

Potent and Selective Benzothiazole-Based Antimitotics with Improved Water Solubility: Design, Synthesis, and Evaluation as Novel Anticancer Agents

Abstract
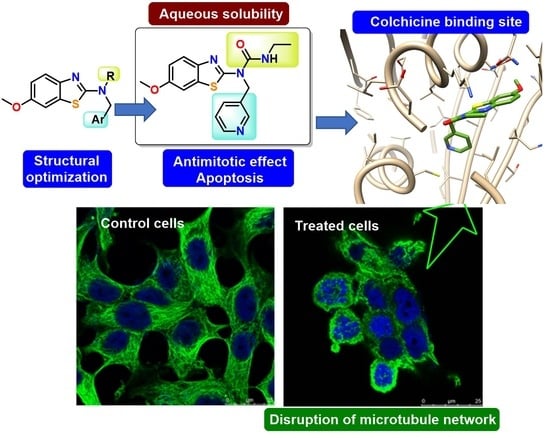
:
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. General Chemical Techniques
2.1.2. Chemical Synthesis and Characterization
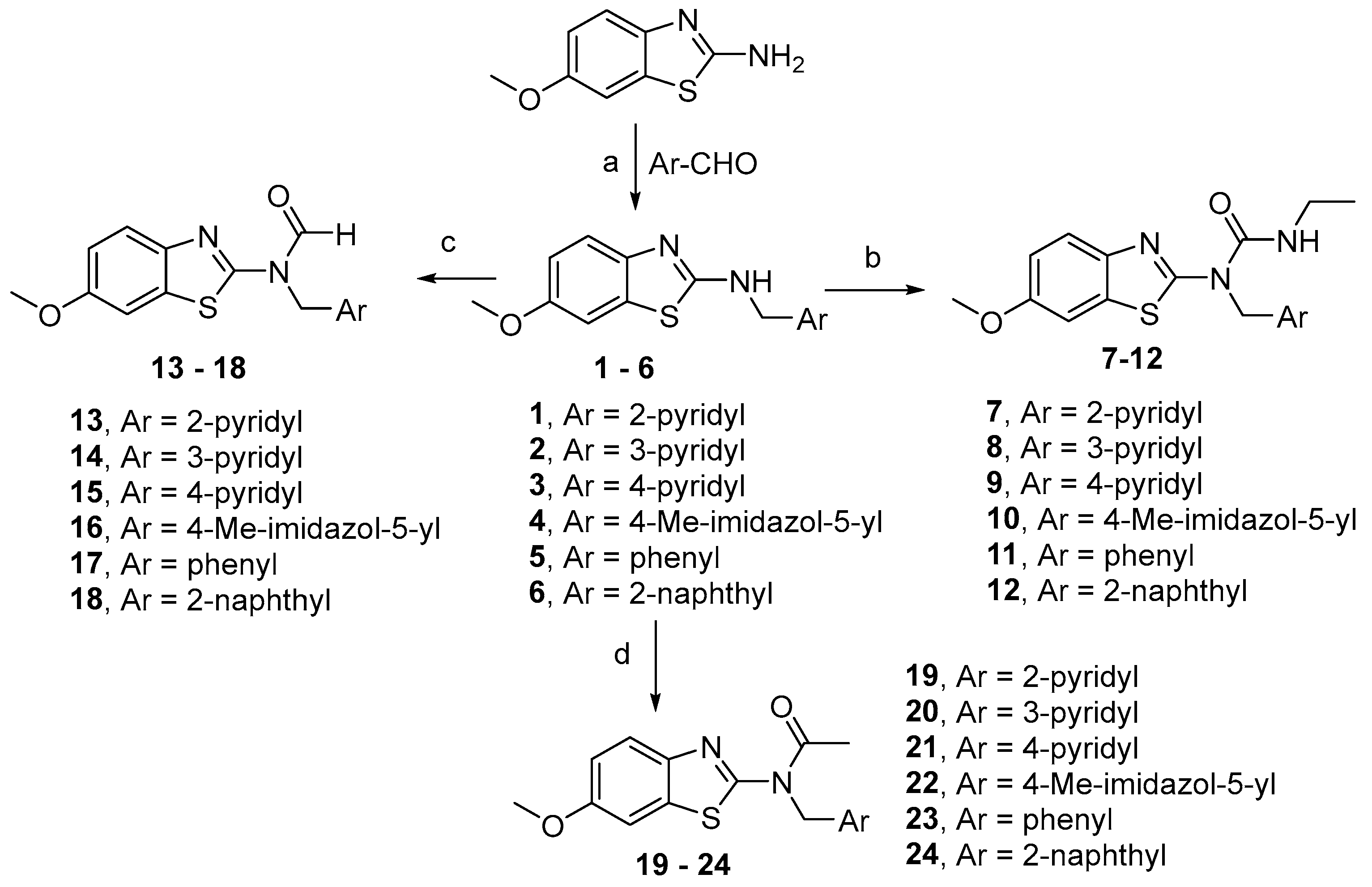
Method A. Reductive Amination (Compounds 1–6)
Method B. Synthesis of Urea Derivatives (Compounds 7–12)
Method C. Synthesis of Formamide Derivatives (Compounds 13–18)
Method D. Synthesis of Acetamide Derivatives (Compounds 19–24)
2.2. Determination of Aqueous Solubility
2.3. Calculation of Properties
2.4. Biology
2.4.1. Cell Culture Conditions
2.4.2. Cell Growth Inhibition Assay
2.4.3. Tubulin Polymerization Inhibition
2.4.4. Immunofluorescence Experiments
2.4.5. Cell Cycle Analysis
2.4.6. Apoptotic Cell Death Quantification
2.4.7. Lactate Dehydrogenase Assay
2.5. Computational Studies
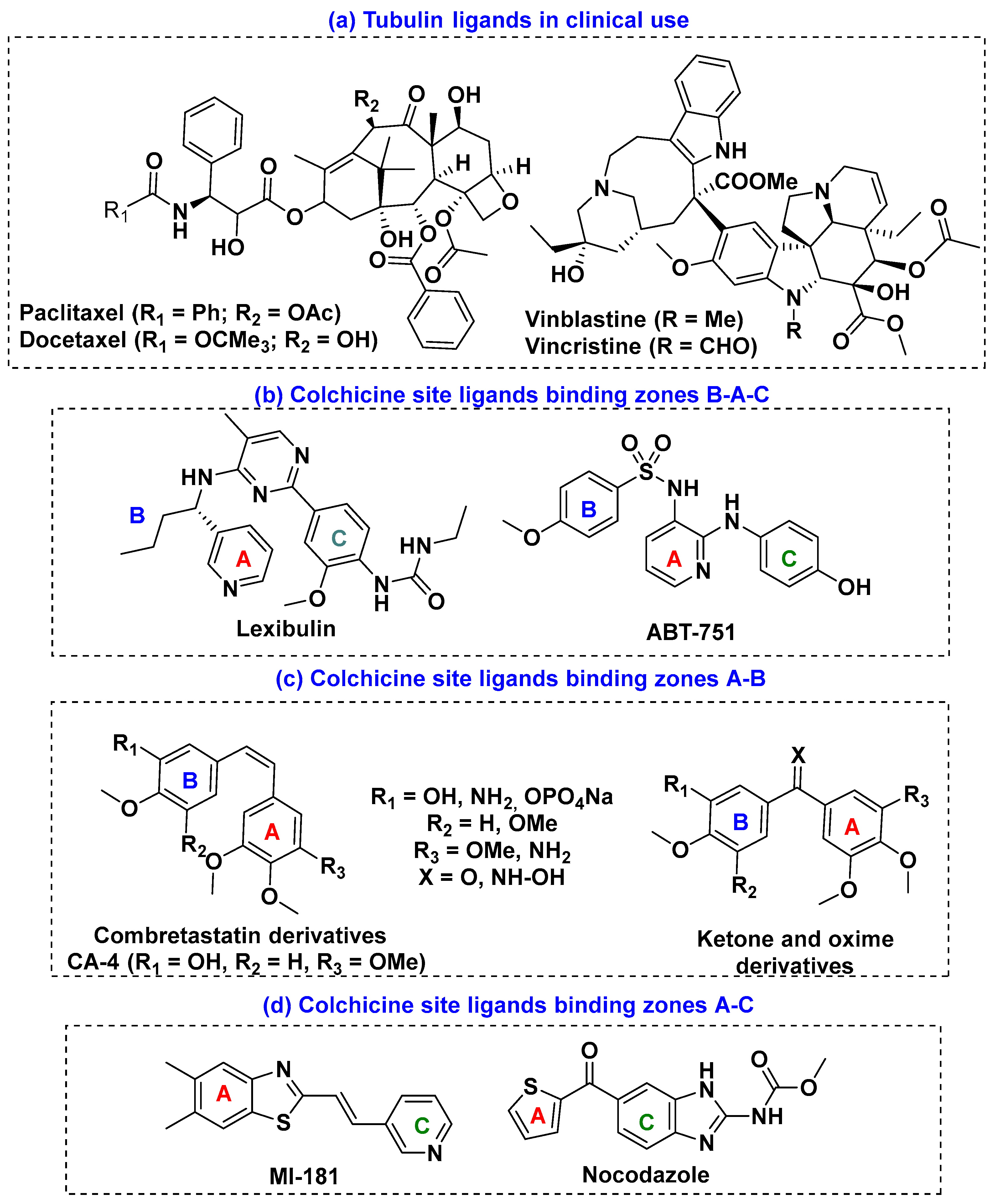
3. Results and Discussion
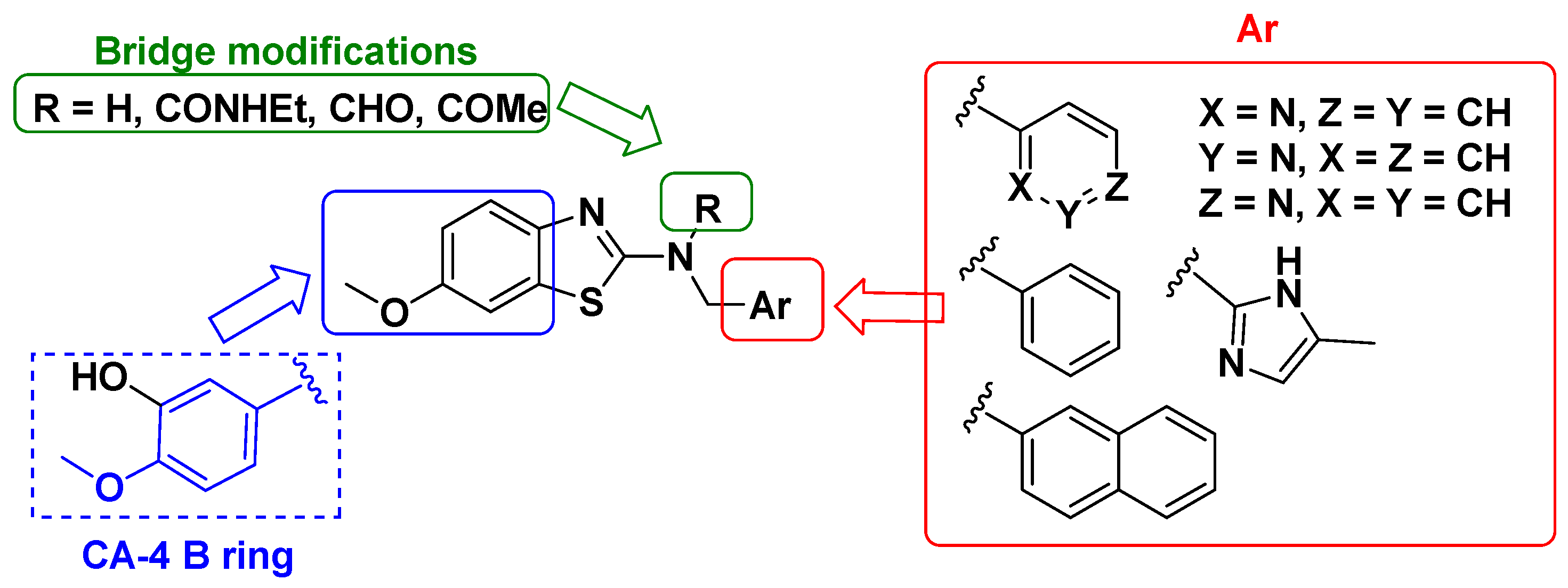
3.1. Chemistry and Properties
3.1.1. Chemical Synthesis
3.1.2. Aqueous Solubility and Calculated Properties
3.2. Biology
3.2.1. Antiproliferative Activity
3.2.2. Sensitivity to MDR Efflux Pumps
3.2.3. Tubulin Polymerization Inhibition (TPI)
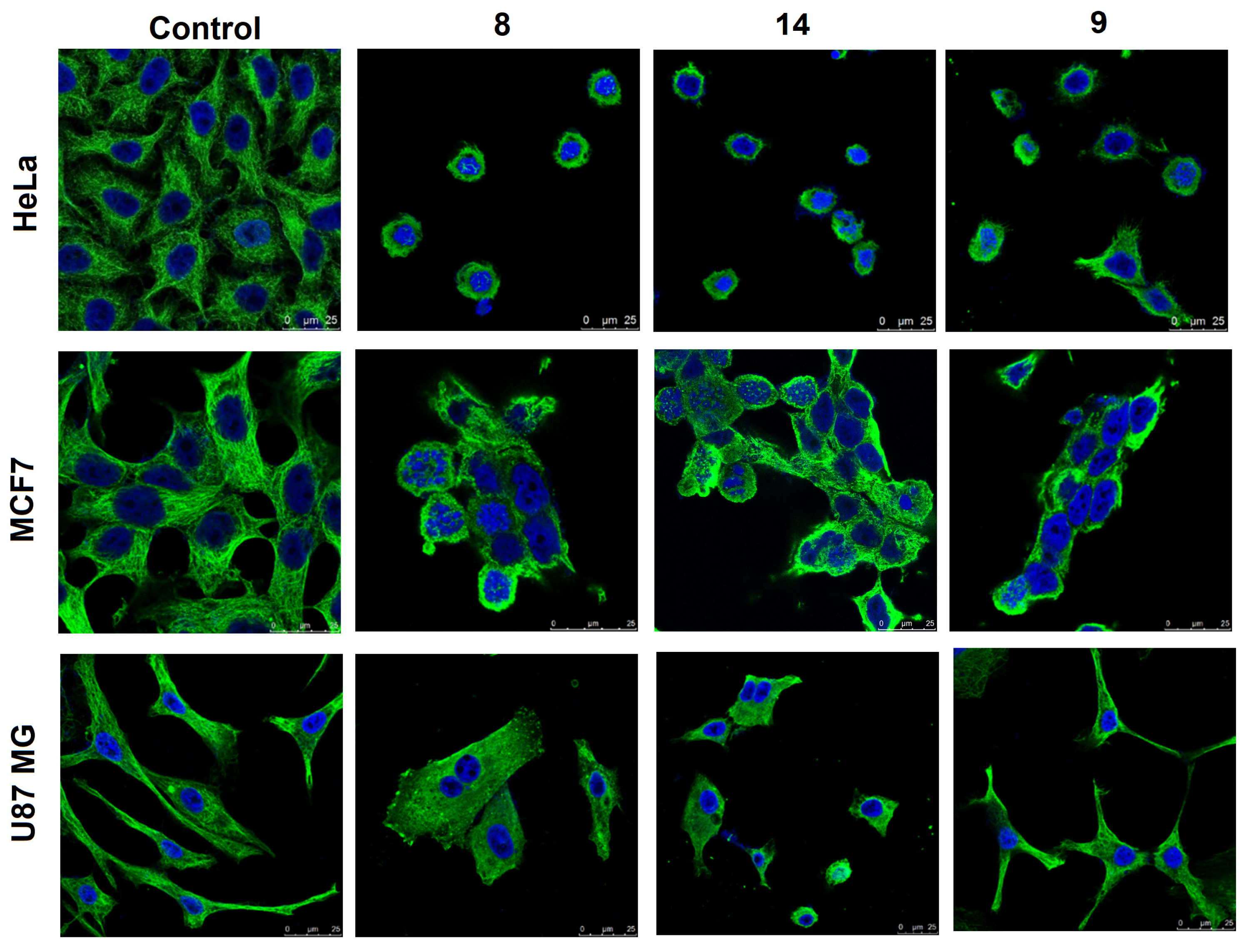
3.2.4. Effects on Cellular Microtubules
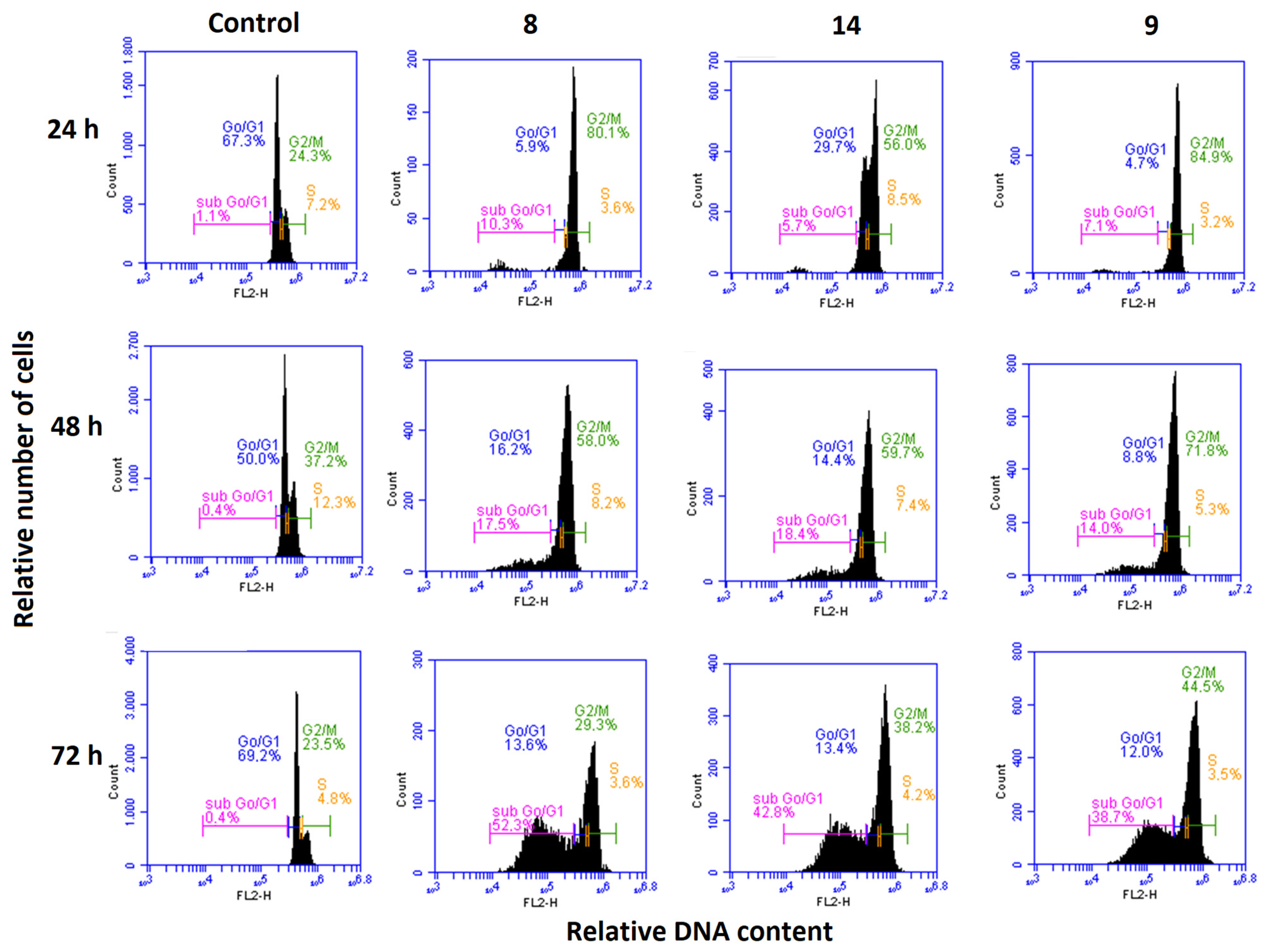
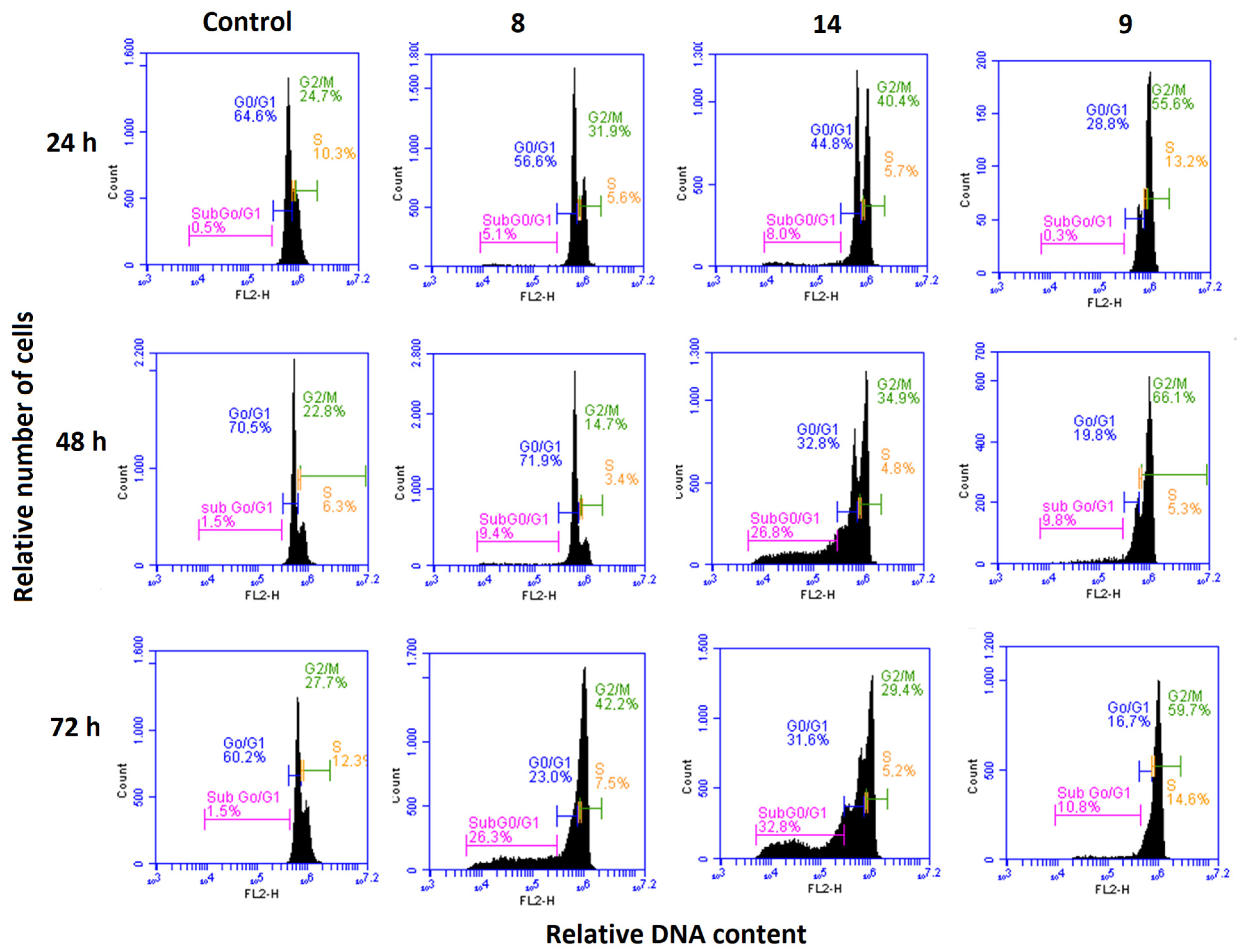
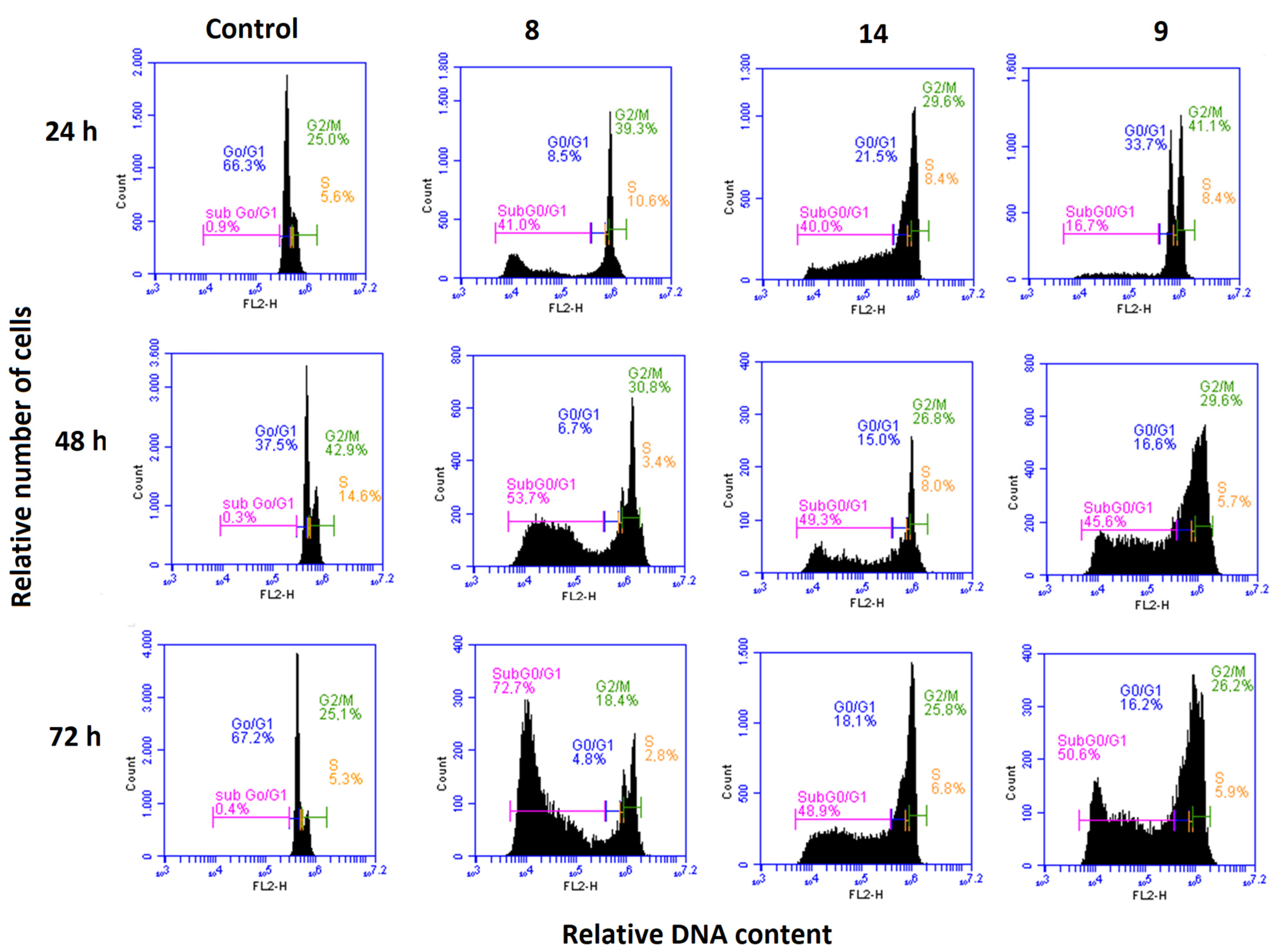
3.2.5. Effects on the Cell Cycle
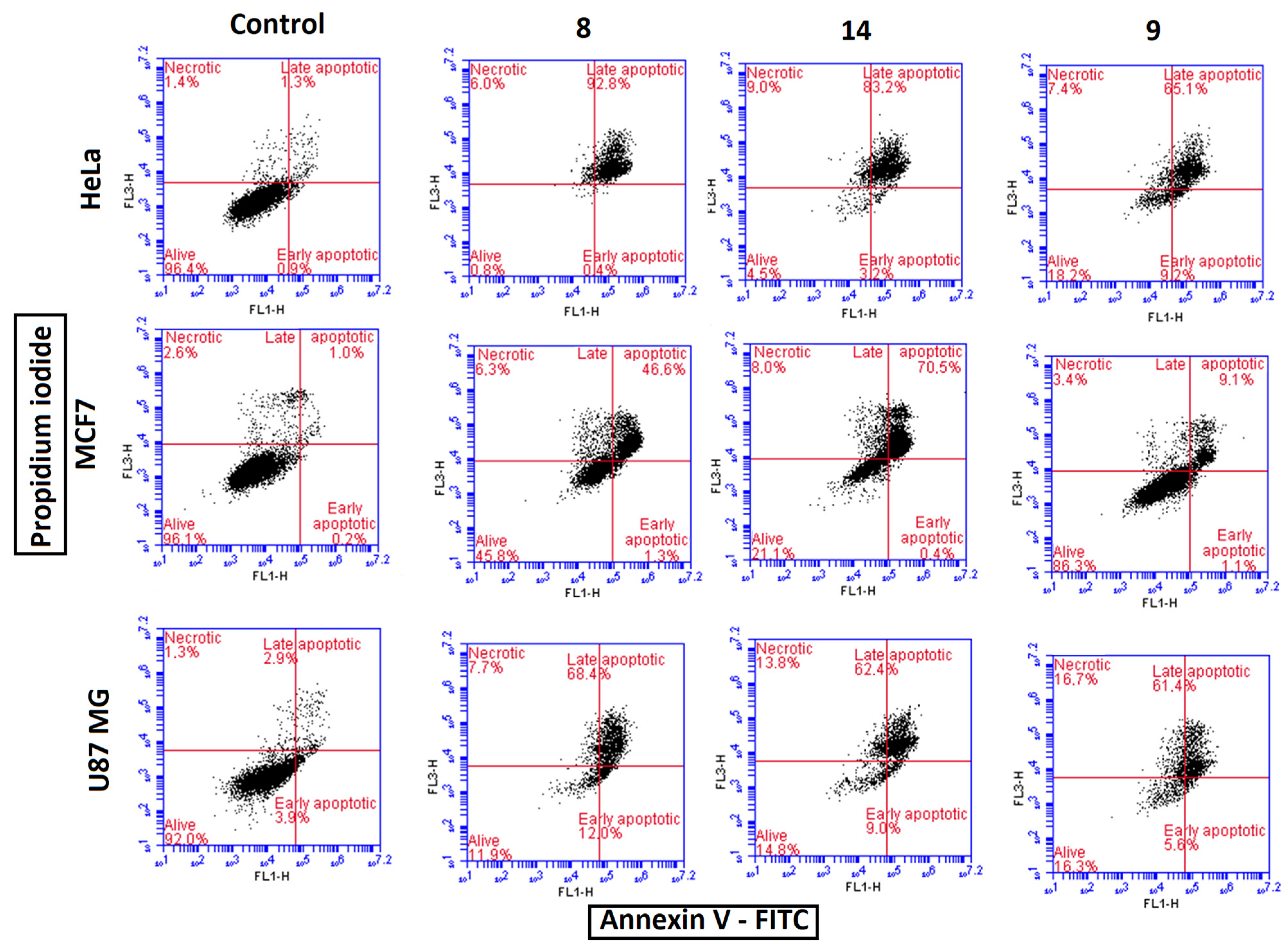
3.2.6. Cell Death Mechanistic Studies
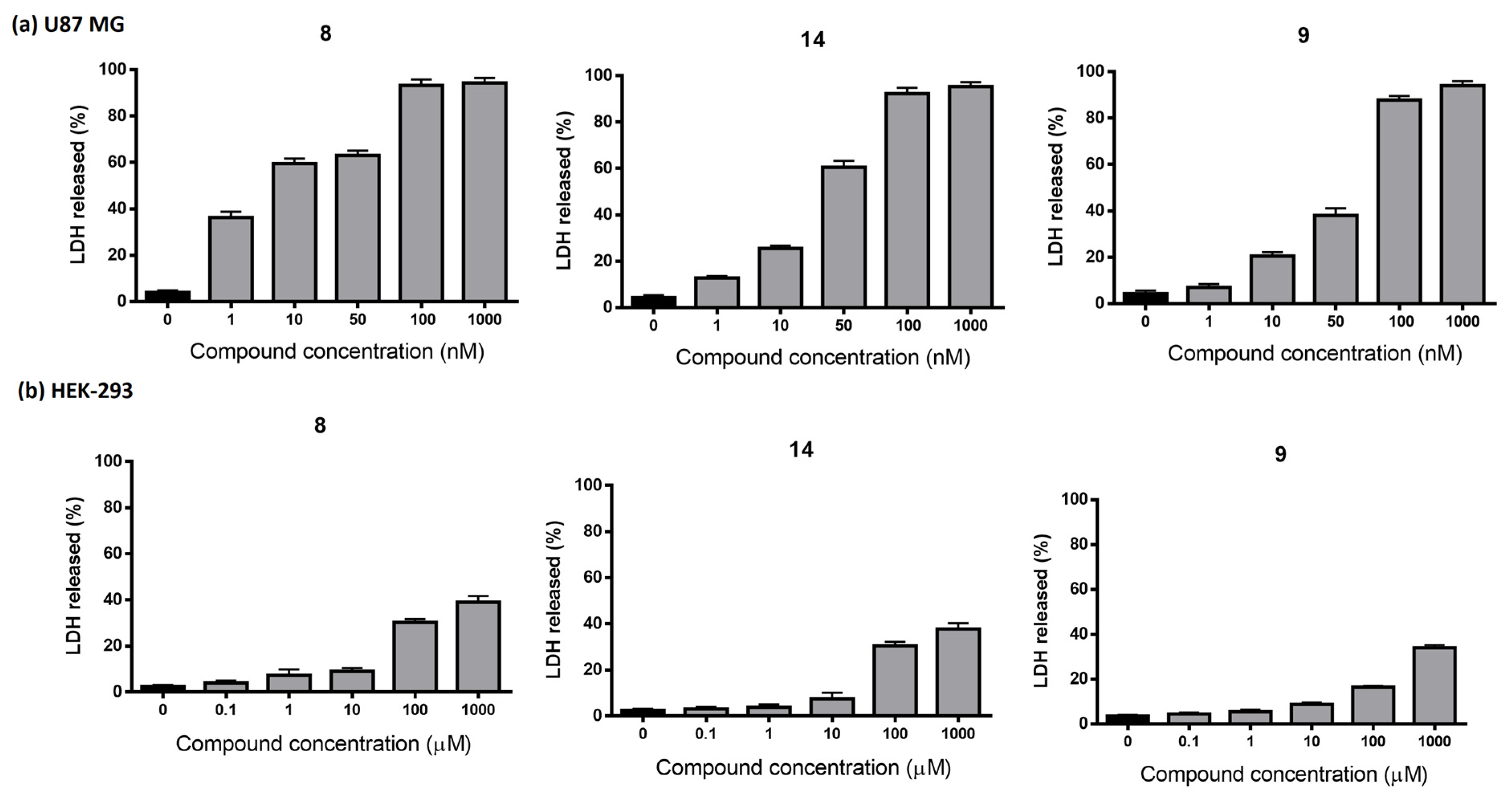
3.2.7. Lactate Dehydrogenase Assays
3.3. Computational Studies
3.3.1. Structural Effects of the Bridge Modifications
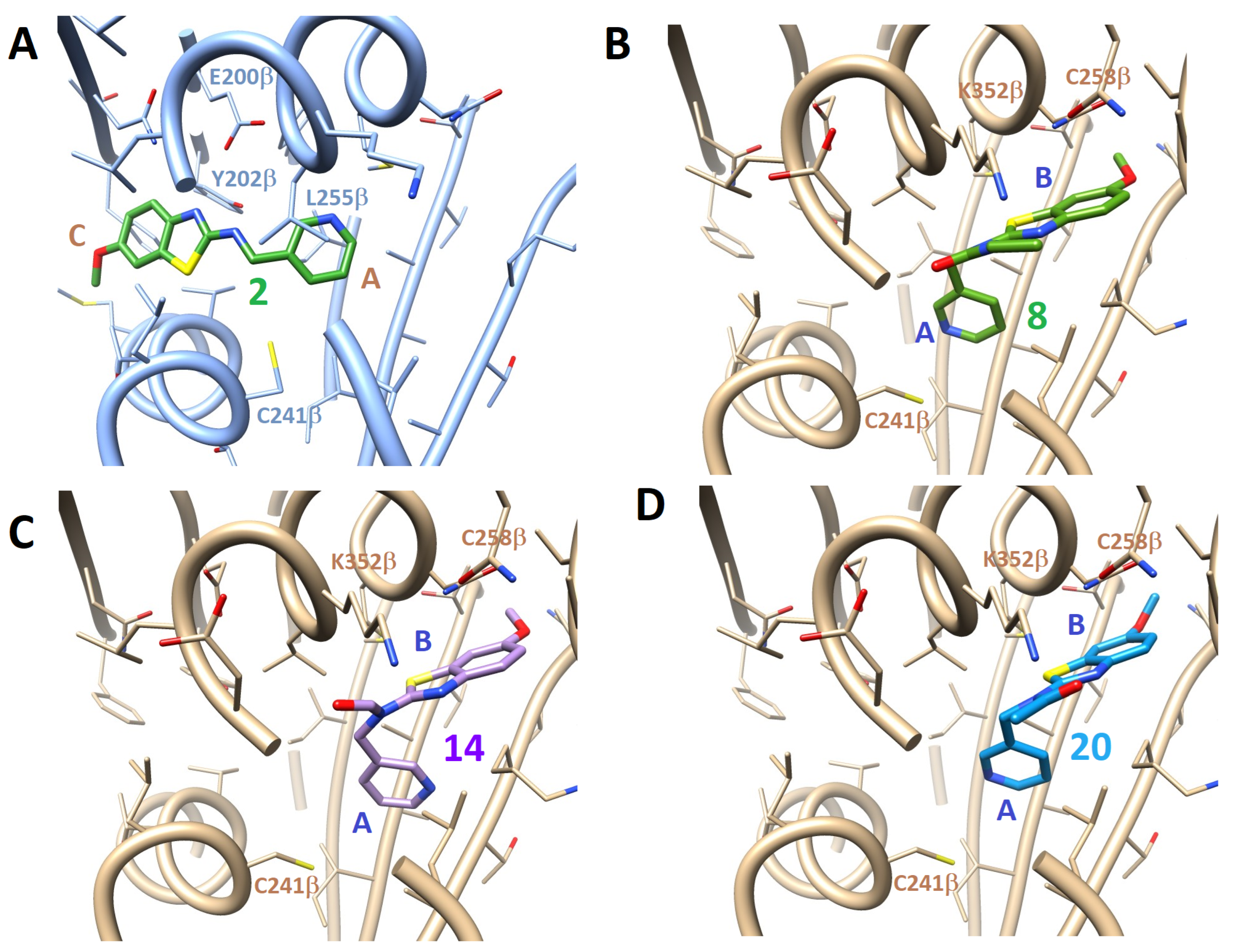
3.3.2. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 11 May 2023).
