
Microbiota Modulation in Blueberry Rhizosphere by Biocontrol Bacteria
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Treatments
2.2. Sampling
2.3. DNA Extraction
2.4. DNA Metabarcoding Library Preparation and Sequencing of Samples
2.5. Bioinformatics Analysis
2.6. Alpha and Beta Diversity
3. Results
3.1. Sensitivity of the Method
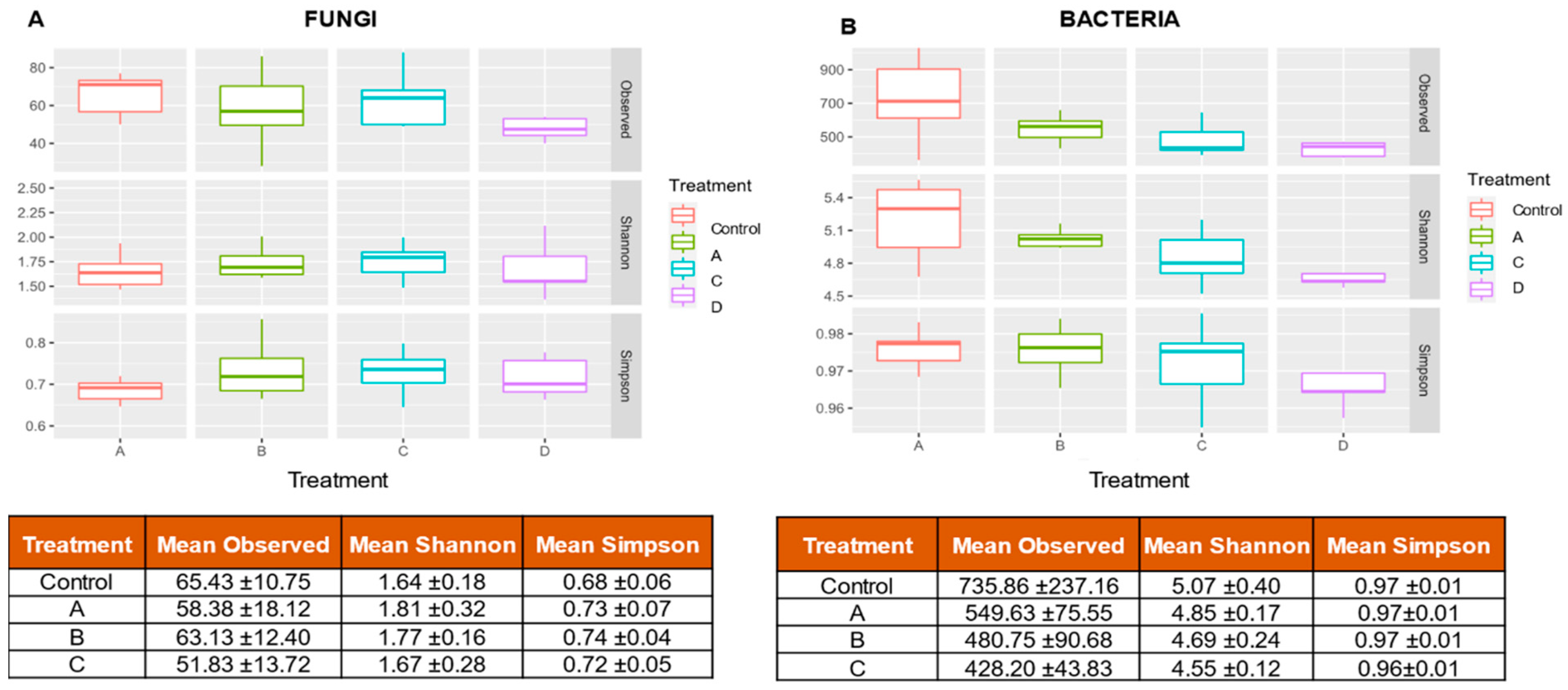
3.2. Alpha Diversity
3.3. Diversity between Conditions
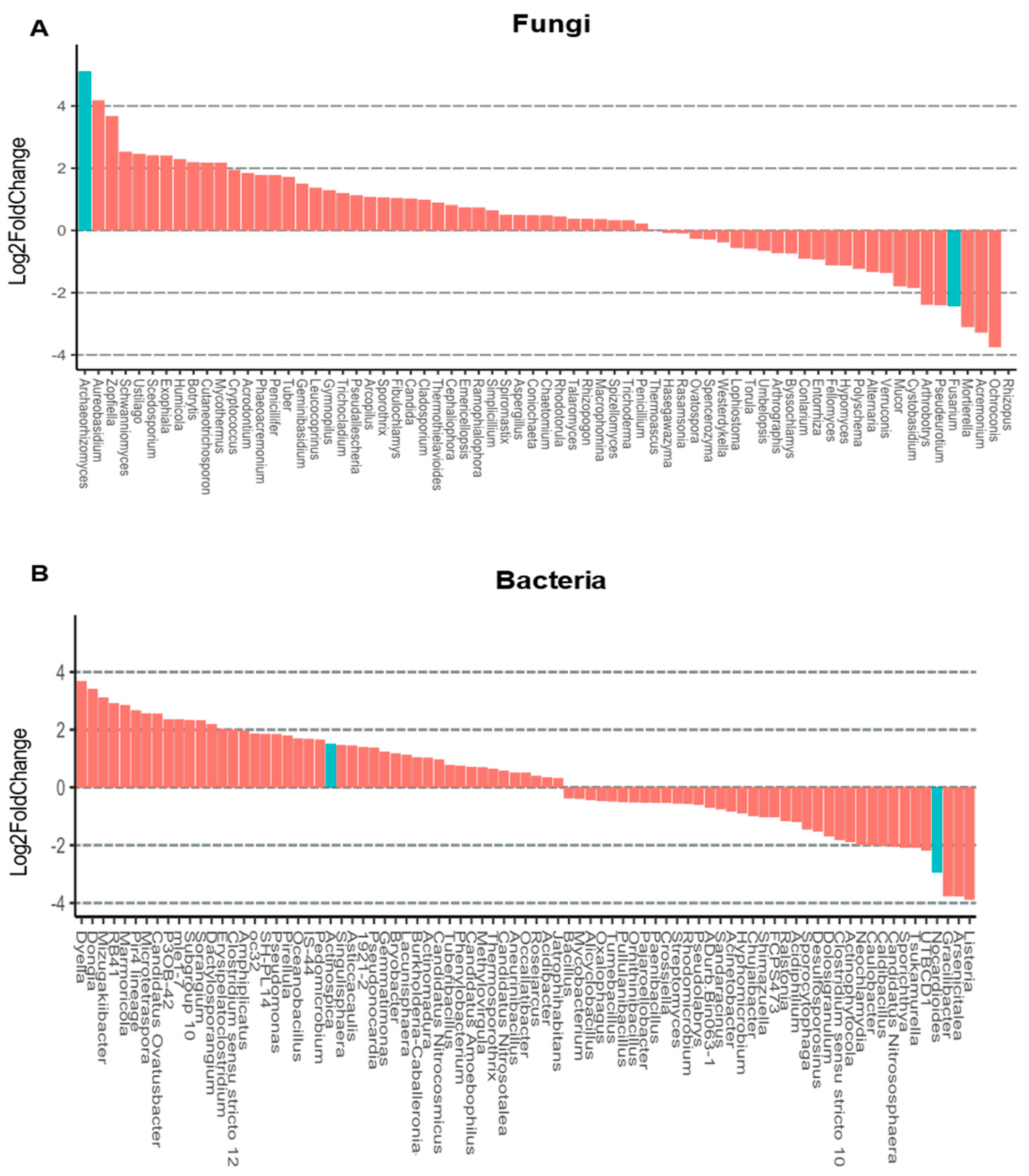
3.4. Rhizosphere Microbiota of Blueberry Crops
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO. FAO (Food and Agriculture Organization of United Nations). Available online: https://www.fao.org/faostat (accessed on 30 August 2022).
