

The Resistance to EGFR-TKIs in Non-Small Cell Lung Cancer: From Molecular Mechanisms to Clinical Application of New Therapeutic Strategies
 , ,
, ,
Abstract
:1. Introduction
1.1. Epidermal Growth Factor Receptor Pathway in NSCLC
1.1.1. Common EGFR mutations
1.1.2. Uncommon EGFR Mutations
1.1.3. Other EGFR Alterations
1.2. Clinical Trials
1.2.1. First-Generation EGFR-TKIs: Gefitinib, Erlotinib, and Icotinib
1.2.2. Second-Generation EGFR-TKIs
1.2.3. Third-Generation EGFR-TKI
1.2.4. EGFR-TKIs Specific for Ins20
1.2.5. EGFR-TKI Treatment Combinations as First-Line Therapy
2. Mechanisms of Resistance
2.1. Intrinsic Resistance
2.1.1. Intrinsic Resistance to First-/Second-Generation EGFR-TKIs
2.1.2. Intrinsic Resistance to Third-Generation EGFR-TKIs
2.2. Acquired Resistance
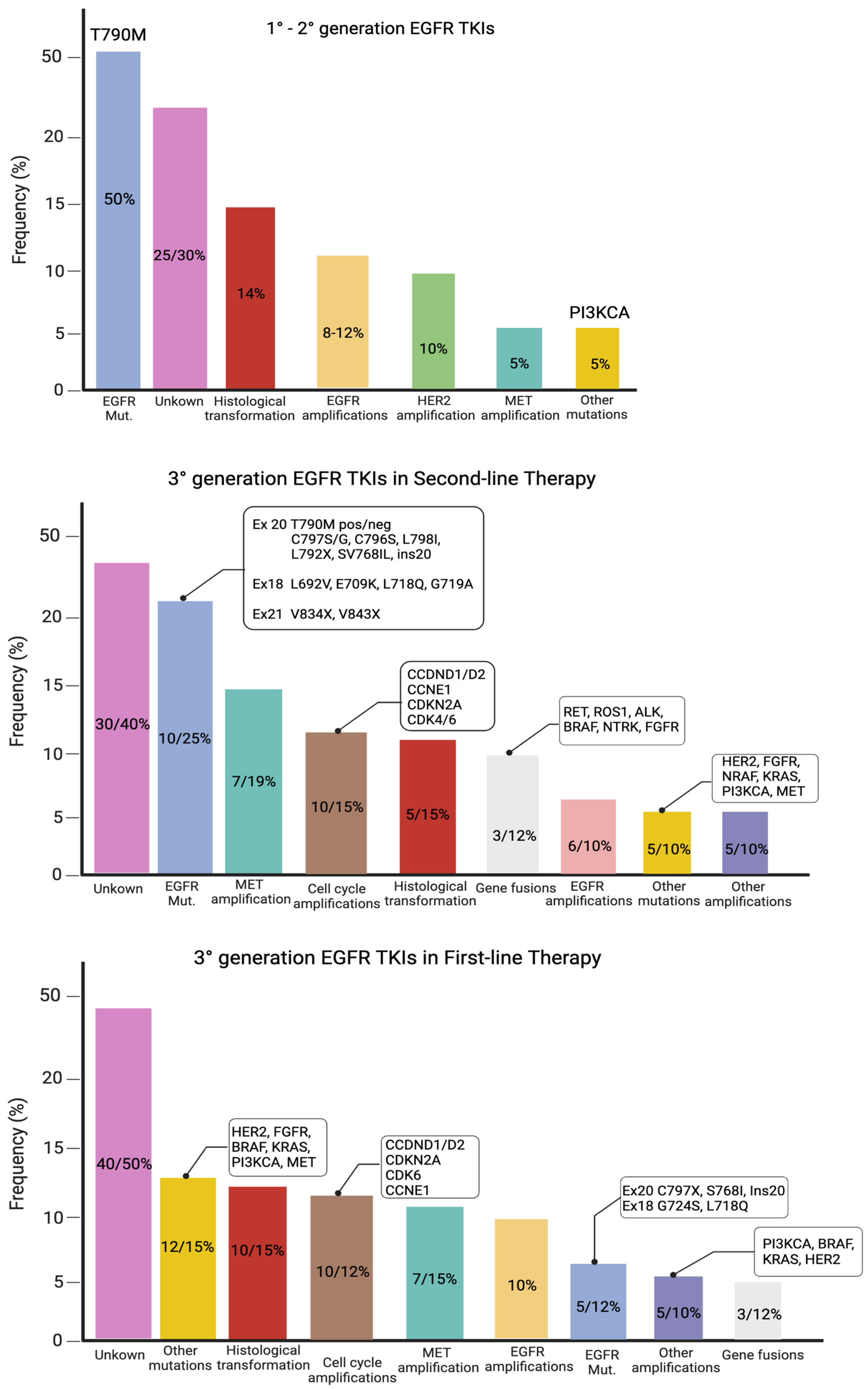
2.2.1. EGFR-Dependent Mechanisms: Secondary EGFR Mutations
EGFR T790 Mutation
Tertiary EGFR Mutations: Resistance to Second-Line Third-Generation EGFR-TKI
Secondary EGFR Mutations: Resistance to First-Line Third-Generation EGFR-TKI
Rare EGFR Mutations
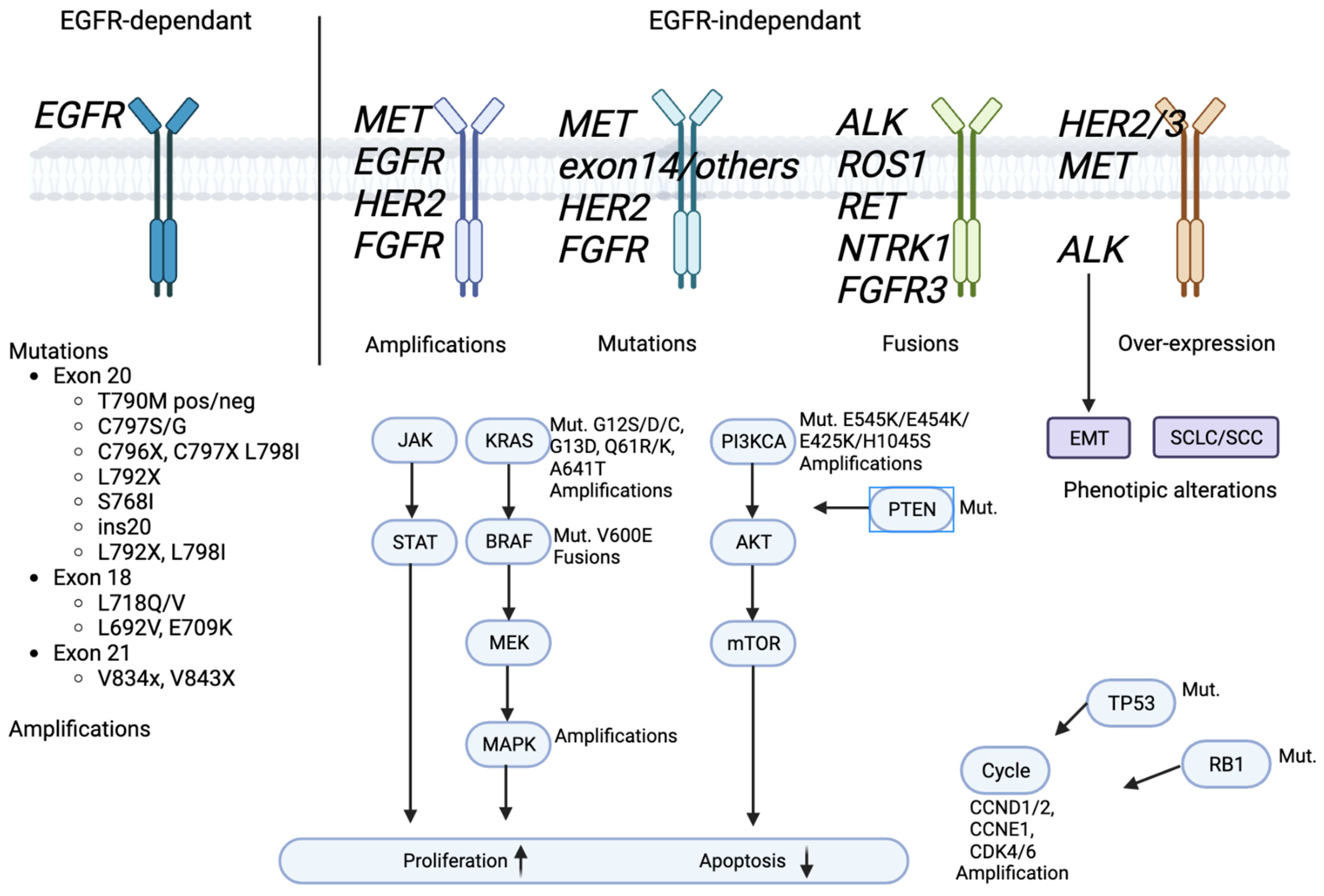
2.2.2. EGFR-Independent Mechanisms: Alternative Pathways
Oncogene Amplification
Rare Gene Mutations
Gene Fusions
Activation of Cell Receptors
Phenotypic Transformation
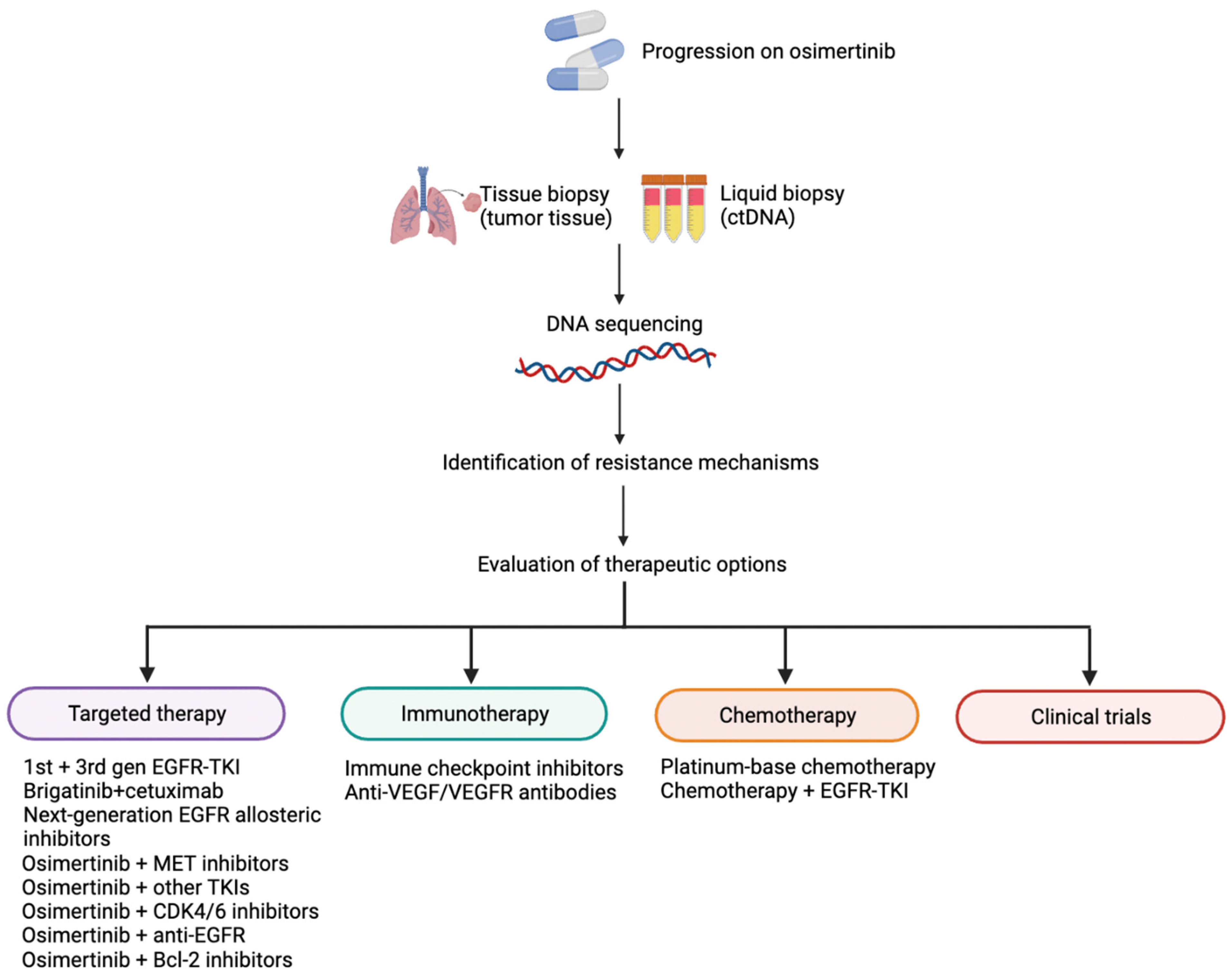
3. Future Perspectives
3.1. Other Third-Generation EGFR-TKIs
3.2. Therapeutic Options in T790M-cis-C797S Mutations
3.3. Next-Generation EGFR Allosteric Inhibitors
3.4. Osimertinib Plus MET Inhibitors
3.5. Osimertinib Plus CDK4/6 Inhibitors
3.6. EGFR-TKIs Plus Anti-EGFR Antibodies
3.7. Third-Generation EGFR-TKI Plus Bcl-2 Inhibitor