- Dolgin, E. Cancer drug approvals and setbacks in 2022. Nat. Cancer 2022, 12, 1406–1408. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Uzui, K.; Shigemi, H.; Negoro, E.; Yoshida, A.; Ueda, T. Aurora B inhibitor barasertib and cytarabine exert a greater-than-additive cytotoxicity in acute myeloid leukemia cells. Cancer Sci. 2013, 7, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, W.S.; Yu, T.; Yi, Y.S.; Park, J.G.; Jeong, D.; Kim, J.H.; Oh, J.S.; Yoon, K.; Kim, J.H.; et al. Novel anti-inflammatory function of NSC95397 by the suppression of multiple kinases. Biochem. Pharmacol. 2014, 88, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Murase, Y.; Ono, H.; Ogawa, K.; Yoshioka, R.; Ishikawa, Y.; Ueda, H.; Akahoshi, K.; Ban, D.; Kudo, A.; Tanaka, S.; et al. Inhibitor library screening identifies ispinesib as a new potential chemotherapeutic agent for pancreatic cancers. Cancer Sci. 2021, 112, 4641–4654. [Google Scholar] [CrossRef]
- Qin, Z.; Li, X.; Zhang, J.; Tang, J.; Han, P.; Xu, Z.; Yu, Y.; Yang, C.; Wang, C.; Xu, T.; et al. Chemotherapy with or without estramustine for treatment of castration-resistant prostate cancer: A systematic review and meta-analysis. Medicine 2016, 95, e4801. [Google Scholar] [CrossRef]
- Zhu, L.; Chen, L. Progress in research on paclitaxel and tumor immunotherapy. Cell. Mol. Biol. Lett. 2019, 13, 24–40. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.H.; Horwitz, S.B. Taxol®: The First Microtubule Stabilizing Agent. Int. J. Mol. Sci. 2017, 18, 1733. [Google Scholar] [CrossRef] [Green Version]
- Haque, A.; Rahman, M.A.; Faizi, M.S.H.; Khan, M.S. Next Generation Antineoplastic Agents: A Review on Structurally Modified Vinblastine (VBL) Analogues. Curr. Med. Chem. 2018, 25, 1650–1662. [Google Scholar] [CrossRef]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, S.; Kaku, K.; Robles, A.J.; Hamel, E.; Mooberry, S.L.; Gangjee, A. Simple monocyclic pyrimidine analogs as microtubule targeting agents binding to the colchicine site. Bioorg. Med. Chem. 2023, 82, 117217. [Google Scholar] [CrossRef]
- Chen, Z.H.; Xu, R.M.; Zheng, G.H.; Jin, Y.Z.; Li, Y.; Chen, X.Y.; Tian, Y.S. Development of Combretastatin A-4 Analogues as Potential Anticancer Agents with Improved Aqueous Solubility. Molecules 2023, 28, 1717. [Google Scholar] [CrossRef]