- Pérez, R.; Laca, A.; Laca, A.; Díaz, M. Environmental Behaviour of Blueberry Production at Small-Scale in Northern Spain and Improvement Opportunities. J. Clean. Prod. 2022, 339, 130594. [Google Scholar] [CrossRef]
- Zhao, L.; Cai, J.; He, W.; Zhang, Y. Macrophomina vaccinii Sp. Nov. Causing Blueberry Stem Blight in China. MycoKeys 2019, 55, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, I.K.; Singh, T.; Machado, C.C.; Sinclair, J.B. Histopathology of Soybean Seed and Seedling Infection by Macrophomina phaseolina. Phytopathology 1986, 76, 532–535. [Google Scholar] [CrossRef]
- Avilés, M.; de los Santos, B.; Borrero, C. Increase of Canker Disease Severity in Blueberries Caused by Neofusicoccum parvum or Lasiodiplodia Theobromae Due to Interaction with Macrophomina phaseolina Root Infection. Eur. J. Plant Pathol. 2021, 159, 655–663. [Google Scholar] [CrossRef]
- Wright, A.F.; Harmon, P.F. Identification of Species in the Botryosphaeriaceae Family Causing Stem Blight on Southern Highbush Blueberry in Florida. Plant Dis. 2010, 94, 966–971. [Google Scholar] [CrossRef] [Green Version]
- Borrero, C.; Castaño, R.; Avilés, M. First Report of Pestalotiopsis clavispora (Neopestalotiopsis clavispora) Causing Canker and Twig Dieback on Blueberry Bushes in Spain. Plant Dis. 2018, 102, 1178. [Google Scholar] [CrossRef]
- De los Santos, B.; Aguado, A.; Borrero, C.; Viejobueno, J.; Avilés, M. First Report of Charcoal Rot, Caused by Macrophomina phaseolina, on Blueberry in Southwestern Spain. Plant Dis. 2019, 103, 2677. [Google Scholar] [CrossRef]
- Viejobueno, V.; de los Santos, B.; Camacho-Sanchez, M.; Aguado, A.; María, C.; Salazar, S.M. Phenotypic Variability and Genetic Diversity of the Pathogenic Fungus Macrophomina phaseolina from Several Hosts and Host-Specialization in Strawberry. Curr. Microbiol. 2022, 189, 1–15. [Google Scholar] [CrossRef]
- Ijaz, S.; Sadaqat, H.A.; Khan, M.N. A Review of the Impact of Charcoal Rot (Macrophomina phaseolina) on Sunflower. J. Agric. Sci. 2013, 151, 222–227. [Google Scholar] [CrossRef]
- Reichert, I. On Research and Cooperation of Mediterranean Phytopathologists. Phytopathol. Mediterr. 1960, 1, 1–4. [Google Scholar]
- Ramezani, M.; Shier, W.T.; Abbas, H.K.; Tonos, J.L.; Baird, R.E.; Sciumbato, G.L. Soybean Charcoal Rot Disease Fungus Macrophomina phaseolina in Mississippi Produces the Phytotoxin (−)-Botryodiplodin but No Detectable Phaseolinone. J. Nat. Prod. 2007, 70, 128–129. [Google Scholar] [CrossRef]
- Dhingra, O.D.; Sinclair, J.B. Biology and Pathology of Macrophomina phaseolina. Australas. Plant Pathol. 1978, 7, 25. [Google Scholar] [CrossRef]
- Mirza, M.S.; Beg, A. Diseases of Sunflower in Pakistan. FAO Bull. Helia 1983, 6, 55–56. [Google Scholar]
- Ross, J.P. Predispositions of Soybeans to Fusarium Wilt by Heterodera Glycines and Meloidogyne Incognita. Phytopathology 1965, 55, 361–364. [Google Scholar]
- Khamari, B.; Beura, S.K.; Sushree, A.; Monalisa, S.P. Assessment of Combined Effect of Macrophomina Phaseolina and Fusarium Oxysporum on Disease Incidence of Sesame (Sesamum Indicum L.). Int. J. Curr. Microbiol. Appl. Sci. 2017, 6, 1135–1139. [Google Scholar] [CrossRef]
- ECHA, (European Chemical Agnecy). Available online: https://echa.europa.eu/es/substance-information/-/substanceinfo/100.007.798 (accessed on 20 December 2021).
