
Translation of Monoclonal Antibodies Pharmacokinetics from Animal to Human Using Physiologically Based Modeling in Open Systems Pharmacology (OSP) Suite: A Retrospective Analysis of Bevacizumab

 , and
, and
Abstract
:1. Introduction
2. Material and Methods
2.1. PK Data
2.2. Software
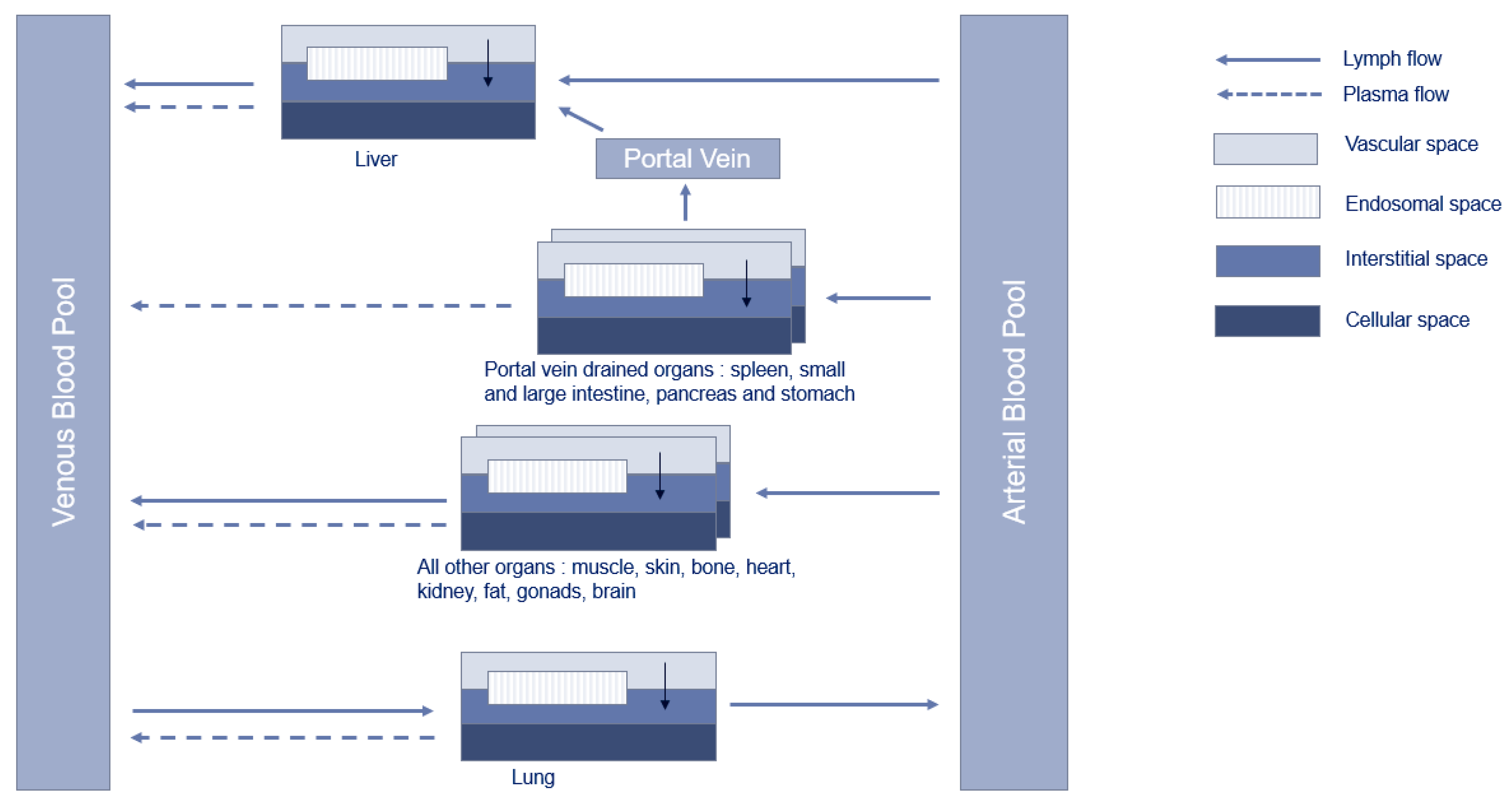
2.3. PBPK Model Structure
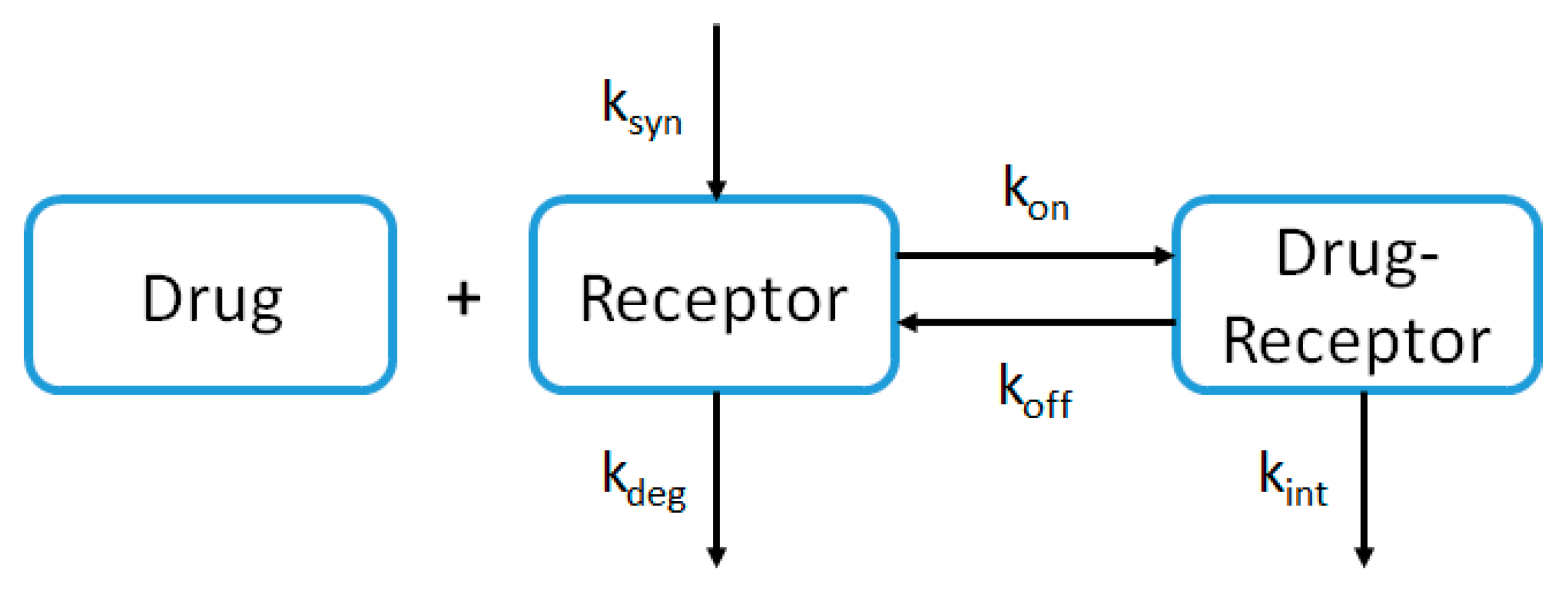
2.4. PBPK Modeling Approach
2.4.1. Non-Human Primate
2.4.2. Healthy Volunteers
2.4.3. Adult Cancer Patients
2.5. Model Evaluation
2.6. Sensitivity Analysis
3. Results
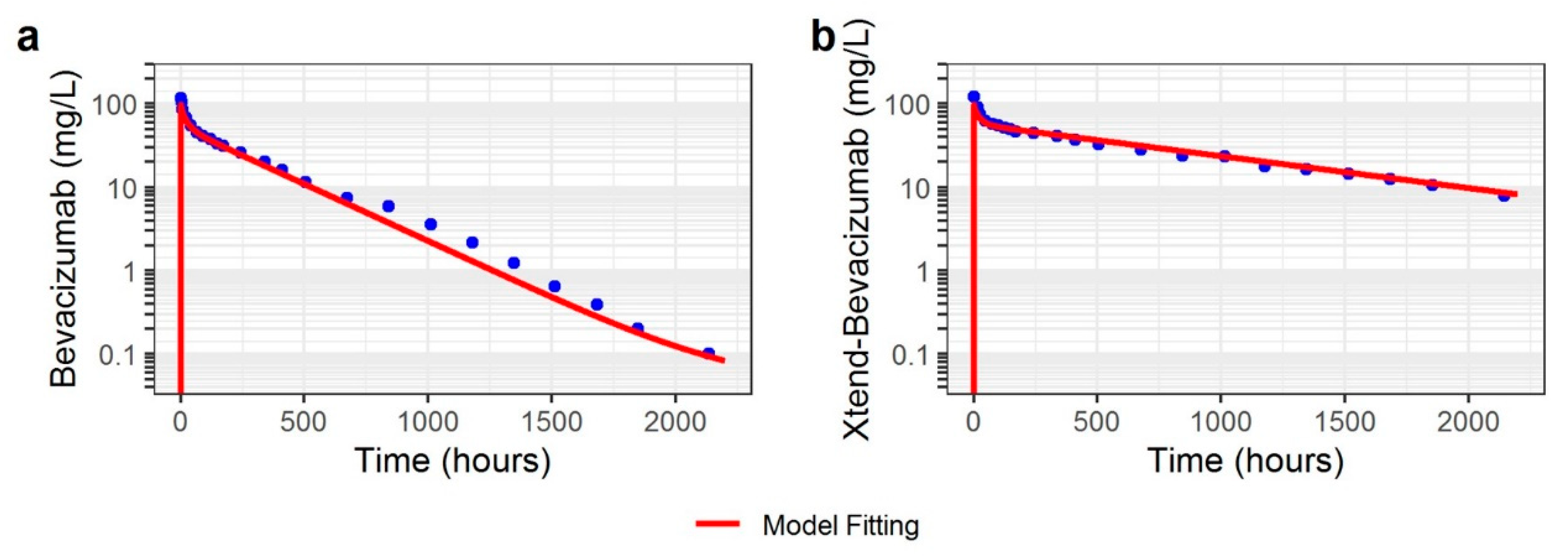
3.1. Non-Human Primate
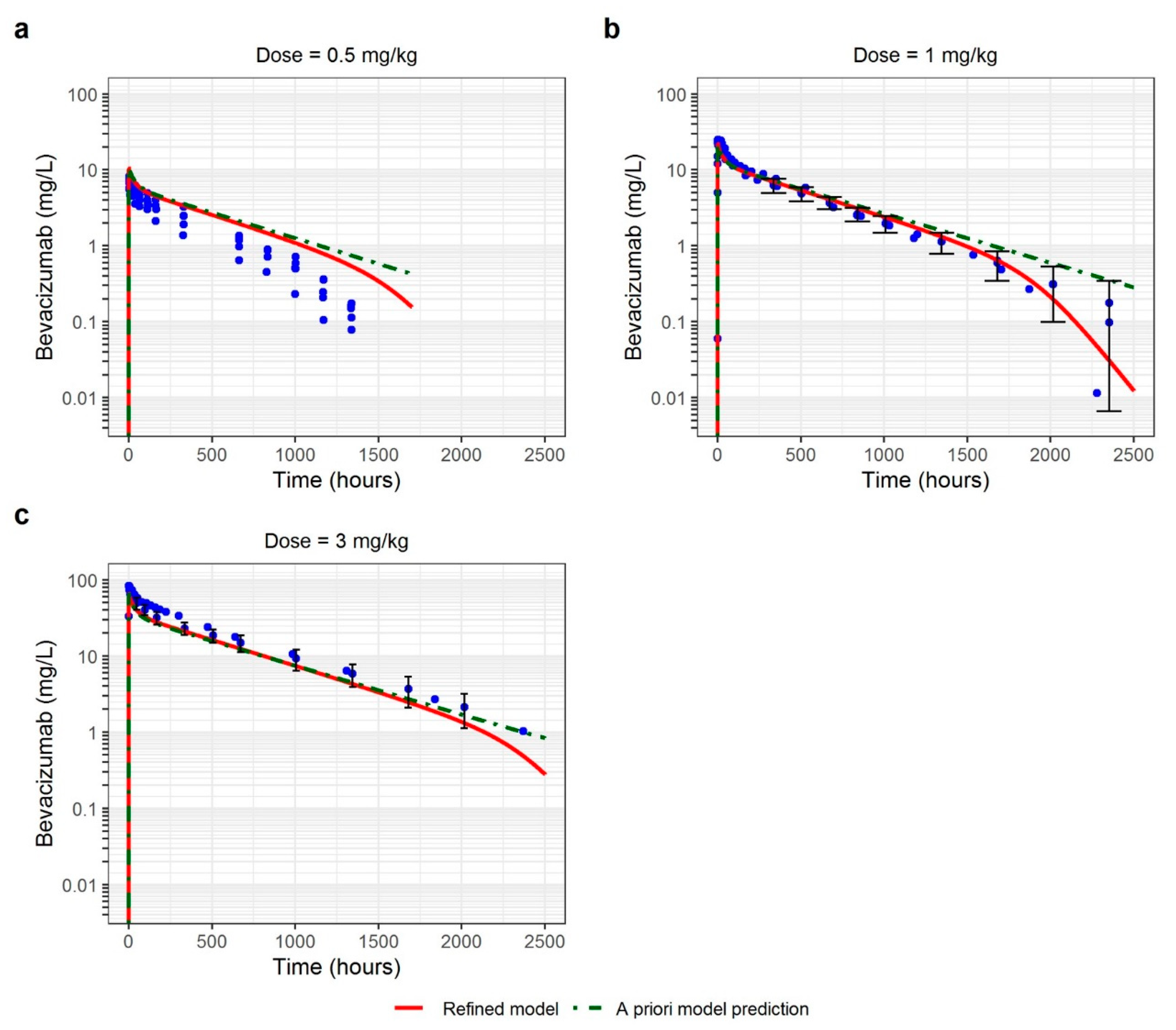
3.2. Healthy Volunteers
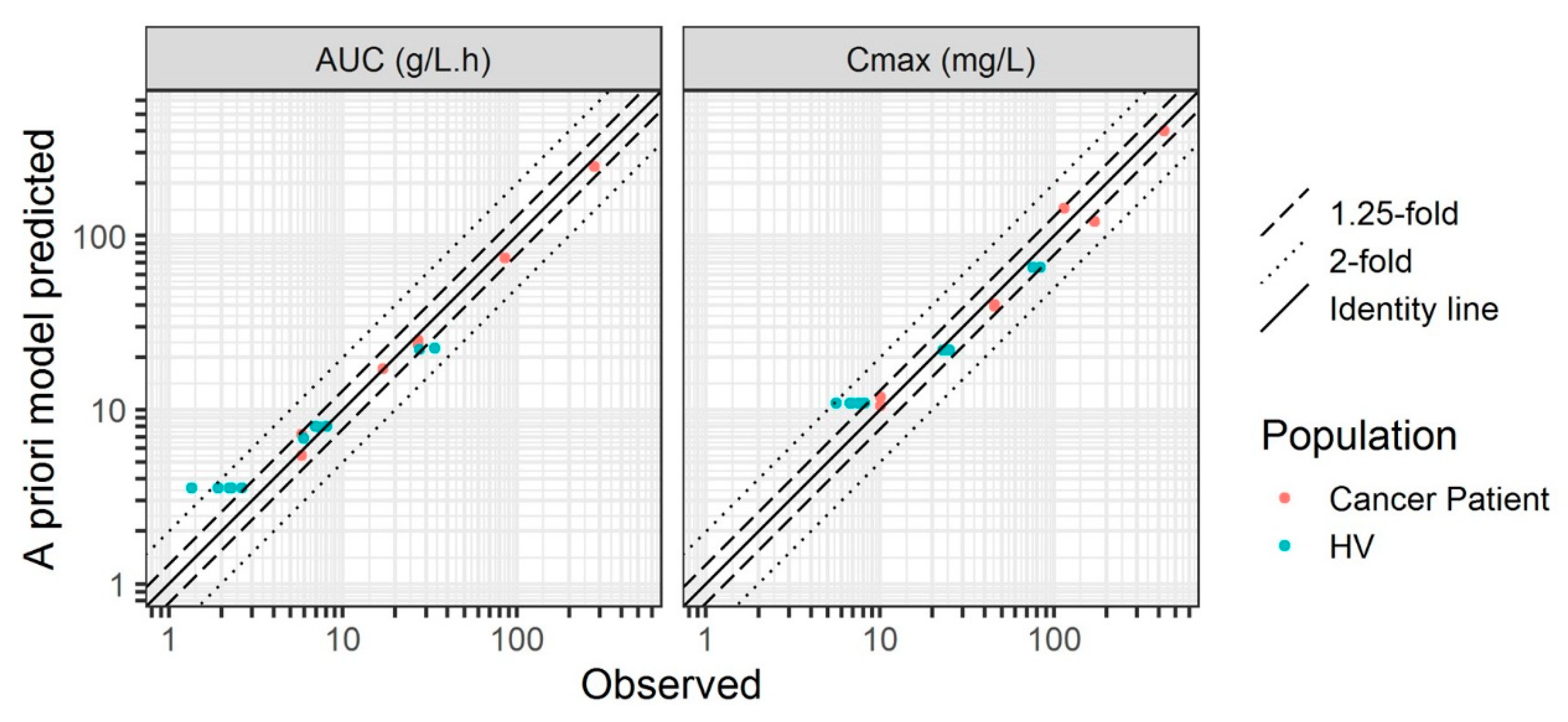
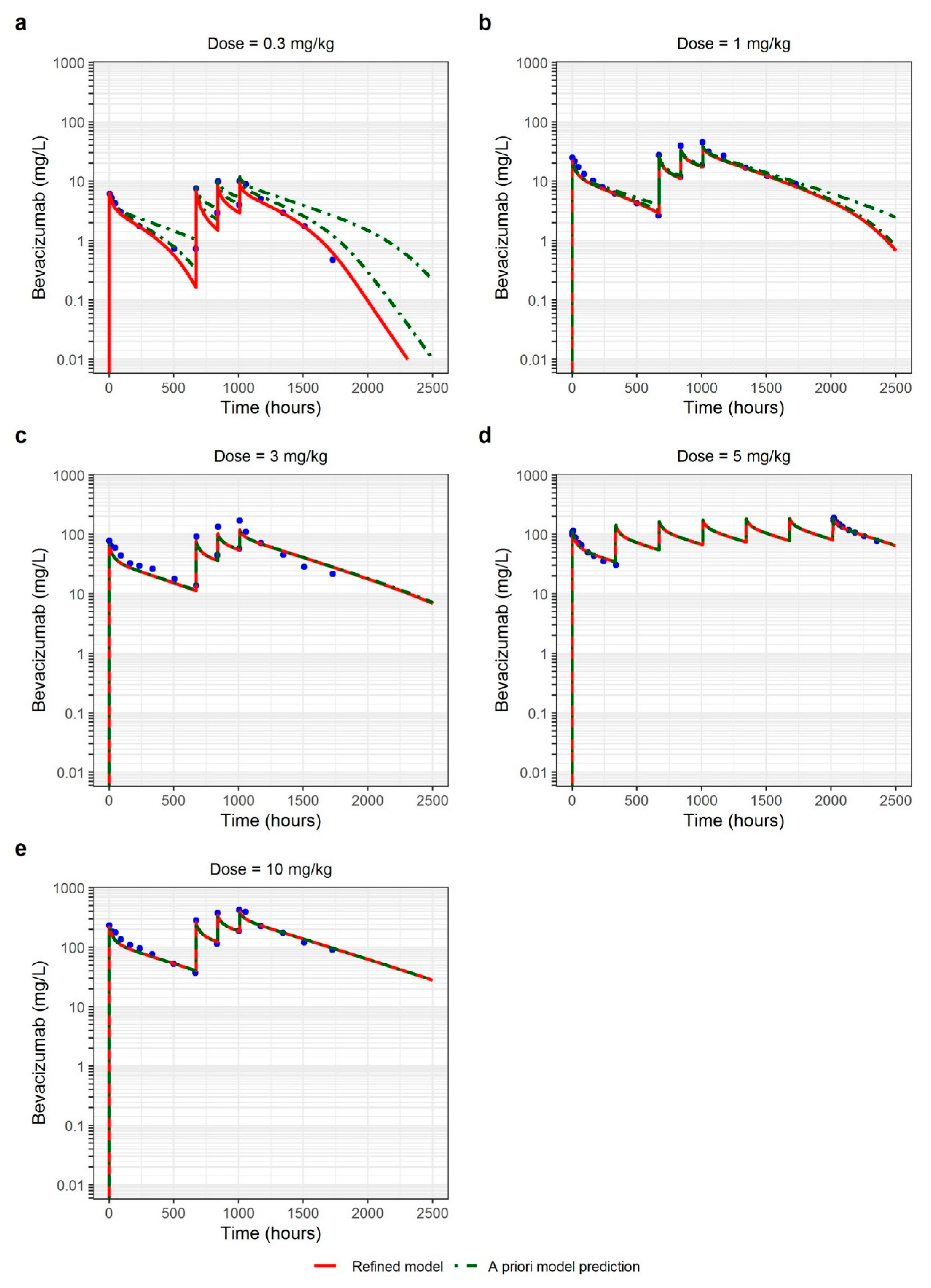
3.3. Adult Cancer Patients
3.4. Sensitivity Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ovacik, M.; Lin, K. Tutorial on Monoclonal Antibody Pharmacokinetics and Its Considerations in Early Development. Clin. Transl. Sci. 2018, 11, 540–552. [Google Scholar] [CrossRef]
- Pasquiers, B.; Benamara, S.; Felices, M.; Nguyen, L.; Declèves, X. Review of the Existing Translational Pharmacokinetics Modeling Approaches Specific to Monoclonal Antibodies (mAbs) to Support the First-In-Human (FIH) Dose Selection. Int. J. Mol. Sci. 2022, 23, 12754. [Google Scholar] [CrossRef] [PubMed]
- Haraya, K.; Tsutsui, H.; Komori, Y.; Tachibana, T. Recent Advances in Translational Pharmacokinetics and Pharmacodynamics Prediction of Therapeutic Antibodies Using Modeling and Simulation. Pharmaceuticals 2022, 15, 508. [Google Scholar] [CrossRef]
- Garg, A.; Balthasar, J.P. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J. Pharmacokinet. Pharmacodyn. 2007, 34, 687–709. [Google Scholar] [CrossRef] [PubMed]
- Glassman, P.M.; Balthasar, J.P. Physiologically-based modeling of monoclonal antibody pharmacokinetics in drug discovery and development. Drug Metab. Pharmacokinet. 