecosystem. Boll. Chim. Farm. 1987, 126, 486–489. [Google Scholar]
- Jiang, B.; Yuan, G.; Wu, J.; Wu, Q.; Li, L.; Jiang, P. Prevotella copri ameliorates cholestasis and liver fibrosis in primary sclerosing cholangitis by enhancing the FXR signalling pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166320. [Google Scholar] [CrossRef]
- Coucke, E.M.; Akbar, H.; Kahloon, A.; Lopez, P.P. Biliary Obstruction. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Wu, T.; Zhang, Z.; Liu, B.; Hou, D.; Liang, Y.; Zhang, J.; Shi, P. Gut microbiota dysbiosis and bacterial community assembly associated with cholesterol gallstones in large-scale study. BMC Genom. 2013, 14, 669. [Google Scholar] [CrossRef]
- Keren, N.; Konikoff, F.M.; Paitan, Y.; Gabay, G.; Reshef, L.; Naftali, T.; Gophna, U. Interactions between the intestinal microbiota and bile acids in gallstones patients. Environ. Microbiol. Rep. 2015, 7, 874–880. [Google Scholar] [CrossRef]
- Wu, R.; Zhang, Y.; Cheng, Q.; Wu, J.; Zhu, Y.; Shi, X.; Qiu, X.; Yang, S.; Wang, S.; Zheng, B.; et al. The effect of biliary obstruction, biliary drainage and bile reinfusion on bile acid metabolism and gut microbiota in mice. Liver Int. Off. J. Int. Assoc. Study Liver 2022, 42, 135–148. [Google Scholar] [CrossRef]
- Hu, H.; Shao, W.; Liu, Q.; Liu, N.; Wang, Q.; Xu, J.; Zhang, X.; Weng, Z.; Lu, Q.; Jiao, L.; et al. Gut microbiota promotes cholesterol gallstone formation by modulating bile acid composition and biliary cholesterol secretion. Nat. Commun. 2022, 13, 252. [Google Scholar] [CrossRef]
- Marzano, M.; Fosso, B.; Colliva, C.; Notario, E.; Passeri, D.; Intranuovo, M.; Gioiello, A.; Adorini, L.; Pesole, G.; Pellicciari, R.; et al. Farnesoid X receptor activation by the novel agonist TC-100 (3α, 7α, 11β-Trihydroxy-6α-ethyl-5β-cholan-24-oic Acid) preserves the intestinal barrier integrity and promotes intestinal microbial reshaping in a mouse model of obstructed bile acid flow. Biomed. Pharmacother. Biomed. Pharmacother. 2022, 153, 113380. [Google Scholar] [CrossRef]
- Yu, L.; Liu, Y.; Wang, S.; Zhang, Q.; Zhao, J.; Zhang, H.; Narbad, A.; Tian, F.; Zhai, Q.; Chen, W. Cholestasis: Exploring the triangular relationship of gut microbiota-bile acid-cholestasis and the potential probiotic strategies. Gut Microbes 2023, 15, 2181930. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, K.; Li, F.; Gu, Z.; Liu, Q.; He, L.; Shao, T.; Song, Q.; Zhu, F.; Zhang, L.; et al. Probiotic Lactobacillus rhamnosus GG Prevents Liver Fibrosis Through Inhibiting Hepatic Bile Acid Synthesis and Enhancing Bile Acid Excretion in Mice. Hepatology 2020, 71, 2050–2066. [Google Scholar] [CrossRef]
- Ren, L.; Song, Q.; Liu, Y.; Zhang, L.; Hao, Z.; Feng, W. Probiotic Lactobacillus rhamnosus GG prevents progesterone metabolite epiallaopregnanolone sulfate-induced hepatic bile acid accumulation and liver injury. Biochem. Biophys. Res. Commun. 2019, 520, 67–72. [Google Scholar] [CrossRef]
- Chen, Q.-W.; Li, Q.-R.; Cao, M.-W.; Yan, J.-H.; Zhang, X.-Z. Hierarchy-Assembled Dual Probiotics System Ameliorates Cholestatic Drug-Induced Liver Injury via Gut-Liver Axis Modulation. Adv. Sci. 2022, 9, e2200986. [Google Scholar] [CrossRef]