- Grillone, K.; Riillo, C.; Rocca, R.; Ascrizzi, S.; Spanò, V.; Scionti, F.; Polerà, N.; Maruca, A.; Barreca, M.; Juli, G.; et al. The New Microtubule-Targeting Agent SIX2G Induces Immunogenic Cell Death in Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 10222. [Google Scholar] [CrossRef]
- Pérez-Pérez, M.-J.; Priego, E.M.; Bueno, O.; Solange Martins, M.; Canela, M.-D.; Liekens, S. Blocking blood flow to solid tumors by destabilizing tubulin: An approach to targeting tumor growth. J. Med. Chem. 2016, 59, 8685–8711. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Miller, D.D.; Li, W. Molecular interactions at the colchicine binding site in tubulin: An X-ray crystallography perspective. Drug Discov. Today 2022, 27, 759–776. [Google Scholar] [CrossRef]
- Álvarez, R.; Medarde, M.; Peláez, P. New ligands of the tubulin colchicine site based on X-ray structures. Curr. Top. Med. Chem. 2014, 14, 2231–2252. [Google Scholar] [CrossRef]
- Massarotti, A.; Coluccia, A.; Silvestri, R.; Sorba, G.; Brancale, A. The tubulin colchicine domain: A molecular modeling perspective. Chem. Med. Chem. 2012, 7, 33–42. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Gigant, B.; Yu, Y.; Wu, Y.; Chen, X.; Lai, Q.; Yang, Z.; Chen, Q.; Yang, J. Structures of a diverse set of colchicine binding site inhibitors in complex with tubulin provide a rationale for drug discovery. FEBS J. 2016, 283, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Hradil, V.P.; Frost, D.J.; Rosenberg, S.H.; Gordon, G.B.; Morgan, S.J.; Gagne, G.D.; Cox, B.F.; Tahir, S.K.; Fox, G.B. ABT-751, a novel tubulin-binding agent, decreases tumor perfusion and disrupts tumor vasculature. Anti-Cancer Drugs 2009, 20, 483–492. [Google Scholar] [CrossRef]
- Young, S.L.; Chaplin, D.J. Combretastatin A4 phosphate: Background and current clinical status. Expert. Opin. Investig. Drugs 2004, 13, 1171–1182. [Google Scholar] [CrossRef]