2019, 34, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Stader, F.; Chan, P.; Shemesh, C.S.; Chen, Y.; Gill, K.L.; Jones, H.M.; Li, L.; Rossato, G.; Wu, B.; et al. Development of a Pediatric Physiologically-Based Pharma-cokinetic Model to Support Recommended Dosing of Atezolizumab in Children with Solid Tumors. Front. Pharmacol. 2022, 13, 974423. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Lien, Y.T.; Vozmediano, V.; Schlender, J.-F.; Eissing, T.; Schmidt, S.; Niederalt, C. Physiologically Based Pharmacokinetic Modeling of Monoclonal Antibodies in Pediatric Populations Using PK-Sim. Front. Pharmacol. 2020, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Glassman, P.M.; Balthasar, J.P. Application of a catenary PBPK model to predict the disposition of “catch and release” anti-PCSK9 antibodies. Int. J. Pharm. 2016, 505, 69–78. [Google Scholar] [CrossRef]
- Glassman, P.M.; Balthasar, J.P. Physiologically-based pharmacokinetic modeling to predict the clinical pharmacokinetics of monoclonal antibodies. J. Pharmacokinet. Pharmacodyn. 2016, 43, 427–446. [Google Scholar] [CrossRef]
- Avastin-Epar-Product-Information_Fr.Pdf [Internet]. Available online: https://www.ema.europa.eu/en/documents/product-information/avastin-epar-product-information_fr.pdf (accessed on 16 January 2023).
- Gordon, M.S.; Margolin, K.; Talpaz, M.; Sledge, G.W., Jr.; Holmgren, E.; Benjamin, R.; Stalter, S.; Shak, S.; Adelman, D.C. Phase I Safety and Pharmacokinetic Study of Recombinant Human Anti-Vascular Endothelial Growth Factor in Patients With Advanced Cancer. J. Clin. Oncol. 2001, 19, 843–850. [Google Scholar] [CrossRef]
- Li, M.; Qiang, W.; Hu, L.; Wang, L.; Cheng, Z. A novel model for the pharmacokinetic studies of bevacizumab and etanercept in healthy volunteers and patients. Biochem. Pharmacol. 2017, 145, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Zalevsky, J.; Chamberlain, A.K.; Horton, H.M.; Karki, S.; Leung, I.W.L.; Sproule, T.J.; A Lazar, G.; Roopenian, D.C.; Desjarlais, J.R. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 2010, 28, 157–159. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Wynne, C.; Xu, C.; Gan, Y.; Wang, C.; Thomas, B.E.; Yu, J.C.; Li, S.; Zhang, L. A Global Phase I Clinical Study Comparing the Safety and Phar-macokinetics of Proposed Biosimilar BAT1706 and Bevacizumab (Avastin®) in Healthy Male Subjects. BioDrugs 2019, 33, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Hettema, W.; Wynne, C.; Lang, B.; Altendorfer, M.; Czeloth, N.; Lohmann, R.; Athalye, S.; Schliephake, D. A randomized, single-blind, Phase I trial (INVICTAN-1) assessing the bioequivalence and safety of BI 695502, a bevacizumab biosimilar candidate, in healthy subjects. Expert Opin. Investig. Drugs 2017, 26, 889–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hummel, M.; Bosje, T.; Shaw, A.; Liu, M.S.; Barve, A.; Kothekar, M.; Socinski, M.A. A pharmacokinetics study of proposed bevacizumab bio-similar MYL-1402O vs EU-bevacizumab and US-bevacizumab. J. Cancer Res. Clin. Oncol. 2022, 148, 487–496. [Google Scholar] [CrossRef]
- Demarchi, M.; Coliat, P.; Barthelemy, P.; Schott, R.; BenAbdelghani, M.; Kim, M.; Hii, J.C.S.; Feyaerts, P.; Ang, F.R.X.; Derde, M.P.; et al. A randomized phase I study comparing the pharmacokinetics of a bevacizumab (HD204) biosimilar to European Union-and United States of America-sourced bevaci-zumab. Handelsman DJ, éditeur. PLoS ONE 2021, 16, e0248222. [Google Scholar] [CrossRef]
- Sinn, A.; García-Alvarado, F.; Gonzalez, V.; Huerga, C.; Bullo, F. A randomized, double blind, single dose, comparative study of the pharmacokinetics, safety and immunogenicity of MB02 (bevacizumab biosimilar) and reference bevacizumab in healthy male volunteers. Br. J. Clin. Pharmacol. 2022, 88, 1063–1073. [Google Scholar] [CrossRef]
- Shin, D.; Lee, Y.J.; Choi, J.; Lee, D.; Park, M.; Petkova, M. A phase I, randomized, single-dose pharmacokinetic study comparing sb8 (bevacizumab biosimilar) with reference bevacizumab in healthy volunteers. Cancer Chemother. Pharmacol. 2020, 86, 567–575. [Google Scholar] [CrossRef]
- Romera, A.; Peredpaya, S.; Shparyk, Y.; Bondarenko, I.; Bariani, G.M.; Abdalla, K.C.; Roca, E.; Franke, F.; Cruz, F.M.; Ramesh, A.; et al. Bevacizumab biosimilar BEVZ92 versus reference bevacizumab in combination with FOLFOX or FOLFIRI as first-line treatment for metastatic colorectal cancer: A multicentre, open-label, randomised controlled trial. Lancet Gastroenterol. Hepatol. 2018, 3, 845–855. [Google Scholar] [CrossRef]