3.8. Chemotherapy
3.9. Immunotherapy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bade, B.C.; Dela Cruz, C.S. Lung Cancer 2020: Epidemiology, Etiology, and Prevention. Clin. Chest Med. 2020, 41, 1–24. [Google Scholar] [CrossRef]
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.A.; Hughes, B.G. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl. Lung Cancer Res. 2015, 4, 36–54. [Google Scholar] [CrossRef] [PubMed]
- Leduc, C.; Merlio, J.P.; Besse, B.; Blons, H.; Debieuvre, D.; Bringuier, P.P.; Monnet, I.; Rouquette, I.; Fraboulet-Moreau, S.; Lemoine, A.; et al. Clinical and molecular characteristics of non-small-cell lung cancer (NSCLC) harboring EGFR mutation: Results of the nationwide French Cooperative Thoracic Intergroup (IFCT) program. Ann. Oncol. 2017, 28, 2715–2724. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, P.; Zhang, C.; Ma, Z. Epidermal growth factor receptor (EGFR): A rising star in the era of precision medicine of lung cancer. Oncotarget 2017, 8, 50209–50220. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Bílek, O.; Holánek, M.; Berkovcová, J.; Horký, O.; Kazda, T.; Čoupková, H.; Špelda, S.; Kristková, L.; Zvaríková, M.; Podhorec, J.; et al. Uncommon EGFR Mutations in Non-Small Cell Lung Cancer and Their Impact on the Treatment. Klin. Onkol. Cas. Ceske A Slov. Onkol. Spol. 2019, 32, 6–12. [Google Scholar] [CrossRef]
- Li, W.Q.; Cui, J.W. Non-small cell lung cancer patients with ex19del or exon 21 L858R mutation: Distinct mechanisms, different efficacies to treatments. J. Cancer Res. Clin. Oncol. 2020, 146, 2329–2338. [Google Scholar] [CrossRef]
