
Targeting EGFR/PI3K/AKT/mTOR Signaling in Hepatocellular Carcinoma

Abstract
:1. Introduction
2. Role of EGFR/PI3K/AKT/mTOR Signaling Pathway in HCC
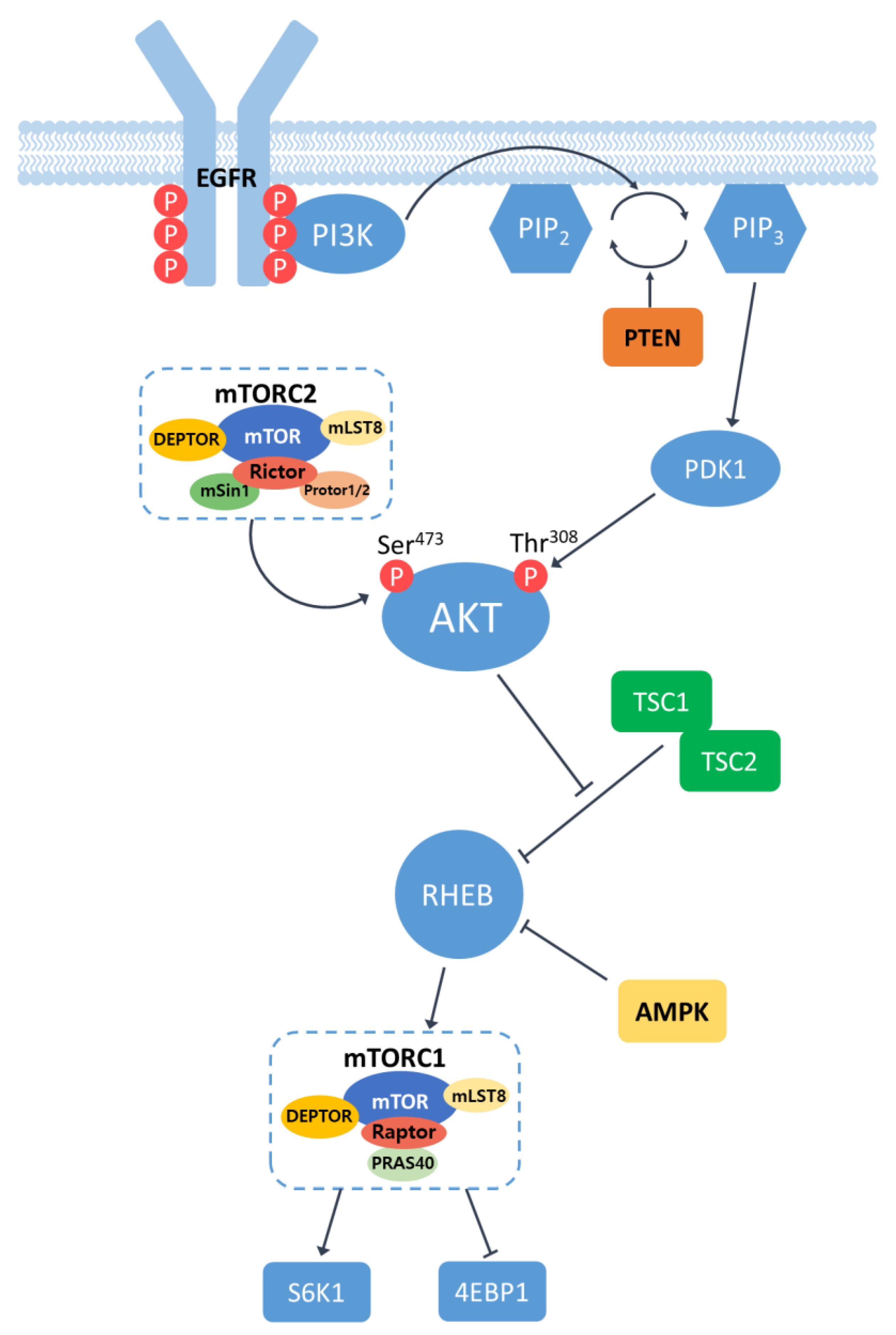
2.1. Overview of EGFR/PI3K/AKT/mTOR Signaling Pathway
2.2. Activation of EGFR/PI3K/AKT/mTOR Signaling Pathway in HCC
3. In Vitro Studies Investigating EGFR/PI3K/AKT/mTOR Signaling in HCC Cell Lines
3.1. Tumorigenic Roles of EGFR/PI3K/AKT/mTOR Signaling in HCC Cells
3.2. Anti-Tumor Effects of Targeting EGFR/PI3K/AKT/mTOR Pathway in HCC Cells
3.3. Targeting EGFR/PI3K/AKT/mTOR Signaling on Sorafenib-Resistant HCC Cells
4. Animal Studies Investigating EGFR/PI3K/AKT/mTOR Signaling in HCC
4.1. Animal Models for HCC Induced by Activated EGFR/PI3K/AKT/mTOR Signaling
4.2. Preclinical Animal Studies Targeting EGFR/PI3K/AKT/mTOR Signaling in HCC
5. Clinical Studies Targeting EGFR/PI3K/AKT/mTOR Signaling
6. Perspectives and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef]
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 2020, 72, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008, 48, 1312–1327. [Google Scholar] [CrossRef] [Green Version]
- Dimri, M.; Satyanarayana, A. Molecular Signaling Pathways and Therapeutic Targets in Hepatocellular Carcinoma. Cancers 2020, 12, 491. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.; Ro, S.W. MAPK/ERK Signaling Pathway in Hepatocellular Carcinoma. Cancers 2021, 13, 3026. [Google Scholar] [CrossRef]
- Park, H.; Park, H.; Baek, J.; Moon, H.; Ro, S.W. Target Therapy for Hepatocellular Carcinoma: Beyond Receptor Tyrosine Kinase Inhibitors and Immune Checkpoint Inhibitors. Biology 2022, 11, 585. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Moten, A.; Lin, H.K. Akt: A new activation mechanism. Cell Res. 2014, 24, 785–786. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Sun, E.J.; Wankell, M.; Palamuthusingam, P.; McFarlane, C.; Hebbard, L. Targeting the PI3K/Akt/mTOR Pathway in Hepatocellular Carcinoma. Biomedicines 2021, 9, 1639. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/mTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front. Oncol. 2022, 12, 819128. [Google Scholar] [CrossRef]
- Yu, L.; Wei, J.; Liu, P. Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin. Cancer Biol. 2022, 85, 69–94. [Google Scholar] [CrossRef]
- Harada, K.; Shiota, G.; Kawasaki, H. Transforming growth factor-alpha and epidermal growth factor receptor in chronic liver disease and hepatocellular carcinoma. Liver 1999, 19, 318–325. [Google Scholar] [CrossRef]
- Ito, Y.; Takeda, T.; Sakon, M.; Tsujimoto, M.; Higashiyama, S.; Noda, K.; Miyoshi, E.; Monden, M.; Matsuura, N. Expression and clinical significance of erb-B receptor family in hepatocellular carcinoma. Br. J. Cancer 2001, 84, 1377–1383. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Huang, Y.; Li, J.; Wang, Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med. Oncol. 2010, 27, 255–261. [Google Scholar] [CrossRef]
- Sieghart, W.; Fuereder, T.; Schmid, K.; Cejka, D.; Werzowa, J.; Wrba, F.; Wang, X.; Gruber, D.; Rasoul-Rockenschaub, S.; Peck-Radosavljevic, M.; et al. Mammalian target of rapamycin pathway activity in hepatocellular carcinomas of patients undergoing liver transplantation. Transplantation 2007, 83, 425–432. [Google Scholar] [CrossRef]
- Villanueva, A.; Chiang, D.Y.; Newell, P.; Peix, J.; Thung, S.; Alsinet, C.; Tovar, V.; Roayaie, S.; Minguez, B.; Sole, M.; et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 2008, 135, 1972–1983.e11. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Li, Z.; Cheng, Q.; Wang, Y.; Qian, L.; Gao, J.; Zhu, J.Y. Genetic alterations and expression of PTEN and its relationship with cancer stem cell markers to investigate pathogenesis and to evaluate prognosis in hepatocellular carcinoma. J. Clin. Pathol. 2019, 72, 588–596. [Google Scholar] [CrossRef]