- González, M.; Ellahioui, Y.; Álvarez, R.; Gallego-Yerga, L.; Caballero, E.; Vicente-Blázquez, A.; Ramudo, L.; Marín, M.; Sanz, C.; Medarde, M.; et al. The Masked Polar Group Incorporation (MPGI) Strategy in Drug Design: Effects of Nitrogen Substitutions on Combretastatin and Isocombretastatin Tubulin Inhibitors. Molecules 2019, 24, 4319. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Blázquez, A.; González, M.; Medarde, M.; Mollinedo, F.; Peláez, R. New indolesulfonamide derivatives targeting the colchicine site of tubulin: Synthesis, anti-tumour activity, structure-activity relationships, and molecular modelling. J. Enzyme Inhib. Med. Chem. 2021, 36, 2025–2044. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, R.; Aramburu, L.; Gajate, C.; Vicente-Blázquez, A.; Mollinedo, F.; Medarde, M.; Peláez, R. Methylsulfanylpyridine based diheteroaryl isocombretastatin analogs as potent anti-proliferative agents. Eur. J. Med. Chem. 2021, 209, 112933. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, E.C.; O’Boyle, N.M. Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals 2020, 13, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez, R.; Aramburu, L.; Gajate, C.; Vicente-Blázquez, A.; Mollinedo, F.; Medarde, M.; Peláez, R. Potent colchicine-site ligands with improved intrinsic solubility by replacement of the 3,4,5-trimethoxyphenyl ring with a 2-methylsulfanyl-6-methoxypyridine ring. Bioorg. Chem. 2020, 98, 103755. [Google Scholar] [CrossRef]
- De la Roche, N.M.; Mühlethaler, T.; Di Martino, R.M.C.; Ortega, J.A.; Gioia, D.; Roy, B.; Prota, A.E.; Steinmetz, M.O.; Cavalli, A. Novel fragment-derived colchicine-site binders as microtubule-destabilizing agents. Eur. J. Med. Chem. 2022, 5, 114614. [Google Scholar] [CrossRef]
- Amoroso, R.; De Lellis, L.; Florio, R.; Moreno, N.; Agamennone, M.; De Filippis, B.; Giampietro, L.; Maccallini, C.; Fernández, I.; Recio, R.; et al. Benzothiazole Derivatives Endowed with Antiproliferative Activity in Paraganglioma and Pancreatic Cancer Cells: Structure-Activity Relationship Studies and Target Prediction Analysis. Pharmaceuticals 2022, 15, 937. [Google Scholar] [CrossRef]
- Ammazzalorso, A.; D’Angelo, A.; Giancristofaro, A.; De Filippis, B.; Di Matteo, M.; Fantacuzzi, M.; Giampietro, L.; Linciano, P.; Maccallini, C.; Amoroso, R. Fibrate-derived N-(methylsulfonyl)amides with antagonistic properties on PPARα. Eur. J. Med. Chem. 2012, 58, 317–322. [Google Scholar] [CrossRef]
