
Genetics of Type 2 Diabetes: Past, Present, and Future


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Approaches in Studies of the Genetics of Diabetes
2.1. Development of Technologies for Genetic Studies
2.2. Candidate Gene Studies and Linkage Analyses
2.3. Genome-Wide Common Variant Association Studies
2.4. Genome-Wide Rare Variants Association Studies
2.5. Polygenic Risk Scores for Type 2 Diabetes
3. Precise Type 2 Diabetes Medicine
3.1. Genetics
3.2. Phenotyping
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- International Diabetes Federation. Available online: https://idf.org/aboutdiabetes/what-is-diabetes/facts-figures.html (accessed on 15 May 2022).
- Genuth, S.; Alberti, K.G.M.M.; Bennett, P.; Buse, J.; DeFronzo, R.; Kahn, R.; Kitzmiller, J.; Knowler, W.C.; Lebovitz, H.; Lernmark, A.; et al. Follow-up report on the diagnosis of diabetes mellitus. Diabetes Care. 2003, 26, 3160–3167. [Google Scholar] [PubMed] [Green Version]
- Laakso, M.; Kuusisto, J. Insulin resistance and hyperglycaemia in cardiovascular disease development. Nat. Rev. Endocrinol. 2014, 10, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Rich, S.S. Mapping genes in diabetes. Genetic epidemiological perspective. Diabetes 1990, 39, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Almgren, P.; Lehtovirta, M.; Isomaa, B.; Sarelin, L.; Taskinen, M.-R.; Lyssenko, V.; Tuomi, T.; Groop, L.; Botnia Study Group. Heritability and familiality of type 2 diabetes and related quantitative traits in the Botnia Study. Diabetologia 2011, 54, 2811–2819. [Google Scholar] [CrossRef] [Green Version]
- Kahn, S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia 2003, 46, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Uusitupa, M.; Khan, T.A.; Viguiliouk, E.; Kahleova, H.; Rivellese, A.A.; Hermansen, K.; Pfeiffer, A.; Thanopoulou, A.; Salas-Salvadó, J.; Schwab, U.; et al. Prevention of type 2 diabetes by lifestyle changes: A systematic review and meta-analysis. Nutrients 2019, 11, 2611. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.M.; Jones, H.; Stephens, J.W. Personalized type 2 diabetes management: An update on recent advances and recommendations. Diabetes Metabol Syndr Obes. 2022, 15, 281–295. [Google Scholar] [CrossRef]
- Guyer, M.S.; Collins, F.S. How is the Human Genome Project doing, and what we have learned so far? Proc. Natl. Acad. Sci. USA 1995, 92, 10841–10848. [Google Scholar] [CrossRef] [Green Version]
- Murray, J.C.; Buetow, K.H.; Weber, J.L.; Ludwigsen, S.; Scherpbier-Heddema, T.; Manion, F.; Quillen, J.; Sheffield, V.C.; Sunden, S.; Duyk, G.M.; et al. A comprehensive human linkage map with centimorgan density. Cooperative Human Linkage Center (CHLC). Science 1994, 265, 2049–2054. [Google Scholar] [CrossRef] [Green Version]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A brief history of human disease genetics. Nature 2020, 577, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vionnet, N.; Stoffel, M.; Takeda, J.; Yasuda, K.; Bell, G.I.; Zouali, H.; Lesage, S.; Velho, G.; Iris, F.; Passa, P.; et al. Nonsense mutation in the glucokinase gene causes early-onset non-insulin-dependent diabetes mellitus. Nature 1992, 356, 721–722. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Furuta, H.; Oda, N.; Kaisaki, P.J.; Menzel, S.; Cox, N.J.; Fajans, S.S.; Signorini, S.; Stoffel, M.; Bell, G.I. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1). Nature 1996, 384, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Oda, N.; Kaisaki, P.J.; Menzel, S.; Furuta, H.; Vaxillaire, M.; Southam, L.; Cox, R.D.; Lathrop, G.M.; Boriraj, V.V.; et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature 1996, 384, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Barroso, I.; Luan, L.; Middelberg, R.P.S.; Harding, A.-H.; Franks, P.W.; Jakes, R.W.; Clayton, D.; Schafer, A.J.; O’Rahilly, S.; Wareham, N.J. Candidate gene association study in type 2 diabetes indicates a role for genes involved in beta-cell function as well as insulin action. PLoS Biol 2003, 1, E20. [Google Scholar] [CrossRef]
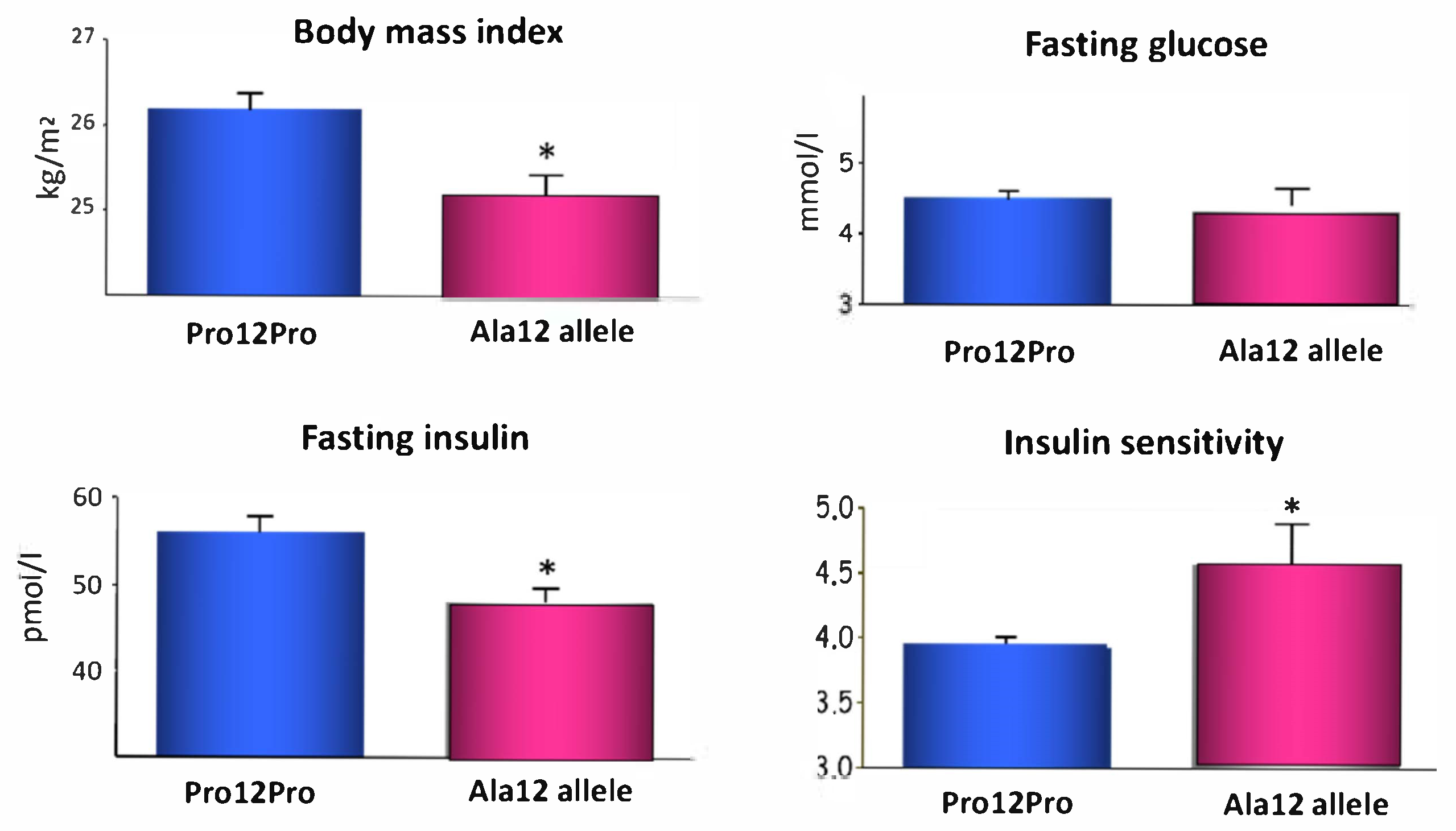
- Deeb, S.S.; Fajas, L.; Nemoto, M.; Pihlajamäki, J.; Mykkänen, L.; Kuusisto, J.; Laakso, M.; Fujimoto, W.; Auwerx, J. A Pro12Ala substitution in PPARγ2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat. Genet. 1998, 20, 284–287. [Google Scholar] [CrossRef]
- Nolan, J.J.; Ludvik, R.; Beerdsen, P.; Joyce, M.; Olefsky, J. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N. Engl. J. Med. 1994, 331, 1188–1193. [Google Scholar] [CrossRef]
- Heikkinen, S.; Argmann, C.; Feige, J.N.; Koutnikova, H.; Champy, M.-F.; Dali-Youcef, N.; Schadt, E.E.; Laakso, M.; Auwerx, J. The Pro12Ala PPARgamma2 variant determines metabolism at the gene-environment interface. Cell Metab. 2009, 9, 88–98. [Google Scholar] [CrossRef]
- Altshuler, D.; Hirschhorn, J.N.; Klannemark, M.; Lindgren, C.M.; Vohl, M.-C.; Nemesh, J.; Lane, C.R.; Schaffner, S.F.; Bolk, S.; Brewer, C.; et al. The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat. Genet. 1998, 20, 284–287. [Google Scholar] [CrossRef]
- Hani, E.H.; Boutin, P.; Durand, E.; Inoue, H.; Permutt, P.A.; Velho, G.; Froguel, P. Missense mutations in the pancreatic islet beta cell inwardly rectifying K+ channel gene (KIR6.2/BIR): A meta-analysis suggests a role in the polygenic basis of type II diabetes mellitus in Caucasians. Diabetologia 1998, 41, 1511–1515. [Google Scholar] [CrossRef] [Green Version]
- Gloyn, A.L.; Weedon, M.N.; Owen, K.R.; Turner, M.J.; Knight, B.A.; Hitman, G.; Walker, M.; Levy, J.C.; Sampson, M.; Halford, S.; et al. Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 2003, 5, 568–572. [Google Scholar] [CrossRef] [Green Version]