- Kawamura, N.; Nagai, H.; Bando, K.; Koyama, M.; Matsumoto, S.; Tajiri, T.; Onda, M.; Fujimoto, J.; Ueki, T.; Konishi, N.; et al. PTEN/MMAC1 mutations in hepatocellular carcinomas: Somatic inactivation of both alleles in tumors. Jpn. J. Cancer Res. 1999, 90, 413–418. [Google Scholar] [CrossRef]
- Yao, Y.J.; Ping, X.L.; Zhang, H.; Chen, F.F.; Lee, P.K.; Ahsan, H.; Chen, C.J.; Lee, P.H.; Peacocke, M.; Santella, R.M.; et al. PTEN/MMAC1 mutations in hepatocellular carcinomas. Oncogene 1999, 18, 3181–3185. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, Y.; Hoon, D.S.; Yamada, T.; Umeshita, K.; Gotoh, M.; Sakon, M.; Nishisho, I.; Monden, M. PTEN/MMAC1 mutation and frequent loss of heterozygosity identified in chromosome 10q in a subset of hepatocellular carcinomas. Jpn. J. Cancer Res. 2000, 91, 287–292. [Google Scholar] [CrossRef]
- Ho, D.W.H.; Chan, L.K.; Chiu, Y.T.; Xu, I.M.J.; Poon, R.T.P.; Cheung, T.T.; Tang, C.N.; Tang, V.W.L.; Lo, I.L.O.; Lam, P.W.Y.; et al. TSC1/2 mutations define a molecular subset of HCC with aggressive behaviour and treatment implication. Gut 2017, 66, 1496–1506. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liao, Y.; He, W.; Zhang, H.; Zuo, D.; Liu, W.; Yang, Z.; Qiu, J.; Yuan, Y.; Li, K.; et al. Elafin promotes tumour metastasis and attenuates the anti-metastatic effects of erlotinib via binding to EGFR in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 113. [Google Scholar] [CrossRef]
- Li, K.S.; Zhu, X.D.; Liu, H.D.; Zhang, S.Z.; Li, X.L.; Xiao, N.; Liu, X.F.; Xu, B.; Lei, M.; Zhang, Y.Y.; et al. NT5DC2 promotes tumor cell proliferation by stabilizing EGFR in hepatocellular carcinoma. Cell Death Dis. 2020, 11, 335. [Google Scholar] [CrossRef]
- Song, J.; Liu, Y.; Liu, F.; Zhang, L.; Li, G.; Yuan, C.; Yu, C.; Lu, X.; Liu, Q.; Chen, X.; et al. The 14-3-3σ protein promotes HCC anoikis resistance by inhibiting EGFR degradation and thereby activating the EGFR-dependent ERK1/2 signaling pathway. Theranostics 2021, 11, 996–1015. [Google Scholar] [CrossRef]
- Zheng, H.; Yang, Y.; Hong, Y.G.; Wang, M.C.; Yuan, S.X.; Wang, Z.G.; Bi, F.R.; Hao, L.Q.; Yan, H.L.; Zhou, W.P. Tropomodulin 3 modulates EGFR-PI3K-AKT signaling to drive hepatocellular carcinoma metastasis. Mol. Carcinog. 2019, 58, 1897–1907. [Google Scholar] [CrossRef]
- Chen, H.; Wong, C.C.; Liu, D.; Go, M.Y.Y.; Wu, B.; Peng, S.; Kuang, M.; Wong, N.; Yu, J. APLN promotes hepatocellular carcinoma through activating PI3K/Akt pathway and is a druggable target. Theranostics 2019, 9, 5246–5260. [Google Scholar] [CrossRef]
- Yu, Y.; Zhao, D.; Li, K.; Cai, Y.; Xu, P.; Li, R.; Li, J.; Chen, X.; Chen, P.; Cui, G. E2F1 mediated DDX11 transcriptional activation promotes hepatocellular carcinoma progression through PI3K/AKT/mTOR pathway. Cell Death Dis. 2020, 11, 273. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhu, M.; Wang, Q.; Hou, Y.; Li, L.; Weng, H.; Zhao, Y.; Chen, D.; Guo, J.; Ding, H.; et al. Publisher Correction: Alpha-fetoprotein inhibits autophagy to promote malignant behaviour in hepatocellular carcinoma cells by activating PI3K/AKT/mTOR signalling. Cell Death Dis. 2019, 10, 214. [Google Scholar] [CrossRef]
- Huang, L.; Zhao, C.; Sun, K.; Yang, D.; Yan, L.; Luo, D.; He, J.; Hu, X.; Wang, R.; Shen, X.; et al. Downregulation of CLDN6 inhibits cell proliferation, migration, and invasion via regulating EGFR/AKT/mTOR signalling pathway in hepatocellular carcinoma. Cell Biochem. Funct. 2020, 38, 541–548. [Google Scholar] [CrossRef]
- Lin, H.; Huang, Z.P.; Liu, J.; Qiu, Y.; Tao, Y.P.; Wang, M.C.; Yao, H.; Hou, K.Z.; Gu, F.M.; Xu, X.F. MiR-494-3p promotes PI3K/AKT pathway hyperactivation and human hepatocellular carcinoma progression by targeting PTEN. Sci. Rep. 2018, 8, 10461. [Google Scholar] [CrossRef] [Green Version]