- Ammazzalorso, A.; De Lellis, L.; Florio, R.; Bruno, I.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Perconti, S.; Verginelli, F.; et al. Cytotoxic effect of a family of peroxisome proliferator-activated receptor antagonists in colorectal and pancreatic cancer cell lines. Chem. Biol. Drug Des. 2017, 90, 1029–1035. [Google Scholar] [CrossRef]
- Ashraf, M.; Shaik, T.B.; Malik, M.S.; Syed, R.; Mallipeddi, P.L.; Vardhan, M.V.P.S.V.; Kamal, A. Design and synthesis of cis-restricted benzimidazole and benzothiazole mimics of combretastatin A-4 as antimitotic agents with apoptosis inducing ability. Bioorg. Med. Chem. Lett. 2016, 26, 4527–4535. [Google Scholar] [CrossRef]
- Shaik, T.B.; Hussaini, S.M.A.; Nayak, V.L.; Sucharitha, M.L.; Malik, M.S.; Kamal, A. Rational design and synthesis of 2-anilinopyridinyl-benzothiazole Schiff bases as antimitotic agents. Bioorg. Med. Chem. Lett. 2017, 27, 2549–2558. [Google Scholar] [CrossRef]
- Subba Rao, A.V.; Swapna, K.; Shaik, S.P.; Lakshma Nayak, V.; Srinivasa Reddy, T.; Sunkari, S.; Shaik, T.B.; Bagul, C.; Kamal, A. Synthesis and biological evaluation of cis-restricted triazole/tetrazole mimics of combretastatin-benzothiazole hybrids as tubulin polymerization inhibitors and apoptosis inducers. Bioorg. Med. Chem. 2017, 25, 977–999. [Google Scholar] [CrossRef]
- Joshi, R.; Mukherjee, D.D.; Chakrabarty, S.; Martin, A.; Jadhao, M.; Chakrabarti, G.; Sarkar, A.; Ghosh, S.K. Unveiling the Potential of Unfused Bichromophoric Naphthalimide to Induce Cytotoxicity by Binding to Tubulin: Breaks Monotony of Naphthalimides as Conventional Intercalators. J. Phys. Chem. B 2018, 122, 3680–3695. [Google Scholar] [CrossRef]