- Grant, S.F.A.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Manolescu, A.; Sainz, J.; Helgason, A.; Stefansson, H.; Emilsson, V.; Helgadottir, A.; et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet. 2006, 38, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Xu, L.; Zhang, L.; Han, Z.; Jiang, Q.; Wang, Z.; Jin, S. Meta-analysis of association between TCF7L2 polymorphism rs7903146 and type 2 diabetes mellitus. BMC Med Genet. 2018, 19, 38. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.; Chen, H.; Roix, J.J.; Kathiresan, S.; Hirschhorn, J.N.; Daly, M.J.; et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007, 316, 1331–1336. [Google Scholar] [CrossRef]
- Scott, L.J.; Mohlke, K.L.; Bonnycastle, L.L.; Willer, C.J.; Li, Y.; Duren, W.L.; Erdos, M.R.; Stringham, H.M.; Chines, P.S.; Jackson, A.U.; et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 2007, 316, 1341–1345. [Google Scholar] [CrossRef] [Green Version]
- Steinthorsdottir, V.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Jonsdottir, T.; Walters, G.B.; Styrkarsdottir, U.; Gretarsdottir, S.; Emilsson, V.; Ghosh, S.; et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat. Genet. 2007, 39, 770–775. [Google Scholar] [CrossRef] [Green Version]
- Zeggini, E.; Weedon, M.N.; Lindgren, C.M.; Frayling, T.M.; Elliott, K.S.; Lango, H.; Timpson, N.J.; Perry, J.R.B.; Rayner, N.W.; Freathy, R.M.; et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007, 316, 1336–1341. [Google Scholar] [CrossRef] [Green Version]
- Flannick, J.; Florez, J.C. Type 2 diabetes: Genetic data sharing to advance complex disease research. Nat. Rev. 2016, 17, 535–549. [Google Scholar] [CrossRef]
- Zeggini, E.; Scott, L.J.; Saxena, R.; Voight, B.F.; Marchini, J.L.; Hu, T.; de Bakker, P.I.W.; Abecasis, G.R.; Almgren, P.; Andersen, G.; et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 2008, 40, 638–645. [Google Scholar] [CrossRef]
- De Bakker, P.I.W.; Ferreira, M.A.R.; Jia, X.; Neale, B.M.N.; Raychaudhuri, S.; Voight, B.F. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum. Mol. Genet. 2008, 17, R122–R128. [Google Scholar] [CrossRef] [Green Version]
- Morris, A.P.; Voight, B.F.; Teslovich, T.M.; Ferreira, F.; Segrè, A.V.; Steinthorsdottir, V.; Strawbridge, R.J.; Khan, H.; Grallert, H.; Mahajan, A.; et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 2012, 44, 981–990. [Google Scholar] [PubMed]
- Voight, B.F.; Scott, L.J.; Steinthorsdottir, V.; Morris, A.P.; Dina, C.; Welch, R.P.; Zeggini, E.; Huth, C.; Aulchenko, Y.S.; Thorleifsson, G.; et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat. Genet. 2010, 42, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Ingelsson, E.; Langenberg, C.; Hivert, M.-F.; Prokopenko, P.; Lyssenko, V.; Dupuis, J.; Mägi, R.; Sharp, S.; Jackson, A.U.; Assimes, T.L.; et al. Detailed physiologic characterization reveals diverse mechanisms for novel genetic Loci regulating glucose and insulin metabolism in humans. Diabetes 2010, 59, 1266–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, R.A.; Scott, L.J.; Mägi, R.; Marullo, L.; Gaulton, K.J.; Kaakinen, M.; Pervjakova, N.; Pers, T.H.; Johnson, A.D.; Eicher, J.D.; et al. An expanded genome-wide association study of type 2 diabetes in Europeans. Diabetes 2017, 66, 2888–2902. [Google Scholar] [CrossRef] [Green Version]