- Dolgormaa, G.; Harimoto, N.; Ishii, N.; Yamanaka, T.; Hagiwara, K.; Tsukagoshi, M.; Igarashi, T.; Watanabe, A.; Kubo, N.; Araki, K.; et al. Mac-2-binding protein glycan isomer enhances the aggressiveness of hepatocellular carcinoma by activating mTOR signaling. Br. J. Cancer 2020, 123, 1145–1153. [Google Scholar] [CrossRef]
- Wang, X.; Zeng, J.; Wang, L.; Zhang, X.; Liu, Z.; Zhang, H.; Dong, J. Overexpression of microRNA-133b is associated with the increased survival of patients with hepatocellular carcinoma after curative hepatectomy: Involvement of the EGFR/PI3K/Akt/mTOR signaling pathway. Oncol. Rep. 2017, 38, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhuang, Y.; Zhang, J.; Chen, M.; Wu, S. METTL14 Inhibits Hepatocellular Carcinoma Metastasis Through Regulating EGFR/PI3K/AKT Signaling Pathway in an m6A-Dependent Manner. Cancer Manag. Res. 2020, 12, 13173–13184. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Liao, D.; Zhang, M.; Zeng, C.; Li, X.; Zhang, R.; Ma, H.; Kang, T. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019, 442, 252–261. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, H.; Li, M.; Wu, H.; Guo, Y.; Chen, J.; Shan, J.; Chen, X.; Shen, J.; Ma, Q.; et al. KIAA1199 promotes sorafenib tolerance and the metastasis of hepatocellular carcinoma by activating the EGF/EGFR-dependent epithelial-mesenchymal transition program. Cancer Lett. 2019, 454, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Liu, J.; Wang, Y.; Dong, J. Co-administration of MDR1 and BCRP or EGFR/PI3K inhibitors overcomes lenvatinib resistance in hepatocellular carcinoma. Front. Oncol. 2022, 12, 944537. [Google Scholar] [CrossRef]
- Buontempo, F.; Ersahin, T.; Missiroli, S.; Senturk, S.; Etro, D.; Ozturk, M.; Capitani, S.; Cetin-Atalay, R.; Neri, M.L. Inhibition of Akt signaling in hepatoma cells induces apoptotic cell death independent of Akt activation status. Investig. New Drugs 2011, 29, 1303–1313. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.; Ai, J.; Fan, Y.; Gao, J.; Liu, W.; Feng, Q.; Liao, W.; Wu, L. NCAPG Promotes The Proliferation Of Hepatocellular Carcinoma Through PI3K/AKT Signaling. Onco Targets Ther. 2019, 12, 8537–8552. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Mayerle, J.; Ziesch, A.; Reiter, F.P.; Gerbes, A.L.; De Toni, E.N. The PI3K inhibitor copanlisib synergizes with sorafenib to induce cell death in hepatocellular carcinoma. Cell Death Discov. 2019, 5, 86. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Luo, Y.; Li, S.; Hong, M.; Wang, Q.; Chi, X.; Yang, C. ISL Induces Apoptosis and Autophagy in Hepatocellular Carcinoma via Downregulation of PI3K/AKT/mTOR Pathway in vivo and in vitro. Drug Des. Dev. Ther. 2020, 14, 4363–4376. [Google Scholar] [CrossRef]
- Ou, D.L.; Lee, B.S.; Lin, L.I.; Liou, J.Y.; Liao, S.C.; Hsu, C.; Cheng, A.L. Vertical blockade of the IGFR-PI3K/Akt/mTOR pathway for the treatment of hepatocellular carcinoma: The role of survivin. Mol. Cancer 2014, 13, 2. [Google Scholar] [CrossRef] [Green Version]
- Grabinski, N.; Ewald, F.; Hofmann, B.T.; Staufer, K.; Schumacher, U.; Nashan, B.; Jücker, M. Combined targeting of AKT and mTOR synergistically inhibits proliferation of hepatocellular carcinoma cells. Mol. Cancer 2012, 11, 85. [Google Scholar] [CrossRef] [Green Version]