- Niederalt, C.; Kuepfer, L.; Solodenko, J.; Eissing, T.; Siegmund, H.-U.; Block, M.; Willmann, S.; Lippert, J. A generic whole body physiologically based pharmacokinetic model for therapeutic proteins in PK-Sim. J. Pharmacokinet. Pharmacodyn. 2018, 45, 235–257. [Google Scholar] [CrossRef] [Green Version]
- Davies, B.; Morris, T. Physiological Parameters in Laboratory Animals and Humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Edginton, A.N.; Schmitt, W.; Willmann, S. Development and Evaluation of a Generic Physiologically Based Pharmacokinetic Model for Children. Clin. Pharmacokinet. 2006, 45, 1013–1034. [Google Scholar] [CrossRef]
- Mordenti, J. Man versus Beast: Pharmacokinetic Scaling in Mammals. J. Pharm. Sci. 1986, 75, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Li, J.; Xu, X.; Wang, C.; Cheng, Y. IBI302, a promising candidate for AMD treatment, targeting both the VEGF and com-plement system with high binding affinity in vitro and effective targeting of the ocular tissue in healthy rhesus monkeys. Exp. Eye Res. 2016, 145, 352–358. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Martin, J.; Ruan, Q.; Rafique, A.; Rosconi, M.P.; Shi, E.; Pyles, E.A.; Yancopoulos, G.D.; Stahl, N.; Wiegand, S.J. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis 2012, 15, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Neuber, T.; Frese, K.; Jaehrling, J.; Jäger, S.; Daubert, D.; Felderer, K.; Hohne, A.; Pabst, S.; Ostendorp, R.; Brocks, B.; et al. Characterization and screening of IgG binding to the ne-onatal Fc receptor. mAbs 2014, 6, 928–942. [Google Scholar] [CrossRef] [Green Version]
- Nowacka, A.; Smuczyński, W.; Rość, D.; Woźniak-Dąbrowska, K.; Śniegocki, M. Serum VEGF-A concentrations in patients with central nervous system (CNS) tumors. PLoS ONE 2018, 13, e0192395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanini, M.O.; Wu, F.T.H.; Mac Gabhann, F.; Popel, A.S. Increase of plasma VEGF after intravenous administration of bevacizumab is predicted by a pharmacokinetic model. Cancer Res. 2010, 70, 9886–9894. [Google Scholar] [CrossRef] [Green Version]