- Gareau, B. Sociology in Global Environmental Governance? Neoliberalism, Protectionism and the Methyl Bromide Controversy in the Montreal Protocol. Environments 2017, 4, 73. [Google Scholar] [CrossRef] [Green Version]
- Guthman, J. Land Access and Costs May Drive Strawberry Growers’ Increased Use of Fumigation. Calif. Agric. 2017, 71, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Holmes, G.J.; Mansouripour, S.M.; Hewavitharana, S.S. Strawberries at the Crossroads: Management of Soilborne Diseases in California without Methyl Bromide. Phytopathology 2020, 110, 956–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, D.; Thakker, J.N.; Dhandhukia, P.C. Portraying Mechanics of Plant Growth Promoting Rhizobacteria (PGPR): A Review. Cogent Food Agric. 2016, 2, 1127500. [Google Scholar] [CrossRef]
- Viejobueno, J.; Albornoz, P.L.; Camacho, M.; de los Santos, B.; Martínez-Zamora, M.G.; Salazar, S.M. Protection of Strawberry Plants against Charcoal Rot Disease (Macrophomina phaseolina) Induced by Azospirillum brasilense. Agronomy 2021, 11, 195. [Google Scholar] [CrossRef]
- Viejobueno, J.; Rodríguez-Berbel, N.; Miranda, L.; de los Santos, B.; Camacho, M. Potential Bacterial Antagonists for the Control of Charcoal Rot (Macrophomina phaseolina) in Strawberry. Horticulturae 2021, 7, 457. [Google Scholar] [CrossRef]
- Liu, H.; Li, J.; Carvalhais, L.C.; Percy, C.D.; Prakash Verma, J.; Schenk, P.M.; Singh, B.K. Evidence for the Plant Recruitment of Beneficial Microbes to Suppress Soil-borne Pathogens. New Phytol. 2021, 229, 2873–2885. [Google Scholar] [CrossRef]
- De Corato, U. Disease-Suppressive Compost Enhances Natural Soil Suppressiveness against Soil-Borne Plant Pathogens: A Critical Review. Rhizosphere 2020, 13, 100192. [Google Scholar] [CrossRef]
- Niu, B.; Wang, W.; Yuan, Z.; Sederoff, R.R.; Sederoff, H.; Chiang, V.L.; Borriss, R. Microbial Interactions Within Multiple-Strain Biological Control Agents Impact Soil-Borne Plant Disease. Front. Microbiol. 2020, 11, 585404. [Google Scholar] [CrossRef]
- Khoshru, B.; Moharramnejad, S.; Gharajeh, N.H.; Asgari Lajayer, B.; Ghorbanpour, M. Plant Microbiome and Its Important in Stressful Agriculture. In Plant Microbiome Paradigm; Springer International Publishing: Cham, Switzerland, 2020; pp. 13–48. [Google Scholar] [CrossRef]
- van Elsas, J.D.; Boersma, F.G.H. A Review of Molecular Methods to Study the Microbiota of Soil and the Mycosphere. Eur. J. Soil Biol. 2011, 47, 77–87. [Google Scholar] [CrossRef]
- Ruiz Gómez, F.J.; Navarro-Cerrillo, R.M.; Pérez-de-Luque, A.; Oβwald, W.; Vannini, A.; Morales-Rodríguez, C. Assessment of Functional and Structural Changes of Soil Fungal and Oomycete Communities in Holm Oak Declined Dehesas through Metabarcoding Analysis. Sci. Rep. 2019, 9, 5315. [Google Scholar] [CrossRef] [Green Version]
- Xu, J. Fungal DNA Barcoding. Genome 2016, 59, 913–932. [Google Scholar] [CrossRef] [Green Version]
- Pauvert, C.; Buée, M.; Laval, V.; Edel-Hermann, V.; Fauchery, L.; Gautier, A.; Lesur, I.; Vallance, J.; Vacher, C. Bioinformatics Matters: The Accuracy of Plant and Soil Fungal Community Data Is Highly Dependent on the Metabarcoding Pipeline. Fungal Ecol. 2019, 41, 23–33. [Google Scholar] [CrossRef]
- Rodríguez-Cárdenas, S. Caracterización de una Colección Bacteriana Aislada de Frutos Rojos en Base a sus Propiedades PGP y de Biocontrol. Ensayo de Promoción del Crecimiento en Plantas de Fresa. Master’s Thesis, University of Pablo de Olavide, Sevilla, Spain, 2020. [Google Scholar]
- Rotolo, C.; De Miccolis Angelini, R.M.; Pollastro, S.; Faretra, F. A TaqMan-Based QPCR Assay for Quantitative Detection of the Biocontrol Agents Bacillus subtilis Strain QST713 and Bacillus amyloliquefaciens Subsp. plantarum Strain D747. BioControl 2016, 61, 91–101. [Google Scholar] [CrossRef]
- Weißbecker, C.; Buscot, F.; Wubet, T. Preservation of Nucleic Acids by Freeze-Drying for next Generation Sequencing Analyses of Soil Microbial Communities. J. Plant Ecol. 2017, 10, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every Base Matters: Assessing Small Subunit RRNA Primers for Marine Microbiomes with Mock Communities, Time Series and Global Field Samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor Revision to V4 Region SSU RRNA 806R Gene Primer Greatly Increases Detection of SAR11 Bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef]
- Earth Microbiome Project. Available online: https://earthmicrobiome.org/protocols-and-standards/16s/ (accessed on 12 December 2021).