- Kahraman, D.C.; Kahraman, T.; Cetin-Atalay, R. Targeting PI3K/Akt/mTOR Pathway Identifies Differential Expression and Functional Role of IL8 in Liver Cancer Stem Cell Enrichment. Mol. Cancer Ther. 2019, 18, 2146–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Song, J.; Zhang, D.; Wu, F.; Tu, J.; Ji, J. Oxysophocarpine suppresses FGFR1-overexpressed hepatocellular carcinoma growth and sensitizes the therapeutic effect of lenvatinib. Life Sci. 2021, 264, 118642. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhang, Y.; Zhang, L.; Ren, X.; Huber-Keener, K.J.; Liu, X.; Zhou, L.; Liao, J.; Keihack, H.; Yan, L.; et al. MK-2206, a novel allosteric inhibitor of Akt, synergizes with gefitinib against malignant glioma via modulating both autophagy and apoptosis. Mol. Cancer Ther. 2012, 11, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.S.; et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Yu, X.; Lin, X.; He, S. Inhibition of mTOR sensitizes breast cancer stem cells to radiation-induced repression of self-renewal through the regulation of MnSOD and Akt. Int. J. Mol. Med. 2016, 37, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Hernandez-Gea, V. Hepatocellular carcinoma: Reasons for phase III failure and novel perspectives on trial design. Clin. Cancer Res. 2014, 20, 2072–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, L.; Liu, L.; Yang, S.; Ren, J.; Lai, P.B.S.; Chen, G.G. New insights into sorafenib resistance in hepatocellular carcinoma: Responsible mechanisms and promising strategies. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 564–570. [Google Scholar] [CrossRef]
- Samarin, J.; Laketa, V.; Malz, M.; Roessler, S.; Stein, I.; Horwitz, E.; Singer, S.; Dimou, E.; Cigliano, A.; Bissinger, M.; et al. PI3K/AKT/mTOR-dependent stabilization of oncogenic far-upstream element binding proteins in hepatocellular carcinoma cells. Hepatology 2016, 63, 813–826. [Google Scholar] [CrossRef]
- Gedaly, R.; Angulo, P.; Chen, C.; Creasy, K.T.; Spear, B.T.; Hundley, J.; Daily, M.F.; Shah, M.; Evers, B.M. The role of PI3K/mTOR inhibition in combination with sorafenib in hepatocellular carcinoma treatment. Anticancer Res. 2012, 32, 2531–2536. [Google Scholar]
- Schneider, P.; Schön, M.; Pletz, N.; Seitz, C.S.; Liu, N.; Ziegelbauer, K.; Zachmann, K.; Emmert, S.; Schön, M.P. The novel PI3 kinase inhibitor, BAY 80-6946, impairs melanoma growth in vivo and in vitro. Exp. Dermatol. 2014, 23, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Dai, N.; Ye, R.; He, Q.; Guo, P.; Chen, H.; Zhang, Q. Capsaicin and sorafenib combination treatment exerts synergistic anti-hepatocellular carcinoma activity by suppressing EGFR and PI3K/Akt/mTOR signaling. Oncol. Rep. 2018, 40, 3235–3248. [Google Scholar] [CrossRef] [PubMed]
- Kenerson, H.L.; Yeh, M.M.; Kazami, M.; Jiang, X.; Riehle, K.J.; McIntyre, R.L.; Park, J.O.; Kwon, S.; Campbell, J.S.; Yeung, R.S. Akt and mTORC1 have different roles during liver tumorigenesis in mice. Gastroenterology 2013, 144, 1055–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Hu, J.; Cao, H.; Pilo, M.G.; Cigliano, A.; Shao, Z.; Xu, M.; Ribback, S.; Dombrowski, F.; Calvisi, D.F.; et al. Loss of Pten synergizes with c-Met to promote hepatocellular carcinoma development via mTORC2 pathway. Exp. Mol. Med. 2018, 50, e417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Che, L.; Li, L.; Pilo, M.G.; Cigliano, A.; Ribback, S.; Li, X.; Latte, G.; Mela, M.; Evert, M.; et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci. Rep. 2016, 6, 20484. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.; Wang, C.; Mattu, S.; Destefanis, G.; Ladu, S.; Delogu, S.; Armbruster, J.; Fan, L.; Lee, S.A.; Jiang, L.; et al. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology 2012, 55, 833–845. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.; Yecies, J.L.; Zhang, H.H.; Howell, J.J.; Nicholatos, J.; Harputlugil, E.; Bronson, R.T.; Kwiatkowski, D.J.; Manning, B.D. Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci. Signal. 2012, 5, ra24. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.D.; Fang, L.; Yu, H.Q.; Zhang, J.; Lin, X.T.; Liu, X.Y.; Wu, D.; Li, G.X.; Huang, D.; Zhang, Y.J.; et al. p53 haploinsufficiency and increased mTOR signalling define a subset of aggressive hepatocellular carcinoma. J. Hepatol. 2021, 74, 96–108. [Google Scholar] [CrossRef]