- Stefanini, M.O.; Wu, F.T.; Mac Gabhann, F.; Popel, A.S. A compartment model of VEGF distribution in blood, healthy and diseased tissues. BMC Syst. Biol. 2008, 2, 77. [Google Scholar] [CrossRef] [Green Version]
- Kut, C.; Mac Gabhann, F.; Popel, A.S. Where is VEGF in the body? A meta-analysis of VEGF distribution in cancer. Br. J. Cancer 2007, 97, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Zajkowska, M.; Lubowicka, E.; Fiedorowicz, W.; Szmitkowski, M.; Jamiołkowski, J.; Ławicki, S. Human Plasma Levels of VEGF-A, VEGF-C, VEGF-D, their Soluble Receptor-VEGFR-2 and Applicability of these Parameters as Tumor Markers in the Diagnostics of Breast Cancer. Pathol. Oncol. Res. 2019, 25, 1477–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.; Schneckener, S.; Ludewig, B.; Kuepfer, L.; Lippert, J. Using Expression Data for Quantification of Active Processes in Physiologically Based Pharmacokinetic Modeling. Drug Metab. Dispos. 2012, 40, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; Helmberg, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2005, 33 (Suppl. S1), D39–D45. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; Helmberg, W.; et al. Database resources of the National Center for Biotechnology. Nucleic Acids Res. 2003, 31, 28–33. [Google Scholar] [CrossRef]
- Liu, Y.N.; Huang, J.; Guo, C.; Yang, S.; Ye, L.; Wu, S.T.; Zhang, X.F.; Yang, X.Y.; Han, C.C.; Pei, Q.; et al. A randomized, double-blind, single-dose study to evaluate the bio-similarity of QL1101 with bevacizumab in healthy male subjects. Cancer Chemother. Pharmacol. 2020, 85, 555–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maharaj, A.R.; Wu, H.; Hornik, C.P.; Arrieta, A.; James, L.; Bhatt-Mehta, V.; Bradley, J.; Muller, W.J.; Al-Uzri, A.; Downes, K.J.; et al. Use of normalized prediction distribution errors for assessing population physiologically-based pharmacokinetic model adequacy. J. Pharmacokinet. Pharmacodyn. 