- Lotta, L.A.; Gulati, P.; Day, F.R.; Payne, F.; Ongen, H.; van de Bunt, M.; Gaulton, K.J.; Eicher, J.D.; Sharp, S.J.; Lua, J.; et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017, 49, 17–26. [Google Scholar] [CrossRef]
- Mahajan, A.; Wessel, J.; Willems, S.M.; Zhao, W.; Robertson, N.R.; Chu, A.Y.; Gan, W.; Kitajima, H.; Taliun, D.; Rayner, N.W. Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat. Genet. 2018, 50, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Vujkovic, M.; Keaton, J.M.; Lynch, J.A.; Miller, D.R.; Zhou, J.; Tcheandjieu, C.; Jennifer E Huffman, J.E.; Assimes, T.L.; Lorenz, K.; Zhu, X.; et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet. 2020, 52, 680–691. [Google Scholar] [CrossRef]
- DeForest, N.; Majithia, A.R. Genetics of type 2 diabetes: Implications from large-scale studies. Curr. Diab. Rep. 2022, 22, 227–235. [Google Scholar] [CrossRef]
- Leong, A.; Wheeler, E. Genetics of HbA1c: A case study in clinical translation. Curr. Opin. Genet. Dev. 2018, 50, 79–85. [Google Scholar] [CrossRef]
- Sarnowski, C.; Hivert, M.-F. Impact of genetic determinants of HbA1c on type 2 diabetes risk and diagnosis. Curr. Diab. Rep. 2018, 18, 52. [Google Scholar] [CrossRef]
- Chen, J.; Spracklen, C.N.l.; Marenne, G.; Varshney, A.; Corbin, L.J.; Luan, J.A.; Willems, S.M.; Wu, Y.; Zhang, X.; Horikoshi, M.; et al. The trans-ancestral genomic architecture of glycemic traits. Nat. Genet. 2021, 53, 840–860. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Bouatia-Naji, N.; Anna L Gloyn, A.; et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 2010, 42, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.K.; Hivert, M.-F.; Scott, R.A.; Grimsby, J.L.; Bouatia-Naji, N.; Chen, H.; Rybin, D.; Liu, C.-T.; Bielak, L.F.; Prokopenko, P.; et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat. Genet. 2012, 44, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.A.; Lagou, V.; Welch, R.P.; Wheeler, E.; Montasser, M.E.; Luan, J.; Mägi, R.; Strawbridge, R.J.; Rehnberg, E.; Gustafsson, S.; et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat. Genet. 2012, 44, 991–1005. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.Y.; Sim, X.; Wu, Y.; Liang, J.; Tabara, Y.; Hu, C.; Hara, K.; Tam, C.H.; Cai, Q.; Zhao, Q.; et al. Genome-wide association meta-analysis identifies novel variants associated with fasting plasma glucose in East Asians. Diabetes 2015, 64, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Horikoshi, M.; Mӓgi, R.; van de Bun, M.; Surakka, I.; Sarin, A.-P.; Mahajan, A.; Letizia Marullo, L.; Thorleifsson, G.; Hӓgg, S.; Hottenga, J.-J.; et al. Discovery and fine-mapping of glycaemic and obesity-related trait loci using high-density imputation. PLoS Genet. 2015, 11, e1005230. [Google Scholar] [CrossRef] [Green Version]
- Strawbridge, R.J.; Dupuis, J.; Prokopenko, I.; Barker, A.; Ahlqvist, E.; Rybin, D.; Petrie, J.R.; Travers, M.E.; Bouatia-Naji, N.; Dimas, A.; et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 2011, 60, 2624–2634. [Google Scholar] [CrossRef] [Green Version]