- Fu, D.J.; Liu, S.M.; Li, F.H.; Yang, J.J.; Li, J. Antiproliferative benzothiazoles incorporating a trimethoxyphenyl scaffold as novel colchicine site tubulin polymerization inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 1050–1059. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Gao, Q.L.; Wu, B.W.; Zhu, T.; Cui, X.X.; Jin, C.J.; Wang, S.Y.; Wang, S.H.; Fu, D.J.; Liu, H.M.; et al. Discovery of tertiary amide derivatives incorporating benzothiazole moiety as anti-gastric cancer agents in vitro via inhibiting tubulin polymerization and activating the Hippo signaling pathway. Eur. J. Med. Chem. 2020, 203, 112618. [Google Scholar] [CrossRef]
- Mohapatra, S.; Gupta, V.; Mondal, P.; Chatterjee, S.; Bhunia, D.; Ghosh, S. A Small Molecule with Bridged Carbonyl and Tri-fluoro-aceto-phenone Groups Impedes Microtubule Dynamics and Subsequently Triggers Cancer Cell Apoptosis. Chem. Med. Chem. 2021, 16, 2703–2714. [Google Scholar] [CrossRef]
- Gao, L.; Meiring, J.C.M.; Kraus, Y.; Wranik, M.; Weinert, T.; Pritzl, S.D.; Bingham, R.; Ntouliou, E.; Jansen, K.I.; Olieric, N.; et al. A Robust, GFP-Orthogonal Photoswitchable Inhibitor Scaffold Extends Optical Control over the Microtubule Cytoskeleton. Cell Chem. Biol. 2021, 28, 228–241. [Google Scholar] [CrossRef]
- Dorléans, A.; Gigant, B.; Ravelli, R.B.; Mailliet, P.; Mikol, V.; Knossow, M. Variations in the colchicine-binding domain provide insight into the structural switch of tubulin. Proc. Natl. Acad. Sci. USA 2009, 106, 13775–13779. [Google Scholar] [CrossRef] [Green Version]
- Vasquez, R.J.; Howell, B.; Yvon, A.M.; Wadsworth, P.; Cassimeris, L. Nanomolar concentrations of nocodazole alter microtubule dynamic instability in vivo and in vitro. Mol. Biol. Cell 1997, 8, 973–985. [Google Scholar] [CrossRef] [Green Version]
- Senese, S.; Lo, Y.C.; Huang, D.; Zangle, T.A.; Gholkar, A.A.; Robert, L.; Homet, B.; Ribas, A.; Summers, M.K.; Teitell, M.A.; et al. Chemical dissection of the cell cycle: Probes for cell biology and anti-cancer drug development. Cell Death Dis. 2014, 5, e1462. [Google Scholar] [CrossRef] [Green Version]
- McNamara, D.E.; Senese, S.; Yeates, T.O.; Torres, J.Z. Structures of potent anticancer compounds bound to tubulin. Protein Sci. 2015, 24, 1164–1172. [Google Scholar] [CrossRef] [Green Version]
- Swiss Institute of Bioinformatics. Available online: http://www.swissadme.ch/ (accessed on 11 May 2023).