- Chen, X.; Calvisi, D.F. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am. J. Pathol. 2014, 184, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Ju, H.L.; Han, K.H.; Lee, J.D.; Ro, S.W. Transgenic mouse models generated by hydrodynamic transfection for genetic studies of liver cancer and preclinical testing of anti-cancer therapy. Int. J. Cancer 2016, 138, 1601–1608. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.; Ro, S.W.; Seo, S.H.; Jeon, Y.; Moon, H.; Kim, D.Y.; Kim, S.U. Genetically Engineered Mouse Models for Liver Cancer. Cancers 2019, 12, 14. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.; Park, H.; Chae, M.J.; Choi, H.J.; Kim, D.Y.; Ro, S.W. Activated TAZ induces liver cancer in collaboration with EGFR/HER2 signaling pathways. BMC Cancer 2022, 22, 423. [Google Scholar] [CrossRef]
- Wang, R.Y.; Chen, L.; Chen, H.Y.; Hu, L.; Li, L.; Sun, H.Y.; Jiang, F.; Zhao, J.; Liu, G.M.; Tang, J.; et al. MUC15 inhibits dimerization of EGFR and PI3K-AKT signaling and is associated with aggressive hepatocellular carcinomas in patients. Gastroenterology 2013, 145, 1436–1448.e12. [Google Scholar] [CrossRef]
- Liu, F.; Pan, Z.; Zhang, J.; Ni, J.; Wang, C.; Wang, Z.; Gu, F.; Dong, W.; Zhou, W.; Liu, H. Overexpression of RHEB is associated with metastasis and poor prognosis in hepatocellular carcinoma. Oncol. Lett. 2018, 15, 3838–3845. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Xu, X.; Liu, M.; Qin, X.; Wu, Q.; Ding, H.; Zhao, Q. DZW-310, a novel phosphoinositide 3-kinase inhibitor, attenuates the angiogenesis and growth of hepatocellular carcinoma cells via PI3K/AKT/mTOR axis. Biochem. Pharmacol. 2022, 201, 115093. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Zhang, J.; Liu, D.; Bao, Y.; Chen, T.; Tang, T.; Lin, J.; Luo, Y.; Jin, Y.; et al. Indole hydrazide compound ZJQ-24 inhibits angiogenesis and induces apoptosis cell death through abrogation of AKT/mTOR pathway in hepatocellular carcinoma. Cell Death Dis. 2020, 11, 926. [Google Scholar] [CrossRef]
- Huynh, H.; Chow, K.H.; Soo, K.C.; Toh, H.C.; Choo, S.P.; Foo, K.F.; Poon, D.; Ngo, V.C.; Tran, E. RAD001 (everolimus) inhibits tumour growth in xenograft models of human hepatocellular carcinoma. J. Cell. Mol. Med. 2009, 13, 1371–1380. [Google Scholar] [CrossRef]
- Ong, L.C.; Song, I.C.; Jin, Y.; Kee, I.H.; Siew, E.; Yu, S.; Thng, C.H.; Huynh, H.; Chow, P.K. Effective inhibition of xenografts of hepatocellular carcinoma (HepG2) by rapamycin and bevacizumab in an intrahepatic model. Mol. Imaging Biol. 2009, 11, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.; Song, L.; Fan, N.; Feng, T.; Liu, L.; Yang, X.; Wang, M.; Li, Y.; Tian, Y.; Zhao, F.; et al. Combination of metformin and sorafenib suppresses proliferation and induces autophagy of hepatocellular carcinoma via targeting the mTOR pathway. Int. J. Oncol. 2017, 50, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Varshney, A.; Panda, J.J.; Singh, A.K.; Yadav, N.; Bihari, C.; Biswas, S.; Sarin, S.K.; Chauhan, V.S. Targeted delivery of microRNA-199a-3p using self-assembled dipeptide nanoparticles efficiently reduces hepatocellular carcinoma in mice. Hepatology 2018, 67, 1392–1407. [Google Scholar] [CrossRef]
- Li, D.; Liu, X.; Lin, L.; Hou, J.; Li, N.; Wang, C.; Wang, P.; Zhang, Q.; Zhang, P.; Zhou, W.; et al. MicroRNA-99a inhibits hepatocellular carcinoma growth and correlates with prognosis of patients with hepatocellular carcinoma. J. Biol. Chem. 2011, 286, 36677–36685. [Google Scholar] [CrossRef] [Green Version]
- Hausch, F.; Kozany, C.; Theodoropoulou, M.; Fabian, A.K. FKBPs and the Akt/mTOR pathway. Cell Cycle 2013, 12, 2366–2370. [Google Scholar] [CrossRef] [Green Version]
- Yip, C.K.; Murata, K.; Walz, T.; Sabatini, D.M.; Kang, S.A. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol. Cell 2010, 38, 768–774. [Google Scholar] [CrossRef] [Green Version]