2020, 47, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Yoshida, K.; Nakada, T.; Kojima, K.; Ogasawara, A.; Nakamaru, Y.; Yamazaki, H. Prediction of Human Distribution Volumes of Compounds in Various Elimination Phases Using Physiologically Based Pharmacokinetic Modeling and Experimental Phar-macokinetics in Animals. Drug Metab. Dispos. Biol. Fate Chem. 2019, 47, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Abduljalil, K.; Cain, T.; Humphries, H.; Rostami-Hodjegan, A. Deciding on Success Criteria for Predictability of Pharmacokinetic Parameters from In Vitro Studies: An Analysis Based on In Vivo Observations. Drug Metab. Dispos. 2014, 42, 1478–1484. [Google Scholar] [CrossRef]
- Shebley, M.; Sandhu, P.; Riedmaier, A.E.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; de Zwart, L.; et al. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef] [Green Version]
- Tranter, E.; Peters, G.; Boyce, M.; Warrington, S. Giving monoclonal antibodies to healthy volunteers in phase 1 trials: Is it safe? Br. J. Clin. Pharmacol. 2013, 76, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Baxter, L.T.; Zhu, H.; Mackensen, D.G.; Butler, W.F.; Jain, R.K. Biodistribution of monoclonal antibodies: Scale-up from mouse to human using a physiologically based pharmacokinetic model. Cancer Res. 1995, 55, 4611–4622. [Google Scholar] [PubMed]
- Davda, J.P.; Jain, M.; Batra, S.K.; Gwilt, P.R.; Robinson, D.H. A physiologically based pharmacokinetic (PBPK) model to characterize and predict the disposition of monoclonal antibody CC49 and its single chain Fv constructs. Int. Immunopharmacol. 2008, 8, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K.; Betts, A.M. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J. Pharmacokinet. Pharmacodyn. 2012, 39, 67–86. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Balthasar, J.P. Evaluation of a Catenary PBPK Model for Predicting the In Vivo Disposition of mAbs Engineered for High-Affinity Binding to FcRn. AAPS J. 2012, 14, 850–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glassman, P.M.; Chen, Y.; Balthasar, J.P. Scale-up of a physiologically-based pharmacokinetic model to predict the disposition of monoclonal antibodies in monkeys. J. Pharmacokinet. Pharmacodyn. 2015, 42, 527–540. [Google Scholar] [CrossRef]