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In PCR Protocols, a Guide to Methods and Applications; Academic Press, Inc.: New York, NY, USA, 1990; pp. 315–322. [Google Scholar] [CrossRef]
- Vierna, J.; Doña, J.; Vizcaíno, A.; Serrano, D.; Jovani, R. PCR Cycles above Routine Numbers Do Not Compromise High-Throughput DNA Barcoding Results. Genome 2017, 60, 868–873. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Dada2 Tutorial with NovaSeq Dataset for Ernakovich Lab. Available online: https://github.com/ErnakovichLab/dada2_ernakovichlab (accessed on 16 November 2021).
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact Sequence Variants Should Replace Operational Taxonomic Units in Marker-Gene Data Analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-Species Living Tree Project (LTP)” Taxonomic Frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [Green Version]
- Kõljalg, U.; Nilsson, H.R.; Schigel, D.; Tedersoo, L.; Larsson, K.-H.; May, T.W.; Taylor, A.F.S.; Jeppesen, T.S.; Frøslev, T.G.; Lindahl, B.D.; et al. The Taxon Hypothesis Paradigm—On the Unambiguous Detection and Communication of Taxa. Microorganisms 2020, 8, 1910. [Google Scholar] [CrossRef]
- Callahan, B.J.; Sankaran, K.; Fukuyama, J.A.; McMurdie, P.J.; Holmes, S.P. Bioconductor Workflow for Microbiome Data Analysis: From Raw Reads to Community Analyses. F1000Research 2016, 5, 1492. [Google Scholar] [CrossRef]
- Vmikk/MetagMisc:, V. Vmikk/MetagMisc: V.0.0.0.9000. Available online: https://doi.org/10.5281/ZENODO.571403 (accessed on 5 December 2021).
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple Statistical Identification and Removal of Contaminant Sequences in Marker-Gene and Metagenomics Data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Thukral, A.K. A Review on Measurement of Alpha Diversity in Biology. Agric. Res. J. 2017, 54, 1–10. [Google Scholar] [CrossRef]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using Lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Whittaker, R.H. Vegetation of the Siskiyou Mountains, Oregon and California. Ecol. Monogr. 1960, 30, 279–338. [Google Scholar] [CrossRef]
- Oksanen, J.; Guillaume Blanchet, F.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. 2020. Available online: https://CRAN.R-project.org/package=vegan (accessed on 6 December 2021).
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Lahti, L.; Shetty, S. Microbiome R Package. 2019. Available online: https://github.com/microbiome/microbiome (accessed on 20 December 2021).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; Available online: https://ggplot2.tidyverse.org (accessed on 20 December 2021).