- Russo, A.; Franchina, T.; Ricciardi, G.; Battaglia, A.; Picciotto, M.; Adamo, V. Heterogeneous Responses to Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors (TKIs) in Patients with Uncommon EGFR Mutations: New Insights and Future Perspectives in this Complex Clinical Scenario. Int. J. Mol. Sci. 2019, 20, 1431. [Google Scholar] [CrossRef]
- Beau-Faller, M.; Prim, N.; Ruppert, A.M.; Nanni-Metéllus, I.; Lacave, R.; Lacroix, L.; Escande, F.; Lizard, S.; Pretet, J.L.; Rouquette, I.; et al. Rare EGFR exon 18 and exon 20 mutations in non-small-cell lung cancer on 10 117 patients: A multicentre observational study by the French ERMETIC-IFCT network. Ann. Oncol. 2014, 25, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin. Cancer Biol. 2020, 61, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Canepa, H.M.; Bailey, A.S.; Nakayama, S.; Yamaguchi, N.; Goldstein, M.A.; Huberman, M.S.; Costa, D.B. Compound EGFR mutations and response to EGFR tyrosine kinase inhibitors. J. Thorac. Oncol. 2013, 8, 45–51. [Google Scholar] [CrossRef]
- Lin, Y.T.; Tsai, T.H.; Wu, S.G.; Liu, Y.N.; Yu, C.J.; Shih, J.Y. Complex EGFR mutations with secondary T790M mutation confer shorter osimertinib progression-free survival and overall survival in advanced non-small cell lung cancer. Lung Cancer 2020, 145, 1–9. [Google Scholar] [CrossRef]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; McCoach, C.E.; McGranahan, N.; Wilson, G.A.; Birkbak, N.J.; Olivas, V.R.; et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 2017, 49, 1693–1704. [Google Scholar] [CrossRef]