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 5, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Shelanski, M.L.; Gaskin, F.; Cantor, C.R. Microtubule assembly in the absence of added nucleotides. Proc. Natl. Acad. Sci. USA 1973, 70, 765–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posadas, I.; López-Hernández, B.; Clemente, M.I.; Jimenez, J.L.; Ortega, P.; de la Mata, J.; Gómez, R.; Muñoz-Fernández, M.A.; Ceña, V. Highly efficient transfection of rat cortical neurons using carbosilane dendrimers unveils a neuroprotective role for HIF-1alpha in early chemical hypoxia-mediated neurotoxicity. Pharm. Res. 2009, 26, 1181–1191. [Google Scholar] [CrossRef]
- González, M.; Alcolea, P.J.; Alvarez, R.; Medarde, M.; Larraga, V.; Peláez, R. New diarylsulfonamide inhibitors of Leishmania infantum amastigotes. Int. J. Parasitol. Drugs Drug Resist. 2021, 16, 45–64. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R. KNIME: The Konstanz Information Miner. In Studies in Classification, Data Analysis, and Knowledge Organization; Springer: Berlin/Heidelberg, Germany, 2007; pp. 319–326. [Google Scholar]
- Velázquez-Libera, J.L.; Duran-Verdugo, F.; Valdes-Jimenez, A.; Núñez-Vivanco, G.; Caballero, J. LigRMSD: A web server for automatic structure matching and RMSD calculations among identical and similar compounds in protein-ligand docking. Bioinformatics 2020, 36, 2912–2914. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Marvin 17.8 ChemAxon. Available online: http://www.chemaxon.com (accessed on 11 May 2023).
- OpenEye Scientific. Available online: https://www.eyesopen.com (accessed on 11 May 2023).
- García-Pérez, C.; Peláez, R.; Theron, R.; López-Perez, J.L. JADOPPT: Java based AutoDock preparing and processing tool. Bioinformatics 2017, 33, 583–585. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.S.; Escobar, L.; Zhu, Y.J.; Cohen, Y.; Ballester, P.; Rebek, J., Jr.; Yu, Y. Relative hydrophilicities of cis and trans formamides. Proc. Natl. Acad. Sci. USA 2019, 116, 19815–19820. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; Decrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef]
- González, M.; Ovejero-Sánchez, M.; Vicente-Blázquez, A.; Álvarez, R.; Herrero, A.B.; Medarde, M.; González-Sarmiento, R.; Peláez, R. Microtubule Destabilizing Sulfonamides as an Alternative to Taxane-Based Chemotherapy. Int. J. Mol. Sci. 2021, 14, 1907. [Google Scholar] [CrossRef]
- González, M.; Ellahioui, Y.; Gallego, L.; Vicente-Blázquez, A.; Álvarez, R.; Medarde, M.; Peláez, R. Novel amino analogs of the trimethoxyphenyl ring in potent colchicine site ligands improve solubility by the masked polar group incorporation (MPGI) strategy. Bioorg. Chem. 2023, 131, 106282. [Google Scholar] [CrossRef]
- González, M.; Ovejero-Sánchez, M.; Vicente-Blázquez, A.; Medarde, M.; González-Sarmiento, R.; Peláez, R. Methoxy and bromo scans on N-(5-methoxyphenyl) methoxybenzenesulphonamides reveal potent cytotoxic compounds, especially against the human breast adenocarcinoma MCF7 cell line. J. Enzyme Inhib. Med. Chem. 2021, 36, 1029–1047. [Google Scholar] [CrossRef]
- Greene, L.M.; O’Boyle, N.M.; Nolan, D.P.; Meegan, M.J.; Zisterer, D.M. The vascular targeting agent Combretastatin-A4 directly induces autophagy in adenocarcinoma-derived colon cancer cells. Biochem. Pharmacol. 2012, 84, 612–624. [Google Scholar] [CrossRef]
- Aprile, S.; Del Grosso, E.; Grosa, G. Identification of the human UDP-glucuronosyltransferases involved in the glucuronidation of combretastatin A-4. Drug Metab. Dispos. 2010, 38, 1141–1146. [Google Scholar] [CrossRef] [Green Version]
- Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef]
- Kagawa, S.; Gu, J.; Honda, T.; McDonnell, T.J.; Swisher, S.G.; Roth, J.A.; Fang, B. Deficiency of caspase-3 in MCF7 cells blocks Bax-mediated nuclear fragmentation but not cell death. Clin. Cancer Res. 2001, 7, 1474–1480. [Google Scholar]
- Vicente-Blázquez, A.; González, M.; Álvarez, R.; Del Mazo, S.; Medarde, M.; Peláez, R. Antitubulin sulfonamides: The successful combination of an established drug class and a multifaceted target. Med. Res. Rev. 2019, 39, 775–830. [Google Scholar] [CrossRef]
- Gallego-Yerga, L.; Ochoa, R.; Lans, I.; Peña-Varas, C.; Alegría-Arcos, M.; Cossio, P.; Ramírez, D.; Peláez, R. Application of ensemble pharmacophore-based virtual screening to the discovery of novel antimitotic tubulin inhibitors. Comput. Struct. Biotechnol. J. 2021, 19, 4360–4372. [Google Scholar] [CrossRef]
- Álvarez, R.; Aramburu, L.; Puebla, P.; Caballero, E.; González, M.; Vicente, A.; Medarde, M.; Peláez, R. Pyridine Based Antitumour Compounds Acting at the Colchicine Site. Curr. Med. Chem. 2016, 23, 1100–1130. [Google Scholar] [CrossRef]
- Álvarez, R.; Puebla, P.; Díaz, J.F.; Bento, A.C.; García-Navas, R.; De la Iglesia-Vicente, J.; Mollinedo, F.; Andreu, J.M.; Medarde, M.; Peláez, R. Endowing indole-based tubulin inhibitors with an anchor for derivatization: Highly potent 3-substituted indolephenstatins and indoleisocombretastatins. J. Med. Chem. 2013, 56, 2813–2827. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stutzle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worldwide Protein Data Bank. Available online: http://www.wwpdb.org/ (accessed on 11 May 2023).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| No. | Ar | R | Solubility 1 (µg/mL) | TPSA 2 (Å2) | Log Po/w 2 |
| 1 | 2-Pyr | H | 48 | 75.28 | 2.83 |
| 2 | 3-Pyr | H | 57 | 75.28 | 2.81 |
| 3 | 4-Pyr | H | 55 | 75.28 | 2.79 |
| 4 | Imid | H | 86 | 91.07 | 2.44 |
| 5 | Ph | H | 5 | 62.39 | 3.64 |
| 6 | Naph | H | 3 | 62.39 | 4.47 |
| 7 | 2-Pyr | CONHEt | 98 | 95.59 | 2.79 |
| 8 | 3-Pyr | CONHEt | 102 | 95.59 | 2.70 |
| 9 | 4-Pyr | CONHEt | 106 | 95.59 | 2.69 |