- Papachristos, A.; Karatza, E.; Kalofonos, H.; Sivolapenko, G. Pharmacogenetics in Model-Based Optimization of Bevacizumab Therapy for Metastatic Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 3753. [Google Scholar] [CrossRef]
- Panoilia, E.; Schindler, E.; Samantas, E.; Aravantinos, G.; Kalofonos, H.P.; Christodoulou, C.; Patrinos, G.P.; Friberg, L.E.; Sivolapenko, G. A pharmacokinetic binding model for bevacizumab and VEGF165 in colorectal cancer patients. Cancer Chemother. Pharmacol. 2015, 75, 791–803. [Google Scholar] [CrossRef]
- Yen, P.; Finley, S.D.; Engel-Stefanini, M.O.; Popel, A.S. A Two-Compartment Model of VEGF Distribution in the Mouse. PLoS ONE 2011, 6, e27514. [Google Scholar] [CrossRef]
- Simón-Yarza, T.; Formiga, F.R.; Tamayo, E.; Pelacho, B.; Prosper, F.; Blanco-Prieto, M.J. Vascular Endothelial Growth Factor-Delivery Systems for Cardiac Repair: An Overview. Theranostics 2012, 2, 541–552. [Google Scholar] [CrossRef]
- Kleinheinz, J.; Jung, S.; Wermker, K.; Fischer, C.; Joos, U. Release kinetics of VEGF165 from a collagen matrix and structural matrix changes in a circulation model. Head Face Med. 2010, 6, 17. [Google Scholar] [CrossRef] [Green Version]
- Han, K.; Peyret, T.; Marchand, M.; Quartino, A.; Gosselin, N.H.; Girish, S.; Allison, D.E.; Jin, J. Population pharmacokinetics of bevacizumab in cancer patients with external validation. Cancer Chemother. Pharmacol. 2016, 78, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lledo-Garcia, R.; Dixon, K.; Shock, A.; Oliver, R. Pharmacokinetic-pharmacodynamic modelling of the anti-FcRn monoclonal antibody rozanolixizumab: Translation from preclinical stages to the clinic. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Modules, Philosophy, and Building Blocks [Internet]. Available online: https://docs.open-systems-pharmacology.org/open-systems-pharmacology-suite/modules-philsophy-building-blocks (accessed on 18 July 2023).