- Labiano, I.; Agirre-Lizaso, A.; Olaizola, P.; Echebarria, A.; Huici-Izagirre, M.; Olaizola, I.; Esparza-Baquer, A.; Sharif, O.; Hijona, E.; Milkiewicz, P.; et al. TREM-2 plays a protective role in cholestasis by acting as a negative regulator of inflammation. J. Hepatol. 2022, 77, 991–1004. [Google Scholar] [CrossRef]
- Gulamhusein, A.F.; Hirschfield, G.M. Primary biliary cholangitis: Pathogenesis and therapeutic opportunities. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 93–110. [Google Scholar] [CrossRef]
- Lindor, K.D.; Bowlus, C.L.; Boyer, J.; Levy, C.; Mayo, M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2019, 69, 394–419. [Google Scholar] [CrossRef] [PubMed]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.H.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Bowlus, C.L.; Gulamhusein, A.; Hameed, B.; Caldwell, S.H.; Shiffman, M.L.; Landis, C.; Muir, A.J.; Billin, A.; Xu, J.; et al. Safety and sustained efficacy of the farnesoid X receptor (FXR) agonist cilofexor over a 96-week open-label extension in patients with PSC. Clin. Gastroenterol. Hepatol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Gulamhusein, A.; Hameed, B.; Caldwell, S.; Shiffman, M.L.; Landis, C.; Eksteen, B.; Agarwal, K.; Muir, A.; Rushbrook, S.; et al. The Nonsteroidal Farnesoid X Receptor Agonist Cilofexor (GS-9674) Improves Markers of Cholestasis and Liver Injury in Patients With Primary Sclerosing Cholangitis. Hepatology 2019, 70, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Schramm, C.; Wedemeyer, H.; Mason, A.; Hirschfield, G.M.; Levy, C.; Kowdley, K.V.; Milkiewicz, P.; Janczewska, E.; Malova, E.S.; Sanni, J.; et al. Farnesoid X receptor agonist tropifexor attenuates cholestasis in a randomised trial in patients with primary biliary cholangitis. JHEP Rep. Innov. Hepatol. 2022, 4, 100544. [Google Scholar] [CrossRef]
- Trauner, M.; Fuchs, C.D. Novel therapeutic targets for cholestatic and fatty liver disease. Gut 2022, 71, 194–209. [Google Scholar] [CrossRef]
- Corpechot, C.; Chazouillères, O.; Rousseau, A.; Le Gruyer, A.; Habersetzer, F.; Mathurin, P.; Goria, O.; Potier, P.; Minello, A.; Silvain, C.; et al. A Placebo-Controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N. Engl. J. Med. 2018, 378, 2171–2181. [Google Scholar] [CrossRef]
- Tanaka, A.; Hirohara, J.; Nakano, T.; Matsumoto, K.; Chazouillères, O.; Takikawa, H.; Hansen, B.E.; Carrat, F.; Corpechot, C. Association of bezafibrate with transplant-free survival in patients with primary biliary cholangitis. J. Hepatol. 2021, 75, 565–571. [Google Scholar] [CrossRef]
- de Vries, E.; Bolier, R.; Goet, J.; Parés, A.; Verbeek, J.; de Vree, M.; Drenth, J.; van Erpecum, K.; van Nieuwkerk, K.; van der Heide, F.; et al. Fibrates for Itch (FITCH) in Fibrosing Cholangiopathies: A Double-Blind, Randomized, Placebo-Controlled Trial. Gastroenterology 2021, 160, 734–743. [Google Scholar] [CrossRef]
- Beuers, U.; Wolters, F.; Oude Elferink, R.P.J. Mechanisms of pruritus in cholestasis: Understanding and treating the itch. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 26–36. [Google Scholar] [CrossRef]