- Walford, G.A.; Gustafsson, S.; Rybin, D.; Stančáková, A.; Chen, H.; Liu, C.-T.; Hong, J.; Jensen, R.A.; Rice, K.; Andrew, P.; et al. Genome-wide association study of the modifed Stumvoll insulin sensitivity index identifies BCL2 and FAM19A2 as novel insulin sensitivity loci. Diabetes 2016, 65, 3200–3211. [Google Scholar] [CrossRef] [Green Version]
- Huyghe, J.R.; Jackson, A.U.; Fogart, M.P.; Buchkovich, M.L.; Stančáková, A.; Stringham, H.M.; Sim, X.; Yang, L.; Fuchsberger, C.; Cederberg, H.; et al. Exome array analysis identifies new loci and low-frequency variants influencing insulin processing and secretion. Nat. Genet. 2013, 45, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Steinthorsdottir, V.; Thorleifsson, G.; Sulem, P.; Helgason, H.; Grarup, N.; Sigurdsson, A.; Helgadottir, H.T.; Johannsdottir, H.; Magnusson, O.T.; Gudjonsson, S.A.; et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat Genet. 2014, 46, 294–298. [Google Scholar] [CrossRef]
- Bonnefond, A.; Clément, N.; Fawcett, K.; Yengo, L.; Vaillant, E.; Guillaume, J.-L.; Dechaume, A.; Payne, F.; Roussel, R.; Czernichow, S.; et al. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat. Genet. 2012, 44, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Estrada, K.; Aukrust, I.; Bjørkhaug, L.; Burtt, N.P.; Mercader, J.M.; García-Ortiz, H.; Huerta-Chagoya, A.; Moreno-Macías, H.; Walford, G.; Flannick, J.; et al. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA 2014, 311, 2305–2314. [Google Scholar] [PubMed] [Green Version]
- Flannick, J.; Thorleifsson, G.; Beer, N.L.; Jacobs, S.B.R.; Grarup, N.; Burtt, N.P.; Mahajan, A.; Fuchsberger, C.; Atzmon, G.; Benediktsson, R.; et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet. 2014, 46, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.; Highland, H.M.; Gasser, J.; Sim, X.; Tukiainen, T.; Fontanillas, P.; Grarup, N.; Rivas, M.A.; Mahajan, A.; Locke, A.E.; et al. A low-frequency inactivating AKT2 variant enriched in the Finnish population is associated with fasting insulin levels and type 2 diabetes risk. Diabetes 2017, 66, 2019–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latva-Rasku, A.; Honka, M.J.; Stančáková, A.; Koistinen, H.A.; Kuusisto, J.; Guan, L.; Manning, A.K.; Stringham, H.; Gloyn, A.L.; Lindgren, C.M.; et al. A partial loss-of-function variant in AKT2 is associated with reduced insulin-mediated glucose uptake in multiple insulin-sensitive tissues: A genotype-based callback positron emission tomography study. Diabetes 2018, 67, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udler, M.S.; McCarthy, M.I.; Florez, J.C.; Mahajan, A. Genetic risk scores for diabetes diagnosis and precision medicine. Endocr Rev. 2019, 40, 1500–1520. [Google Scholar] [CrossRef] [Green Version]