| 10 | Imid | CONHEt | 178 | 111.38 | 2.29 |
| 11 | Ph | CONHEt | 16 | 82.70 | 3.46 |
| 12 | Naph | CONHEt | 7 | 82.70 | 4.31 |
| 13 | 2-Pyr | CHO | 90 | 83.56 | 2.50 |
| 14 | 3-Pyr | CHO | 95 | 83.56 | 2.51 |
| 15 | 4-Pyr | CHO | 93 | 83.56 | 2.51 |
| 16 | Imid | CHO | 165 | 99.35 | 2.16 |
| 17 | Ph | CHO | 12 | 70.67 | 3.23 |
| 18 | Naph | CHO | 6 | 70.67 | 4.15 |
| 19 | 2-Pyr | COMe | 87 | 83.56 | 2.58 |
| 20 | 3-Pyr | COMe | 93 | 83.56 | 2.54 |
| 21 | 4-Pyr | COMe | 90 | 83.56 | 2.54 |
| 22 | Imid | COMe | 171 | 99.35 | 2.25 |
| 23 | Ph | COMe | 13 | 70.67 | 3.29 |
| 24 | Naph | COMe | 8 | 70.67 | 4.19 |
| MI181 | 5.6 | 54.02 | 4.04 | ||
| CA-4 | 1.1 | 57.15 | 3.27 | ||
| ABT-751 | 40 | 108.93 | 2.27 | ||
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | Ar | R | HeLa | HepG2 | MCF7 | U87MG | T98G | GL261 | LNCaP | PC-3 | HT-29 | HEK-293 |
| 1 | 2-Pyr | H | 1.78 | 2.40 | 1.53 | 1.61 | 2.40 | 2.17 | 1.52 | 2.11 | 3.51 | >10 |
| 2 | 3-Pyr | H | 1.27 | 1.19 | 1.17 | 1.32 | 2.11 | 1.91 | 1.43 | 1.55 | 2.23 | >10 |
| 3 | 4-Pyr | H | 1.45 | 1.23 | 1.74 | 1.51 | 2.27 | 2.03 | 2.01 | 3.31 | 3.39 | >10 |
| 4 | Imid | H | 9.33 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| 5 | Ph | H | 4.36 | 5.78 | 6.31 | 8.31 | >10 | 7.25 | 5.97 | >10 | >10 | >10 |
| 6 | Naph | H | 3.71 | 4.51 | 4.23 | 6.54 | >10 | 6.25 | 4.32 | >10 | >10 | >10 |
| 7 | 2-Pyr | CONHEt | 0.275 | 0.198 | 0.251 | 0.062 | 0.125 | 0.096 | 0.247 | 0.330 | 0.303 | 8.81 |
| 8 | 3-Pyr | CONHEt | 0.085 | 0.094 | 0.034 | 0.009 | 0.052 | 0.025 | 0.088 | 0.161 | 0.208 | 7.97 |
| 9 | 4-Pyr | CONHEt | 0.209 | 0.103 | 0.203 | 0.065 | 0.181 | 0.077 | 0.221 | 0.313 | 0.311 | 8.03 |
| 10 | Imid | CONHEt | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| 11 | Ph | CONHEt | 2.71 | 2.45 | 2.04 | 2.91 | >10 | 2.93 | 2.37 | >10 | >10 | >10 |
| 12 | Naph | CONHEt | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| 13 | 2-Pyr | CHO | 0.331 | 0.321 | 0.221 | 0.254 | 0.327 | 0.195 | 0.315 | 0.423 | 0.557 | 9.31 |
| 14 | 3-Pyr | CHO | 0.107 | 0.083 | 0.015 | 0.037 | 0.075 | 0.035 | 0.093 | 0.195 | 0.322 | 8.22 |
| 15 | 4-Pyr | CHO | 0.311 | 0.239 | 0.207 | 0.214 | 0.257 | 0.260 | 0.285 | 0.412 | 0.471 | 8.51 |
| 16 | Imid | CHO | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| 17 | Ph | CHO | 3.40 | 5.55 | 5.02 | 5.27 | >10 | 6.41 | 4.01 | >10 | >10 | >10 |
| 18 | Naph | CHO | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| 19 | 2-Pyr | COMe | 1.55 | 1.88 | 2.03 | 1.41 | 2.44 | 1.38 | 2.07 | 3.01 | 2.54 | >10 |
| 20 | 3-Pyr | COMe | 1.03 | 0.813 | 0.911 | 0.632 | 0.825 | 0.622 | 1.39 | 1.62 | 1.52 | 9.90 |
| 21 | 4-Pyr | COMe | 1.22 | 1.49 | 1.97 | 1.27 | 2.30 | 1.59 | 1.81 | 2.63 | 2.23 | >10 |
| 22 | Imid | COMe | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| 23 | Ph | COMe | 3.90 | 4.29 | 6.30 | 7.51 | >10 | 6.33 | 4.95 | >10 | >10 | >10 |
| 24 | Naph | COMe | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| ABT-751 | 0.388 | 0.327 | 0.180 | 0.375 | 0.421 | 0.309 | 0.325 | 0.422 | 0.213 | 1.23 | ||
| TMZ | >10 | >10 | >10 | 135 | 241 | 179 | >10 | >10 | >10 | >10 | ||
| DTX | 0.051 | 0.055 | 0.013 | 0.022 | 0.059 | 0.035 | 0.010 | 0.020 | 0.307 | 1.55 | ||
| |||||||
|---|---|---|---|---|---|---|---|
| No. | Ar | R | IC50 1 | No. | Ar | R | IC50 1 |
| 1 | 2-Pyr | H | 3.47 | 13 | 2-Pyr | CHO | 0.540 |
| 2 | 3-Pyr | H | 2.15 | 14 | 3-Pyr | CHO | 0.307 |
| 3 | 4-Pyr | H | 3.27 | 15 | 4-Pyr | CHO | 0.459 |
| 4 | Imid | H | >10 | 16 | Imid | CHO | >10 |
| 5 | Ph | H | >10 | 17 | Ph | CHO | >10 |
| 6 | Naph | H | >10 | 18 | Naph | CHO | >10 |
| 7 | 2-Pyr | CONHEt | 0.315 | 19 | 2-Pyr | COMe | 2.93 |
| 8 | 3-Pyr | CONHEt | 0.215 | 20 | 3-Pyr | COMe | 1.48 |
| 9 | 4-Pyr | CONHEt | 0.305 | 21 | 4-Pyr | COMe | 2.55 |
| 10 | Imid | CONHEt | >10 | 22 | Imid | COMe | >10 |
| 11 | Ph | CONHEt | >10 | 23 | Ph | COMe | >10 |
| 12 | Naph | CONHEt | >10 | 24 | Naph | COMe | >10 |
| ABT-751 | 0.250 | TMZ | >10 | ||||
| No. | Ar | R | % TPI | IC50 | No. | Ar | R | % TPI 1 | IC50 2 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2-Pyr | H | 37 | >10 | 13 | 2-Pyr | CHO | 80 | 5.0 |
| 2 | 3-Pyr | H | 42 | >10 | 14 | 3-Pyr | CHO | 87 | 4.1 |
| 3 | 4-Pyr | H | 29 | >10 | 15 | 4-Pyr | CHO | 85 | 4.5 |
| 4 | Imid | H | 0 | >10 | 16 | Imid | CHO | 0 | >10 |
| 5 | Ph | H | 0 | >10 | 17 | Ph | CHO | 4 | >10 |
| 6 | Naph | H | 7 | >10 | 18 | Naph | CHO | 0 | >10 |
| 7 | 2-Pyr | CONHEt | 87 | 4.2 | 19 | 2-Pyr | COMe | 43 | >10 |
| 8 | 3-Pyr | CONHEt | 100 | 2.9 | 20 | 3-Pyr | COMe | 57 | 8.1 |
| 9 | 4-Pyr | CONHEt | 96 | 3.8 | 21 | 4-Pyr | COMe | 41 | >10 |
| 10 | Imid | CONHEt | 0 | >10 | 22 | Imid | COMe | 0 | >10 |
| 11 | Ph | CONHEt | 6 | >10 | 23 | Ph | COMe | 3 | >10 |
| 12 | Naph | CONHEt | 0 | >10 | 24 | Naph | COMe | 0 | >10 |
| ABT-751 | 4.4 | MI-181 | 4.6 | ||||||
| CA-4 | 2.8 | ||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallego-Yerga, L.; Ceña, V.; Peláez, R. Potent and Selective Benzothiazole-Based Antimitotics with Improved Water Solubility: Design, Synthesis, and Evaluation as Novel Anticancer Agents. Pharmaceutics 2023, 15, 1698. https://doi.org/10.3390/pharmaceutics15061698
Gallego-Yerga L, Ceña V, Peláez R. Potent and Selective Benzothiazole-Based Antimitotics with Improved Water Solubility: Design, Synthesis, and Evaluation as Novel Anticancer Agents. Pharmaceutics. 2023; 15(6):1698. https://doi.org/10.3390/pharmaceutics15061698
Chicago/Turabian StyleGallego-Yerga, Laura, Valentín Ceña, and Rafael Peláez. 2023. "Potent and Selective Benzothiazole-Based Antimitotics with Improved Water Solubility: Design, Synthesis, and Evaluation as Novel Anticancer Agents" Pharmaceutics 15, no. 6: 1698. https://doi.org/10.3390/pharmaceutics15061698