- Perrone, F.; Craparo, E.F.; Cemazar, M.; Kamensek, U.; Drago, S.E.; Dapas, B.; Scaggiante, B.; Zanconati, F.; Bonazza, D.; Grassi, M.; et al. Targeted delivery of siRNAs against hepatocellular carcinoma-related genes by a galactosylated polyaspartamide copolymer. J. Control. Release 2021, 330, 1132–1151. [Google Scholar] [CrossRef]
- Scarabel, L.; Perrone, F.; Garziera, M.; Farra, R.; Grassi, M.; Musiani, F.; Russo Spena, C.; Salis, B.; De Stefano, L.; Toffoli, G.; et al. Strategies to optimize siRNA delivery to hepatocellular carcinoma cells. Expert. Opin. Drug Deliv. 2017, 14, 797–810. [Google Scholar] [CrossRef]
- Farra, R.; Musiani, F.; Perrone, F.; Čemažar, M.; Kamenšek, U.; Tonon, F.; Abrami, M.; Ručigaj, A.; Grassi, M.; Pozzato, G.; et al. Polymer-Mediated Delivery of siRNAs to Hepatocellular Carcinoma: Variables Affecting Specificity and Effectiveness. Molecules 2018, 23, 777. [Google Scholar] [CrossRef] [Green Version]
- Ji, L.; Lin, Z.; Wan, Z.; Xia, S.; Jiang, S.; Cen, D.; Cai, L.; Xu, J.; Cai, X. miR-486-3p mediates hepatocellular carcinoma sorafenib resistance by targeting FGFR4 and EGFR. Cell Death Dis. 2020, 11, 250. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Fujita, K.; Gong, J.; Nakahara, M.; Iwama, H.; Liu, S.; Yoneyama, H.; Morishita, A.; Nomura, T.; Tani, J.; et al. Aspirin inhibits hepatocellular carcinoma cell proliferation in vitro and in vivo via inducing cell cycle arrest and apoptosis. Oncol. Rep. 2020, 44, 457–468. [Google Scholar] [CrossRef]
- Wang, L.; Yao, J.; Shi, X.; Hu, L.; Li, Z.; Song, T.; Huang, C. MicroRNA-302b suppresses cell proliferation by targeting EGFR in human hepatocellular carcinoma SMMC-7721 cells. BMC Cancer 2013, 13, 448. [Google Scholar] [CrossRef] [Green Version]
- Lang, Q.; Ling, C. MiR-124 suppresses cell proliferation in hepatocellular carcinoma by targeting PIK3CA. Biochem. Biophys. Res. Commun. 2012, 426, 247–252. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, X.; Xiong, L.; Yu, L.; Li, Z.; Guo, Q.; Li, Z.; Li, B.; Lin, N. Comprehensive analysis of microRNA-regulated protein interaction network reveals the tumor suppressive role of microRNA-149 in human hepatocellular carcinoma via targeting AKT-mTOR pathway. Mol. Cancer 2014, 13, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damodaran, S.; Zhao, F.; Deming, D.A.; Mitchell, E.P.; Wright, J.J.; Gray, R.J.; Wang, V.; McShane, L.M.; Rubinstein, L.V.; Patton, D.R.; et al. Phase II Study of Copanlisib in Patients With Tumors With PIK3CA Mutations: Results From the NCI-MATCH ECOG-ACRIN Trial (EAY131) Subprotocol Z1F. J. Clin. Oncol. 2022, 40, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.H.; Li, J.; Wei, L.; Doyle, A.; Marshall, J.L.; Schaaf, L.J.; Phelps, M.A.; Villalona-Calero, M.A.; Bekaii-Saab, T. Results of an abbreviated phase-II study with the Akt Inhibitor MK-2206 in Patients with Advanced Biliary Cancer. Sci. Rep. 2015, 5, 12122. [Google Scholar] [CrossRef] [Green Version]
- Matter, M.S.; Decaens, T.; Andersen, J.B.; Thorgeirsson, S.S. Targeting the mTOR pathway in hepatocellular carcinoma: Current state and future trends. J. Hepatol. 2014, 60, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.W.; Kim, S.H.; Yoon, K.C.; Lee, J.M.; Cho, J.H.; Hong, S.K.; Yi, N.J.; Han, S.S.; Park, S.J.; Suh, K.S. Sirolimus Prolongs Survival after Living Donor Liver Transplantation for Hepatocellular Carcinoma Beyond Milan Criteria: A Prospective, Randomised, Open-Label, Multicentre Phase 2 Trial. J. Clin. Med. 2020, 9, 3264. [Google Scholar] [CrossRef]
- Tian, L.Y.; Smit, D.J.; Jücker, M. The Role of PI3K/AKT/mTOR Signaling in Hepatocellular Carcinoma Metabolism. Int. J. Mol. Sci. 2023, 24, 2652. [Google Scholar] [CrossRef]