- Adeyemo, A.; Balaconis, M.K.; Darnes, D.R.; Fatumo, S.; Moreno, P.G.; Hodonsky, C.H.; Inouye, M.; Kanai, M.; Kato, K.; Bartha, M.; et al. Polygenic risk score task force of the international common disease alliance. Nat. Med. 2021, 27, 1876–1884. [Google Scholar]
- Inouye, M.; Abraham, G.; Nelson, C.P.; Wood, A.M.; Sweeting, M.J.; Dudbridge, F.; Lai, F.Y.; Kaptoge, S.; Brozynska, M.; Wang, T.; et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. J. Am. Coll. Cardiol. 2018, 72, 1883–1893. [Google Scholar] [CrossRef]
- Padilla-Martínez, F.; Collin, F.; Kwasniewski, M.; Kretowski, A. Systematic review of polygenic risk scores for type 1 and type 2 diabetes. Int. J. Mol. Sci. 2020, 21, 1703. [Google Scholar] [CrossRef] [Green Version]
- Lyssenko, V.; Jonsson, A.; Almgren, P.; Pulizzi, N.; Isomaa, B.; Tuomi, T.; Berglund, G.; Altshuler, D.; Nilsson, P.; Groop, L.; et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N. Engl. J. Med. 2008, 359, 2220–2232. [Google Scholar] [CrossRef] [Green Version]
- Meigs, J.B.; Shrader, P.; Sullivan, L.M.; McAteer, J.B.; Fox, C.S.; Dupuis, J.; Manning, A.K.; Florez, J.C.; Wilson, P.W.; D’Agostino, R.B., Sr.; et al. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N. Engl. J. Med. 2008, 359, 2208–2219. [Google Scholar] [CrossRef] [PubMed]
- Lango, H.; Palmer, C.N.; Morris, A.D.; Zeggini, E.; Hattersley, A.T.; McCarthy, M.I.; Frayling, T.M.; Weedon, M.N.; UK Type 2 Diabetes Genetics Consortium. Assessing the combined impact of 18 common genetic variants of modest effect sizes on type 2 diabetes risk. Diabetes 2008, 57, 3129–3135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassy, J.L.; Hivert, M.-F.; Porneala, B.; Dauriz, M.; Florez, J.C.; Dupuis, J.; Siscovick, D.S.; Fornage, M.; Rasmussen-Torvik, L.J.; Bouchard, C.; et al. Polygenic type 2 diabetes prediction at the limit of common variant detection. Diabetes 2014, 63, 2172–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stančáková, A.; Kuulasmaa, T.; Kuusisto, J.; Mohlke, K.L.; Collins, F.S.; Boehnke, M.; Laakso, M. Genetic risk scores in the prediction of plasma glucose, impaired insulin secretion, insulin resistance and incident type 2 diabetes in the METSIM study. Diabetologia 2017, 60, 1722–1730. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Wheeler, B.; Sampson, J.; Hartge, P.; Chanock, S.J.; Park, J.H. Projecting the performance of risk prediction based on polygenic analyses of genome-wide association studies. Nat. Genet. 2013, 45, 400–405. [Google Scholar] [CrossRef] [Green Version]
- Wand, H.; Lambert, S.A.; Tamburro, C.; Iacocca, M.I.; O’Sullivan, J.W.; Sillari, C.; Kullo, I.J.; Rowley, R.; Dron, J.S.; Brockman, D.; et al. Improving reporting standards for polygenic scores in risk prediction studies. Nature 2021, 591, 211–219. [Google Scholar] [CrossRef]
- Dennis, J.M. Precision Medicine in type 2 diabetes: Using individualized prediction models to optimize selection of treatment. Diabetes 2020, 69, 2075–2085. [Google Scholar] [CrossRef]
- Collins, F.S.; Varmus, H. A new initiative on Precision Medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [Green Version]