- Khot, A.; Tibbitts, J.; Rock, D.; Shah, D.K. Development of a Translational Physiologically Based Pharmacokinetic Model for Anti-body-Drug Conjugates: A Case Study with T-DM1. AAPS J. 2017, 19, 1715–1734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xiang, Y.; Wang, L.; Wang, F.; Li, G.; Zhuang, X. Translational pharmacokinetics of a novel bispecific antibody against Ebola virus (MBS77E) from animal to human by PBPK modeling & simulation. Int. J. Pharm. 2022, 626, 122160. [Google Scholar] [CrossRef]
- Betts, A.; Haddish-Berhane, N.; Shah, D.K.; van der Graaf, P.H.; Barletta, F.; King, L.; Root, A.; Hoper, A.; Chen, X. A Translational Quantitative Systems Pharmacology Model for CD3 Bispecific Molecules: Application to Quantify T Cell-Mediated Tumor Cell Killing by P-Cadherin LP DART®. AAPS J. 2019, 21, 66. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, G.; Wang, X.; Agrawal, S.; Gupta, M.; Roy, A.; Feng, Y. Model-Based Population Pharmacokinetic Analysis of Nivolumab in Patients With Solid Tumors. CPT Pharmacomet. Syst. Pharmacol. 2016, 6, 58–66. [Google Scholar] [CrossRef]
- Li, H.; Yu, J.; Liu, C.; Liu, J.; Subramaniam, S.; Zhao, H.; Blumenthal, G.M.; Turner, D.C.; Li, C.; Ahamadi, M.; et al. Time dependent pharmacokinetics of pembrolizumab in patients with solid tumor and its correlation with best overall response. J. Pharmacokinet. Pharmacodyn. 2017, 44, 403–414. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Number of Subjects | Bevacizumab Dose | Study Description |
|---|---|---|---|
| Cynomolgus monkey | 2 (bevacizumab) 2 (xtend-bevacizumab) | 4 mg/kg Single dose | PK study in cynomolgus monkey, Mean profiles Zalevksy et al., 2010 [13] |
| HV | 5 | 0.5 mg/kg Single dose | PK study of bevacizumab, Individual profiles in Chinese HV Li et al., 2017 [12] |
| HV | 43 | 1 mg/kg Single dose | Phase I study in HV, Mean (sd) profiles Wu et al., 2019 [14] |
| HV | 30 | 1 mg/kg Single dose | Phase I study in HV, mean profiles Hettema et al., 2017 [15] |
| HV | 37 | 1 mg/kg Single dose | Phase I study in HV, Mean profiles Hummel et al., 2022 [16] |
| HV | 39 | 1 mg/kg Single dose | Phase I study in HV, mean profiles Demarchi et al., 2021 [17] |
| HV | 38 | 3 mg/kg Single dose | Phase I study in HV, Mean profiles Sinn et al., 2022 [18] |
| HV | 40 | 3 mg/kg Single Dose | Phase I study in HV, Mean (sd) profiles Shin et al., 2020 [19] |
| Adult cancer patients | 5 subjects per dose | 0.3, 1, 3, 10 mg/kg Repeated dose | Phase I study in cancer patients, Mean profiles Gordon et al., 2001 [11] |
| Adult cancer patients | 61 | 5 mg/kg Repeated dose | Biosimilar study in patients, Mean profiles Romera et al., 2018 [20] |
| Parameter | Cynomolgus Monkey | Healthy Volunteers | Adult Cancer Patients | |||
|---|---|---|---|---|---|---|
| Bevacizumab | Xtend-Bevacizumab | A Priori Predicted | Refined | A Priori Predicted | Refined | |
| KD-FcRn (nmol/L) | 450 | 41 | 900 | 940 | 940 | 940 |
| KD,target a (nmol/L) | 0.032 [25] | 0.032 [25] | 0.058 [26] | 0.058 [26] | 0.058 [26] | 0.058 [26] |
| koff a (day−1) | 3.5 [25] | 3.5 [25] | 3.5 [26] | 2.7 [26] | 2.7 [26] | 2.7 [26] |
| VEGF-A (nmol/L) | 0 to 3 (min-max) | 0 to 3 (min-max) | 1.5 | 0.3 | 0.6 to 3 (min-max) | 3.86 |
| kdeg (day−1) | - | - | - | 0.18 | 0.18 | 0.18 |
| kint (day−1) | - | - | - | 0.043 | 0.043 | 0.043 |
| Population | Dose (mg/kg) | AFE | AAFE |
|---|---|---|---|
| Adult Cancer Patients | 0.3 | 0.89 | 1.38 |
| 1 | 0.75 | 1.39 | |
| 3 | 0.86 | 1.27 | |
| 5 | 1.04 | 1.17 | |
| 10 | 0.85 | 1.23 | |
| Healthy Volunteers | 0.5 | 1.94 | 1.94 |
| 1 | 1.05 | 1.30 | |
| 3 | 0.72 | 1.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasquiers, B.; Benamara, S.; Felices, M.; Ternant, D.; Declèves, X.; Puszkiel, A. Translation of Monoclonal Antibodies Pharmacokinetics from Animal to Human Using Physiologically Based Modeling in Open Systems Pharmacology (OSP) Suite: A Retrospective Analysis of Bevacizumab. Pharmaceutics 2023, 15, 2129. https://doi.org/10.3390/pharmaceutics15082129
Pasquiers B, Benamara S, Felices M, Ternant D, Declèves X, Puszkiel A. Translation of Monoclonal Antibodies Pharmacokinetics from Animal to Human Using Physiologically Based Modeling in Open Systems Pharmacology (OSP) Suite: A Retrospective Analysis of Bevacizumab. Pharmaceutics. 2023; 15(8):2129. https://doi.org/10.3390/pharmaceutics15082129
Chicago/Turabian StylePasquiers, Blaise, Salih Benamara, Mathieu Felices, David Ternant, Xavier Declèves, and Alicja Puszkiel. 2023. "Translation of Monoclonal Antibodies Pharmacokinetics from Animal to Human Using Physiologically Based Modeling in Open Systems Pharmacology (OSP) Suite: A Retrospective Analysis of Bevacizumab" Pharmaceutics 15, no. 8: 2129. https://doi.org/10.3390/pharmaceutics15082129