- Bardgett, R.D.; van der Putten, W.H. Belowground Biodiversity and Ecosystem Functioning. Nature 2014, 515, 505–511. [Google Scholar] [CrossRef]
- van der Heijden, M.G.A.; Bardgett, R.D.; van Straalen, N.M. The Unseen Majority: Soil Microbes as Drivers of Plant Diversity and Productivity in Terrestrial Ecosystems. Ecol. Lett. 2008, 11, 296–310. [Google Scholar] [CrossRef]
- Crous, P.W.; Schoch, C.L.; Hyde, K.D.; Wood, A.R.; Gueidan, C.; de Hoog, G.S.; Groenewald, J.Z. Phylogenetic Lineages in the Capnodiales. Stud. Mycol. 2009, 64, 17–47. [Google Scholar] [CrossRef]
- Liu, C.; Lin, H.; He, P.; Li, X.; Geng, Y.; Tuerhong, A.; Dong, Y. Peat and Bentonite Amendments Assisted Soilless Revegetation of Oligotrophic and Heavy Metal Contaminated Nonferrous Metallic Tailing. Chemosphere 2022, 287, 132101. [Google Scholar] [CrossRef]
- Kepler, R.M.; Maul, J.E.; Rehner, S.A. Managing the Plant Microbiome for Biocontrol Fungi: Examples from Hypocreales. Curr. Opin. Microbiol. 2017, 37, 48–53. [Google Scholar] [CrossRef]
- Zhang, Y.; Crous, P.W.; Schoch, C.L.; Hyde, K.D. Pleosporales. Fungal Divers. 2012, 53, 1–221. [Google Scholar] [CrossRef]
- Hashmi, I.; Bindschedler, S.; Junier, P. Firmicutes. In Beneficial Microbes in Agro-Ecology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 363–396. [Google Scholar] [CrossRef]
- Pérez, B.A.; Murillo, F.; Divo de Sesar, M.; Wright, E.R. Occurrence of Fusarium solani on Blueberry in Argentina. Plant Dis. 2007, 91, 1053. [Google Scholar] [CrossRef]
- Rosling, A.; Cox, F.; Cruz-Martinez, K.; Ihrmark, K.; Grelet, G.-A.; Lindahl, B.D.; Menkis, A.; James, T.Y. Archaeorhizomycetes: Unearthing an Ancient Class of Ubiquitous Soil Fungi. Science 2011, 333, 876–879. [Google Scholar] [CrossRef]
- Ayangbenro, A.S.; Babalola, O.O. Reclamation of Arid and Semi-Arid Soils: The Role of Plant Growth-Promoting Archaea and Bacteria. Curr. Plant Biol. 2021, 25, 100173. [Google Scholar] [CrossRef]
- Zhao, F.; Zhang, Y.; Dong, W.; Zhang, Y.; Zhang, G.; Sun, Z.; Yang, L. Vermicompost Can Suppress Fusarium Oxysporum f. Sp. Lycopersici via Generation of Beneficial Bacteria in a Long-Term Tomato Monoculture Soil. Plant Soil 2019, 440, 491–505. [Google Scholar] [CrossRef]
- Lazcano, C.; Boyd, E.; Holmes, G.; Hewavitharana, S.; Pasulka, A.; Ivors, K. The Rhizosphere Microbiome Plays a Role in the Resistance to Soil-Borne Pathogens and Nutrient Uptake of Strawberry Cultivars under Field Conditions. Sci. Rep. 2021, 11, 3188. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Ma, G.; Chen, S.; Lin, C.; Zhao, X. Microbial Network and Soil Properties Are Changed in Bacterial Wilt-Susceptible Soil. Appl. Environ. Microbiol. 2019, 85, e00162-19. [Google Scholar] [CrossRef] [Green Version]
- Gupta, C.P.; Sharma, A.; Dubey, R.C.; Maheshwari, D.K. Pseudomonas aeruginosa (GRC1) as a Strong Antagonist of Macrophomina phaseolina and Fusarium oxysporum. Cytobios 1999, 1999, 183–189. [Google Scholar]
- Dawar, S.; Wahab, S.; Tariq, M.; Zaki, M.J. Application of Bacillus Species in the Control of Root Rot Diseases of Crop Plants. Arch. Phytopathol. Plant Prot. 2010, 43, 412–418. [Google Scholar] [CrossRef]
- Bhattacharyya, D.; Basu, S.; Chattapadhyay, J.P.; Bose, S.K. Biocontrol of Macrophomina Root-Rot Disease of Jute by an Antagonistic Organism, Aspergillus versicolor. Plant Soil 1985, 87, 435–438. [Google Scholar] [CrossRef]