- Regnell, S.; Lernmark, Å. Early prediction of autoimmune (type 1) diabetes. Diabetologia 2017, 60, 1370–1381. [Google Scholar] [CrossRef] [Green Version]
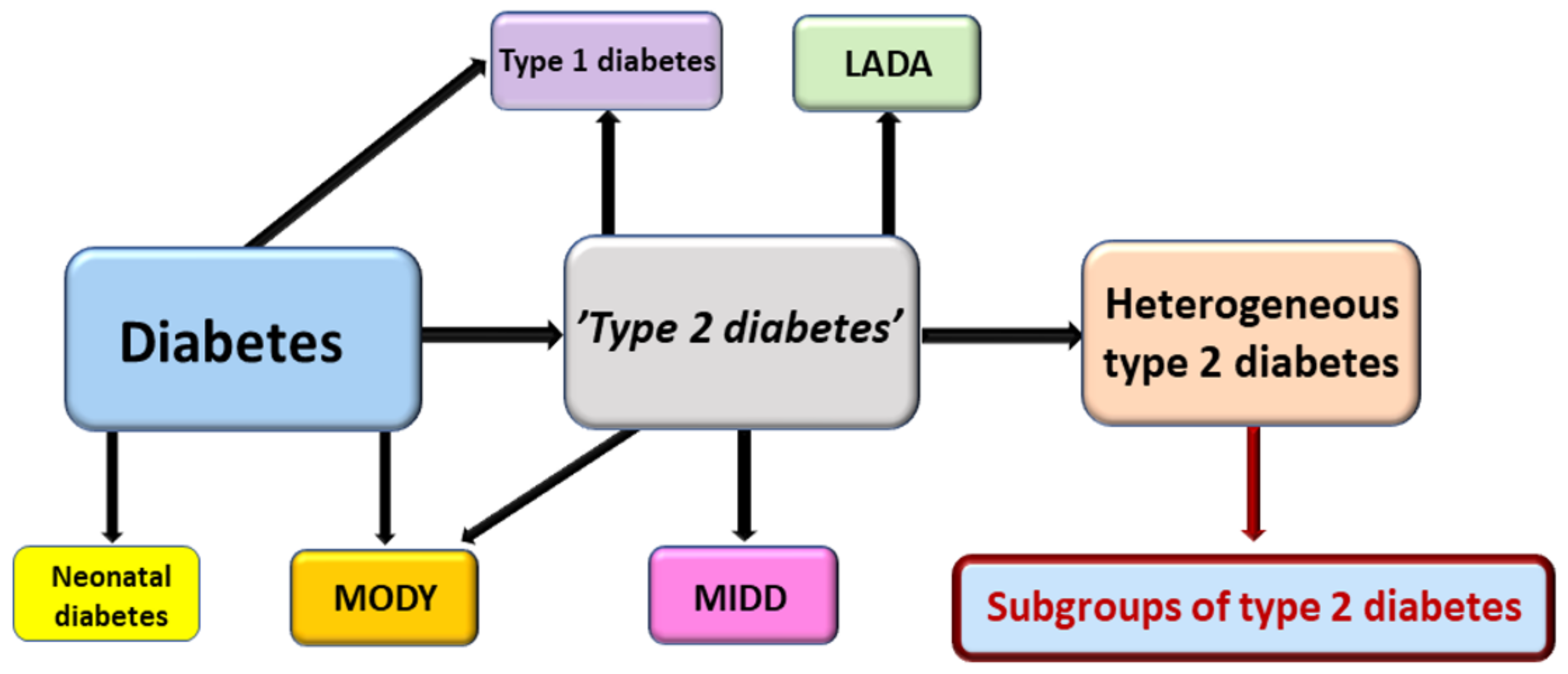
- Li, L.; Cheng, W.-Y.; Glicksberg, B.S.; Gottesman, O.; Tamler, T.; Chen, R.; Bottinger, E.P.; Dudley, J.T. Identification of type 2 diabetes subgroups through topological analysis of patient similarity. Sci. Transl. Med. 2015, 7, 311ra174. [Google Scholar]
- Ahlqvist, E.; Storm, P.; Käräjämäki, A.; Martinell, M.; Dorkhan, M.; Carlsson, A.; Vikman, P.; Prasad, R.B.; Mansour Aly, D.; Almgren, P. Novel subgroups of adult-onset diabetes and their association with outcomes: A data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018, 6, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Aly, D.M.; Dwivedi, O.P.; Prasad, R.B.; Käräjämäki, A.; Hjort, R.; Thangam, M.; Åkerlund, M.; Mahajan, A.; Udler, M.S.; Florez, J.C. Genome-wide association analyses highlight etiological differences underlying newly defined subtypes of diabetes. Nat. Genet. 2021, 53, 1534–1542. [Google Scholar]
- Kim, D.S.; Gloyn, A.L.; Knowles, J.W. Genetics of type 2 diabetes: Opportunities for precision medicine: JACC Focus Seminar. J. Am. Coll. Cardiol. 2021, 78, 496–512. [Google Scholar] [CrossRef] [PubMed]
- Udler, M.S.; Kim, J.; von Grotthuss, M.; Bonàs-Guarch, S.; Cole, J.B.; Chiou, J.; Anderson, C.D.; on behalf of METASTROKE and the ISGC; Boehnke, M.; Laakso, M.; et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: A soft clustering analysis. PLoS Med. 2018, 15, e1002654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaghootkar, H.; Scott, R.A.; White, C.C.; Zhang, W.; Speliotes, E.; Munroe, P.B.; Ehret, G.B.; Bis, J.C.; Fox, C.S.; Walker, M.; et al. Genetic evidence for a normal-weight “metabolically obese” phenotype linking insulin resistance, hypertension, coronary artery disease, and type 2 diabetes. Diabetes 2014, 63, 4369–4377. [Google Scholar] [CrossRef] [Green Version]
- DiCorpo, D.; LeClair, J.; Cole, J.B.; Sarnowski, C.; Ahmadizar, F.; Bielak, L.F.; Blokstra, A.; Bottinger, E.P.; Chaker, L.; Chen, Y.-D.; et al. Type 2 diabetes partitioned polygenic scores associate with disease outcomes in 454,193 individuals across 13 cohorts. Diabetes Care 2022, 45, 674–683. [Google Scholar] [CrossRef]
- Wagner, R.; Heni, M.; Tabák, A.G.; Machann, J.; Schick, F.; Randrianarisoa, E.; Hrabě de Angelis, M.; Birkenfeld, A.L.; Stefan, N.; Peter, A.; et al. Pathophysiology-based subphenotyping of individuals at elevated risk for type 2 diabetes. Nat. Med. 2021, 27, 49–57. [Google Scholar] [CrossRef]
- Wesolowska-Andersen, A.; Brorsson, C.A.; Bizzotto, R.; Mari, A.; Tura, A.; Koivula, R.; Mahajan, A.; Vinuela, A.; Fernandez Tajes, J.; Sapna Sharma, S.; et al. Four groups of type 2 diabetes contribute to the etiological and clinical heterogeneity in newly diagnosed individuals: An IMI DIRECT study. Cell Rep Med. 2022, 3, 100477. [Google Scholar] [CrossRef]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.Z.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Sørensen, T.I.A.; Metz, S.; Kilpeläinen, T.O. Do gene–environment interactions have implications for the precision prevention of type 2 diabetes? Diabetologia, 2022; online ahead of print. [Google Scholar] [CrossRef]