- Cervello, M.; McCubrey, J.A.; Cusimano, A.; Lampiasi, N.; Azzolina, A.; Montalto, G. Targeted therapy for hepatocellular carcinoma: Novel agents on the horizon. Oncotarget 2012, 3, 236–260. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.B.; Garrett-Mayer, E.; Anis, M.; Anderton, K.; Bentz, T.; Edwards, A.; Brisendine, A.; Weiss, G.; Siegel, A.B.; Bendell, J.; et al. A Randomized Phase II Open-Label Multi-Institution Study of the Combination of Bevacizumab and Erlotinib Compared to Sorafenib in the First-Line Treatment of Patients with Advanced Hepatocellular Carcinoma. Oncology 2018, 94, 329–339. [Google Scholar] [CrossRef]
- Kelley, R.K.; Joseph, N.M.; Nimeiri, H.S.; Hwang, J.; Kulik, L.M.; Ngo, Z.; Behr, S.C.; Onodera, C.; Zhang, K.; Bocobo, A.G.; et al. Phase II Trial of the Combination of Temsirolimus and Sorafenib in Advanced Hepatocellular Carcinoma with Tumor Mutation Profiling. Liver Cancer 2021, 10, 561–571. [Google Scholar] [CrossRef]
- Chan, S.L.; Yeo, W. Development of systemic therapy for hepatocellular carcinoma at 2013: Updates and insights. World J. Gastroenterol. 2014, 20, 3135–3145. [Google Scholar] [CrossRef]
- Lu, X.; Paliogiannis, P.; Calvisi, D.F.; Chen, X. Role of the Mammalian Target of Rapamycin Pathway in Liver Cancer: From Molecular Genetics to Targeted Therapies. Hepatology 2021, 73 (Suppl. S1), 49–61. [Google Scholar] [CrossRef]
- Avramović, N.; Mandić, B.; Savić-Radojević, A.; Simić, T. Polymeric Nanocarriers of Drug Delivery Systems in Cancer Therapy. Pharmaceutics 2020, 12, 298. [Google Scholar] [CrossRef] [Green Version]
- Mu, H.; Lin, K.X.; Zhao, H.; Xing, S.; Li, C.; Liu, F.; Lu, H.Z.; Zhang, Z.; Sun, Y.L.; Yan, X.Y.; et al. Identification of biomarkers for hepatocellular carcinoma by semiquantitative immunocytochemistry. World J. Gastroenterol. 2014, 20, 5826–5838. [Google Scholar] [CrossRef] [PubMed]
- Baumhoer, D.; Tornillo, L.; Stadlmann, S.; Roncalli, M.; Diamantis, E.K.; Terracciano, L.M. Glypican 3 expression in human nonneoplastic, preneoplastic, and neoplastic tissues: A tissue microarray analysis of 4,387 tissue samples. Am. J. Clin. Pathol. 2008, 129, 899–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Drug | Target | HCC Cell Line | Phenotype | Reference |
|---|---|---|---|---|
| erlotinib | EGFR | MHCC97H, PLC/RLF/5 | reduced cell proliferation | [32] |
| Huh7 | reduced migration | [44] | ||
| AG1478 | EGFR | HepG2 | reduced cell proliferation and invasion | [38] |
| gefitinib | EGFR | Huh7 | induced apoptosis | [45] |
| GW2974 | EGFR | HepG2 | reduced cell proliferation, induced apoptosis | [41] |
| copanlisib | PI3K | Huh7, HepG2 | induced apoptosis, inhibited cell growth, inducing cell cycle arrest | [45,48] |
| LY294002 | PI3K | MHCC97-H | induced apoptosis | [49] |
| Huh7, Mahlavu | suppressed cell growth, induced apoptosis | [46] | ||
| wortmannin | PI3K | Huh7, Mahlavu | suppressed cell growth, induced apoptosis | [46] |
| HepG2 | induced apoptosis | [46] | ||
| 740Y-P | PI3K | SMMC-7721, MHCC-97H | reduced cell proliferation, induced apoptosis | [47] |
| MK2206 | AKT | Hep3B, Huh7, PLC/RLF/5 | growth-inhibitory, induced apoptosis | [50] |
| HepG2 | anti-proliferative | [51] | ||
| AKT inhibitor VIII | AKT | Huh7, Mahlavu | suppressed cell growth, induced apoptosis | [46] |
| rapamycin | mTOR | Hep3B | prevent enrichment of CSCs | [52] |
| RAD001 (everolimus) | mTOR | Hep3B, Huh7, PLC/RLF/5 | growth-inhibitory, induced apoptosis | [50] |
| BEZ235 | PI3K/mTOR | Hep3B, Huh7, PLC/RLF/5 | growth-inhibitory, induced apoptosis | [50] |
| lenvatinib | AKT/mTOR | Hep3B, HepG2 | reduced cell proliferation, migration | [53] |
| Gene | Mouse Model | Phenotype/Tumor Type | Ref. |
|---|---|---|---|
| Pten | Alb-Cre; Ptenfl/fl | ballooning, steatosis/ICC, HCC | [63] |
| sgPten sgPten/c-Met | lipid accumulation/no tumor lipid accumulation/HCA, HCC | [64] | |