- Blons, H.; Oudart, J.B.; Merlio, J.P.; Debieuvre, D.; de Fraipont, F.; Audigier-Valette, C.; Escande, F.; Hominal, S.; Bringuier, P.P.; Fraboulet-Moreau, S.; et al. PTEN, ATM, IDH1 mutations and MAPK pathway activation as modulators of PFS and OS in patients treated by first line EGFR TKI, an ancillary study of the French Cooperative Thoracic Intergroup (IFCT) Biomarkers France project. Lung Cancer 2021, 151, 69–75. [Google Scholar] [CrossRef]
- Canale, M.; Petracci, E.; Delmonte, A.; Bronte, G.; Chiadini, E.; Ludovini, V.; Dubini, A.; Papi, M.; Baglivo, S.; De Luigi, N.; et al. Concomitant TP53 Mutation Confers Worse Prognosis in EGFR-Mutated Non-Small Cell Lung Cancer Patients Treated with TKIs. J. Clin. Med. 2020, 9, 1047. [Google Scholar] [CrossRef]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef]
- Kohsaka, S.; Petronczki, M.; Solca, F.; Maemondo, M. Tumor clonality and resistance mechanisms in EGFR mutation-positive non-small-cell lung cancer: Implications for therapeutic sequencing. Future Oncol. 2019, 15, 637–652. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Thress, K.S.; Alden, R.S.; Lawrance, R.; Paweletz, C.P.; Cantarini, M.; Yang, J.C.; Barrett, J.C.; Jänne, P.A. Association Between Plasma Genotyping and Outcomes of Treatment with Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 3375–3382. [Google Scholar] [CrossRef] [PubMed]
- Vaclova, T.; Grazini, U.; Ward, L.; O’Neill, D.; Markovets, A.; Huang, X.; Chmielecki, J.; Hartmaier, R.; Thress, K.S.; Smith, P.D.; et al. Clinical impact of subclonal EGFR T790M mutations in advanced-stage EGFR-mutant non-small-cell lung cancers. Nat. Commun. 2021, 12, 1780. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients with EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef]