- Yang, Q.; Vijayakumar, A.; Kahn, B.B. Metabolites as regulators of insulin sensitivity and metabolism. Nat. Rev. Mol. Cell Biol. 2018, 19, 654–672. [Google Scholar] [CrossRef] [PubMed]
- Laakso, M. Biomarkers for type 2 diabetes. Mol. Metab. 2019, 27S, S139–S146. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, D.I. Mendelian randomization and type 2 diabetes. Cardiovasc. Drugs Therapy. 2016, 30, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbin, L.J.; Richmond, R.C.; Wade, K.H.; Burgess, S.; Bowden, J.; Smith, G.D.; Timpson, N.J. BMI as a modifiable risk factor for type 2 diabetes: Refining and understanding causal estimates using Mendelian Randomization. Diabetes 2016, 65, 3002–3007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dale, C.E.; Fatemifar, G.; Palmer, T.M.; White, J.; Prieto-Merino, D.; Zabaneh, D.; Engmann, J.E.L.; Shah, T.; Wong, A.; Warren, H.R.; et al. Causal associations of adiposity and body fat distribution with coronary heart disease, stroke subtypes, and type 2 diabetes mellitus: A Mendelian randomization analysis. Circulation 2017, 135, 2373–2388. [Google Scholar] [CrossRef] [Green Version]
- Izundegui, D.G.; Matthew Nayor, M. Metabolomics of type 1 and type 2 diabetes: Insights into risk prediction and mechanisms. Curr. Diabetes Rep. 2022, 22, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Suhre, K.; Meisinger, C.; Döring, A.; Altmaier, E.; Belcredi, P.; Gieger, C.; Chang, D.; Milburn, M.M.; Gall, W.E.; Klaus, M.; et al. Metabolic footprint of diabetes: A multiplatform metabolomics study in an epidemiological setting. PLoS ONE 2010, 5, e13953. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Jia, W. Multi-omics profiling: The way towards precision medicine in metabolic diseases. J. Mol. Cell Biol. 2021, 13, 576–593. [Google Scholar] [CrossRef]
- Suhre, K.; McCarthy, M.I.; Schwenk, J.M. Genetics meets proteomics: Perspectives for large population-based studies. Nat. Rev. Genet. 2021, 22, 9–37. [Google Scholar] [CrossRef]
- Hattersley, A.T.; Patel, K.A. Precision diabetes: Learning from monogenic diabetes. Diabetologia 2017, 60, 769–777. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laakso, M.; Fernandes Silva, L. Genetics of Type 2 Diabetes: Past, Present, and Future. Nutrients 2022, 14, 3201. https://doi.org/10.3390/nu14153201
Laakso M, Fernandes Silva L. Genetics of Type 2 Diabetes: Past, Present, and Future. Nutrients. 2022; 14(15):3201. https://doi.org/10.3390/nu14153201
Chicago/Turabian StyleLaakso, Markku, and Lilian Fernandes Silva. 2022. "Genetics of Type 2 Diabetes: Past, Present, and Future" Nutrients 14, no. 15: 3201. https://doi.org/10.3390/nu14153201