- Khan, I.H.; Javaid, A. In Vitro Screening of Aspergillus Spp. for Their Biocontrol Potential against Macrophomina phaseolina. J. Plant Pathol. 2021, 103, 1195–1205. [Google Scholar] [CrossRef]
- Houbraken, J.; Kocsubé, S.; Visagie, C.M.; Yilmaz, N.; Wang, X.-C.; Meijer, M.; Kraak, B.; Hubka, V.; Bensch, K.; Samson, R.A.; et al. Classification of Aspergillus, Penicillium, Talaromyces and Related Genera (Eurotiales): An Overview of Families, Genera, Subgenera, Sections, Series and Species. Stud. Mycol. 2020, 95, 5–169. [Google Scholar] [CrossRef]
- Fahima, T.; Henis, Y. Quantitative Assessment of the Interaction between the Antagonistic Fungus Talaromyces flavus and the Wilt Pathogen Verticillium dahliae on Eggplant Roots. Plant Soil 1995, 176, 129–137. [Google Scholar] [CrossRef]
- Khalmuratova, I.; Kim, H.; Nam, Y.J.; Oh, Y.; Jeong, M.J.; Choi, H.R.; You, Y.H.; Choo, Y.S.; Lee, I.J.; Shin, J.H.; et al. Diversity and Plant Growth Promoting Capacity of Endophytic Fungi Associated with Halophytic Plants from the West Coast of Korea. Mycobiology 2015, 43, 373–383. [Google Scholar] [CrossRef] [Green Version]
- McLaren, D.L.; Huang, H.C.; Rimmer, S.R.; Kokko, E.G. Ultrastructural Studies on Infection of Sclerotia of Sclerotinia sclerotiorum by Talaromyces flavus. Can. J. Bot. 1989, 67, 2199–2205. [Google Scholar] [CrossRef]
- Black, M.; Moolhuijzen, P.; Chapman, B.; Barrero, R.; Howieson, J.; Hungria, M.; Bellgard, M. The Genetics of Symbiotic Nitrogen Fixation: Comparative Genomics of 14 Rhizobia Strains by Resolution of Protein Clusters. Genes 2012, 3, 138–166. [Google Scholar] [CrossRef] [Green Version]
- Masuda, Y.; Yamanaka, H.; Xu, Z.-X.; Shiratori, Y.; Aono, T.; Amachi, S.; Senoo, K.; Itoh, H. Diazotrophic Anaeromyxobacter Isolates from Soils. Appl. Environ. Microbiol. 2020, 86, e00956-20. [Google Scholar] [CrossRef] [PubMed]
- Kavitha, A.; Prabhakar, P.; Narasimhulu, M.; Vijayalakshmi, M.; Venkateswarlu, Y.; Venkateswara Rao, K.; Balaraju Subba Raju, V. Isolation, Characterization and Biological Evaluation of Bioactive Metabolites from Nocardia Levis MK-VL_113. Microbiol. Res. 2010, 165, 199–210. [Google Scholar] [CrossRef]
- Nisrina, L.; Effendi, Y.; Pancoro, A. Revealing the Role of Plant Growth Promoting Rhizobacteria in Suppressive Soils against Fusarium Oxysporum f.Sp. Cubense Based on Metagenomic Analysis. Heliyon 2021, 7, e07636. [Google Scholar] [CrossRef]
- Ghodhbane-Gtari, F.; Nouioui, I.; Hezbri, K.; Lundstedt, E.; D’Angelo, T.; McNutt, Z.; Laplaze, L.; Gherbi, H.; Vaissayre, V.; Svistoonoff, S.; et al. The Plant-Growth-Promoting Actinobacteria of the Genus Nocardia Induces Root Nodule Formation in Casuarina glauca. Antonie Leeuwenhoek 2019, 112, 75–90. [Google Scholar] [CrossRef]
- Luo, L.; Wang, L.; Deng, L.; Mei, X.; Liu, Y.; Huang, H.; Du, F.; Zhu, S.; Yang, M. Enrichment of Burkholderia in the Rhizosphere by Autotoxic Ginsenosides to Alleviate Negative Plant-Soil Feedback. Microbiol. Spectr. 2021, 9, e01400-21. [Google Scholar] [CrossRef]
- Kumar, U.; Kumar Nayak, A.; Shahid, M.; Gupta, V.V.S.R.; Panneerselvam, P.; Mohanty, S.; Kaviraj, M.; Kumar, A.; Chatterjee, D.; Lal, B.; et al. Continuous Application of Inorganic and Organic Fertilizers over 47 Years in Paddy Soil Alters the Bacterial Community Structure and Its Influence on Rice Production. Agric. Ecosyst. Environ. 2018, 262, 65–75. [Google Scholar] [CrossRef]
