


Hematol. Rep., Volume 14, Issue 4 (December 2022) – 13 articles
Cover Story (view full-size image):
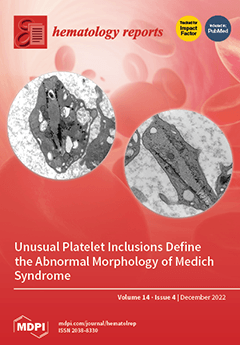
Medich syndrome is a rare macrothrombocytopenia with unusual scroll-like platelet inclusions in cross-sections; they are tubular or cone-shaped longitudinally and may deform a cell’s discoid shape. At the periphery of the inclusions, numerous layers of membranes and aggregates of glycogen are also present. First described by Dr. James G. White in 2004, the condition also includes a deficiency of alpha granules and occasional giant alpha granules, as seen in chromosome 11q deletion syndrome. Of five cases reported to date, three presented with thrombocytopenia at birth, another at two weeks, and one at a year of age. All have been transfusion-dependent, and the diagnosis of Medich syndrome took years to establish. There is no specific genetic mutation identified yet for the syndrome. View this paper
- Issues are regarded as officially published after their release is announced to the table of contents alert mailing list.
- You may sign up for e-mail alerts to receive table of contents of newly released issues.
- PDF is the official format for papers published in both, html and pdf forms. To view the papers in pdf format, click on the "PDF Full-text" link, and use the free Adobe Reader to open them.
Previous Issue
Next Issue