- Bowlus, C.L.; Galambos, M.R.; Aspinall, R.J.; Hirschfield, G.M.; Jones, D.E.J.; Dörffel, Y.; Gordon, S.C.; Harrison, S.A.; Kremer, A.E.; Mayo, M.J.; et al. A phase II, randomized, open-label, 52-week study of seladelpar in patients with primary biliary cholangitis. J. Hepatol. 2022, 77, 353–364. [Google Scholar] [CrossRef]
- Schattenberg, J.M.; Pares, A.; Kowdley, K.V.; Heneghan, M.A.; Caldwell, S.; Pratt, D.; Bonder, A.; Hirschfield, G.M.; Levy, C.; Vierling, J.; et al. A randomized placebo-controlled trial of elafibranor in patients with primary biliary cholangitis and incomplete response to UDCA. J. Hepatol. 2021, 74, 1344–1354. [Google Scholar] [CrossRef]
- Boyer-Diaz, Z.; Aristu-Zabalza, P.; Andrés-Rozas, M.; Robert, C.; Ortega-Ribera, M.; Fernández-Iglesias, A.; Broqua, P.; Junien, J.-L.; Wettstein, G.; Bosch, J.; et al. Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. J. Hepatol. 2021, 74, 1188–1199. [Google Scholar] [CrossRef]
- Talukdar, S.; Kharitonenkov, A. FGF19 and FGF21: In NASH we trust. Mol. Metab. 2021, 46, 101152. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Chazouillères, O.; Drenth, J.P.; Thorburn, D.; Harrison, S.A.; Landis, C.S.; Mayo, M.J.; Muir, A.J.; Trotter, J.F.; Leeming, D.J.; et al. Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: A multicenter, randomized, double-blind, placebo-controlled phase II trial. J. Hepatol. 2019, 70, 483–493. [Google Scholar] [CrossRef]
- Wang, X.X.; Wang, D.; Luo, Y.; Myakala, K.; Dobrinskikh, E.; Rosenberg, A.Z.; Levi, J.; Kopp, J.B.; Field, A.; Hill, A.; et al. FXR/TGR5 Dual Agonist Prevents Progression of Nephropathy in Diabetes and Obesity. J. Am. Soc. Nephrol. 2018, 29, 118–137. [Google Scholar] [CrossRef]
- Ito, K.; Okumura, A.; Takeuchi, J.S.; Watashi, K.; Inoue, R.; Yamauchi, T.; Sakamoto, K.; Yamashita, Y.; Iguchi, Y.; Une, M.; et al. Dual Agonist of Farnesoid X Receptor and Takeda G Protein-Coupled Receptor 5 Inhibits Hepatitis B Virus Infection In Vitro and In Vivo. Hepatology 2021, 74, 83–98. [Google Scholar] [CrossRef]
- Trivedi, P.J.; Muir, A.J.; Levy, C.; Bowlus, C.L.; Manns, M.P.; Lu, X.; Crans, G.; Chung, C.; Subramanian, G.M.; Myers, R.P.; et al. Inter- and Intra-individual Variation, and Limited Prognostic Utility, of Serum Alkaline Phosphatase in a Trial of Patients With Primary Sclerosing Cholangitis. Clin. Gastroenterol. Hepatol. 2021, 19, 1248–1257. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Q.; Yuan, X.; Zeng, Y.; Wang, J.; Zhang, Y.; Xue, C.; Li, L. Crosstalk between Gut Microbiota and Bile Acids in Cholestatic Liver Disease. Nutrients 2023, 15, 2411. https://doi.org/10.3390/nu15102411
Shi Q, Yuan X, Zeng Y, Wang J, Zhang Y, Xue C, Li L. Crosstalk between Gut Microbiota and Bile Acids in Cholestatic Liver Disease. Nutrients. 2023; 15(10):2411. https://doi.org/10.3390/nu15102411
Chicago/Turabian StyleShi, Qingmiao, Xin Yuan, Yifan Zeng, Jinzhi Wang, Yaqi Zhang, Chen Xue, and Lanjuan Li. 2023. "Crosstalk between Gut Microbiota and Bile Acids in Cholestatic Liver Disease" Nutrients 15, no. 10: 2411. https://doi.org/10.3390/nu15102411