- Franke-Whittle, I.H.; Manici, L.M.; Insam, H.; Stres, B. Rhizosphere Bacteria and Fungi Associated with Plant Growth in Soils of Three Replanted Apple Orchards. Plant Soil 2015, 395, 317–333. [Google Scholar] [CrossRef]
- Schleper, C.; Nicol, G.W. Ammonia-Oxidising Archaea—Physiology, Ecology and Evolution. Adv. Microb. Physiol. 2010, 57, 1–41. [Google Scholar] [CrossRef]
- Zhang, H.; Sekiguchi, Y.; Hanada, S.; Hugenholtz, P.; Kim, H.; Kamagata, Y.; Nakamura, K. Gemmatimonas aurantiaca Gen. Nov., Sp. Nov., a Gram-Negative, Aerobic, Polyphosphate-Accumulating Micro-Organism, the First Cultured Representative of the New Bacterial Phylum Gemmatimonadetes Phyl. Nov. Int. J. Syst. Evol. Microbiol. 2003, 53, 1155–1163. [Google Scholar] [CrossRef]
- Asaf, S.; Numan, M.; Khan, A.L.; Al-Harrasi, A. Sphingomonas: From Diversity and Genomics to Functional Role in Environmental Remediation and Plant Growth. Crit. Rev. Biotechnol. 2020, 40, 138–152. [Google Scholar] [CrossRef]
- Grady, E.N.; MacDonald, J.; Liu, L.; Richman, A.; Yuan, Z.-C. Current Knowledge and Perspectives of Paenibacillus: A Review. Microb. Cell Fact. 2016, 15, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Wang, J.; You, Y.; Wang, R.; Chu, S.; Chi, Y.; Hayat, K.; Hui, N.; Liu, X.; Zhang, D.; et al. When Nanoparticle and Microbes Meet: The Effect of Multi-Walled Carbon Nanotubes on Microbial Community and Nutrient Cycling in Hyperaccumulator System. J. Hazard. Mater. 2022, 423, 126947. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Wang, H.; Yu, Z.; Zhang, S.; Qi, X.; Sun, L.; Wang, Z.; Zhang, M.; Ahmed, T.; Li, B. Effect of Two Kinds of Fertilizers on Growth and Rhizosphere Soil Properties of Bayberry with Decline Disease. Plants 2021, 10, 2386. [Google Scholar] [CrossRef]
- Child, R.; Miller, C.D.; Liang, Y.; Narasimham, G.; Chatterton, J.; Harrison, P.; Sims, R.C.; Britt, D.; Anderson, A.J. Polycyclic Aromatic Hydrocarbon-Degrading Mycobacterium Isolates: Their Association with Plant Roots. Appl. Microbiol. Biotechnol. 2007, 75, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Ueki, A.; Kaku, N.; Ueki, K. Role of Anaerobic Bacteria in Biological Soil Disinfestation for Elimination of Soil-Borne Plant Pathogens in Agriculture. Appl. Microbiol. Biotechnol. 2018, 102, 6309–6318. [Google Scholar] [CrossRef] [PubMed]
- Sahin, N. Oxalotrophic Bacteria. Res. Microbiol. 2003, 154, 399–407. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | SEQUENCE (5′–3′) | |
|---|---|---|
| Bacterial 16S | 515F-Y | GTGYCAGCMGCCGCGGTAA |
| 806R | GGACTACNVGGGTWTCTAAT | |
| Fungal ITS2 | ITS3 | GCATCGATGAAGAACGCAGC |
| ITS4R | TCCTCCGCTTATTGATATGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez-Mena, S.; Camacho, M.; de los Santos, B.; Miranda, L.; Camacho-Sanchez, M. Microbiota Modulation in Blueberry Rhizosphere by Biocontrol Bacteria. Microbiol. Res. 2022, 13, 809-824. https://doi.org/10.3390/microbiolres13040057
Rodriguez-Mena S, Camacho M, de los Santos B, Miranda L, Camacho-Sanchez M. Microbiota Modulation in Blueberry Rhizosphere by Biocontrol Bacteria. Microbiology Research. 2022; 13(4):809-824. https://doi.org/10.3390/microbiolres13040057
Chicago/Turabian StyleRodriguez-Mena, Sara, María Camacho, Berta de los Santos, Luis Miranda, and Miguel Camacho-Sanchez. 2022. "Microbiota Modulation in Blueberry Rhizosphere by Biocontrol Bacteria" Microbiology Research 13, no. 4: 809-824. https://doi.org/10.3390/microbiolres13040057