| AKT | HA-myr-AKT1 HA-myr-AKT1/V5-c-Met | hepatic steatosis, proliferation/HCC rapid liver tumor growth | [65] |
| myr-AKT1 myr-AKT1/N-RasG12V | hepatocyte proliferation/HCA (12 w), HCC (6 m) spotty and pale color/nodular lesions (~4 w) | [66] | |
| Tsc1/Tsc2 | Alb-Cre; Tsc1fl/fl | dysplastic foci, nodules, hepatomas/HCC | [67] |
| Alb-Cre; Tsc1fl/fl | moderate tumor incidence rate/HCC | [68] | |
| Alb-Cre; Tsc1fl/fl Alb-Cre; Tsc1fl/fl/Ptenfl/fl | no steatosis/HCC, ICC hepatomegaly (early), large tumor (14 w)/HCC | [63] |
| Agent | Target | Administration Route | HCC Cell Line Transplanted | Phenotype | Ref. |
|---|---|---|---|---|---|
| MUC15 | EGFR | NA | HCCLM3 | delayed tumor formation, higher survival rate | [73] |
| RHEB-shRNA | RHEB | NA | SMMC-7721 | decrease in tumor growth | [74] |
| DZW-310 | PI3K | OA | Hep3B | decrease in tumor growth | [75] |
| MK2206 | AKT | IP | Hep3B, Huh7 | decrease in tumor growth | [50] |
| ZJQ-24 | AKT/mTOR | OA | HepG2 | Tumor cell death/reduced tumor growth | [76] |
| RAD001 | mTOR | OA | Patient-derived HCCs | decrease in tumor growth | [77] |
| rapamycin | mTOR | OA | HepG2 | no effects | [78] |
| rapamycin + bevacizumab | mTOR + VEGF | OA + IP | HepG2 | decreased tumor size/increased survival | [78] |
| metformin | mTOR | IP | Bel-7402 | decrease in tumor growth | [79] |
| miRNA-199a-3p | mTOR | IV | Huh7 | decreased tumor growth/increased survival | [80] |
| miRNA-99a | mTOR | IT | SMMC-LTNM | reduction in tumor size | [81] |
| Agent | Target | Phase | Clinical Outcomes | Adverse Events (Side Effects) | NCT # |
|---|---|---|---|---|---|
| Copanlisib | pan-class I PI3K | II | discontinuation due to disease progression | hyperglycemia (63%), fatigue (40%), diarrhea (37%), hypertension (33%), nausea (33%), maculopapular rash (30%) | 02465060 |
| MK-2206 | AKT | II | early termination for discouraging results | anemia (73.33%), hyperglycemia (60.00%), hypoalbuminemia (46.67%), hyperbilirubinaemia (13.33%) | 01239355 |
| everolimus (RAD001) | mTOR | I/II | only 2 patients (8%) responded to treatment | hyperglycemia (42.86%), diarrhea (39.29%), hyponatremia (32.14%) | 00516165 |
| sirolimus (rapamycin) | mTOR | II | MOS of 21.1 m vs. 14.1 m for control (survival benefit over control) | dyslipidaemia, oral mucositis, diarrhea | 01374750 |
| onatasertib (CC-223) | mTOR | II | preliminary antitumor activity | diarrhea (60.38%), hyperglycaemia (60.38%), thrombocytopenia (30.19%), hyperbilirubinaemia (11.32%) | 03591965 |
| AZD8055 | mTOR | I/II | not yet determined | increased aspartate aminotransferase (22%), fatigue (16%) | 00999882 |
| rapamycin + bevacizumab | mTOR + VEGF | I | no survival benefit over bevacizumab-only treatment | hyperglycaemia (83%), thrombocytopenia (75%), fatigue (46%), mucositis (46%), anorexia (42%), Diarrhea (33%) | 00467194 |
| erlotinib + bevacizumab | EGFR + VEGF | II | no improvement over sorafenib-treated group | increased alkaline phosphatase (38.30%), hypoalbuminemia (29.79%) | 00881751 |
| temsirolimus + sorafenib | mTOR + RTK | II | no survival benefit over sorafenib-only group | hypophosphatemia (60.71%), diarrhea (28.57%), anemia (10.71%) | 01687673 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bang, J.; Jun, M.; Lee, S.; Moon, H.; Ro, S.W. Targeting EGFR/PI3K/AKT/mTOR Signaling in Hepatocellular Carcinoma. Pharmaceutics 2023, 15, 2130. https://doi.org/10.3390/pharmaceutics15082130
Bang J, Jun M, Lee S, Moon H, Ro SW. Targeting EGFR/PI3K/AKT/mTOR Signaling in Hepatocellular Carcinoma. Pharmaceutics. 2023; 15(8):2130. https://doi.org/10.3390/pharmaceutics15082130
Chicago/Turabian StyleBang, Jieun, Mihyeon Jun, Soyun Lee, Hyuk Moon, and Simon Weonsang Ro. 2023. "Targeting EGFR/PI3K/AKT/mTOR Signaling in Hepatocellular Carcinoma" Pharmaceutics 15, no. 8: 2130. https://doi.org/10.3390/pharmaceutics15082130