- Beau-Faller, M.; Pencreach, E.; Leduc, C.; Blons, H.; Merlio, J.-P.; Bringuier, P.-P.; de Fraipont, F.; Escande, F.; Lemoine, A.; Ouafik, L.H.; et al. Independent prognostic value of ultra-sensitive quantification of tumor pre-treatment T790M subclones in EGFR mutated non-small cell lung cancer (NSCLC) treated by first/second generation TKI, depends on variant allele frequency (VAF): Results of the French cooperative thoracic intergroup (IFCT) biomarkers France project. Lung Cancer 2020, 140, 19–26. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Mitsudomi, T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: Perspectives for individualized treatment strategy. Cancer Sci. 2016, 107, 1179–1186. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Kobayashi, K.; Maemondo, M.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann. Oncol. 2013, 24, 54–59. [Google Scholar] [CrossRef]
- Miyauchi, E.; Inoue, A.; Kobayashi, K.; Maemondo, M.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Saijo, Y.; Yoshizawa, H.; et al. Efficacy of chemotherapy after first-line gefitinib therapy in EGFR mutation-positive advanced non-small cell lung cancer-data from a randomized Phase III study comparing gefitinib with carboplatin plus paclitaxel (NEJ002). Jpn. J. Clin. Oncol. 2015, 45, 670–676. [Google Scholar] [CrossRef]
- Fukuoka, M.; Wu, Y.L.; Thongprasert, S.; Sunpaweravong, P.; Leong, S.S.; Sriuranpong, V.; Chao, T.Y.; Nakagawa, K.; Chu, D.T.; Saijo, N.; et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J. Clin. Oncol. 2011, 29, 2866–2874. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Yoshioka, H.; Shimokawa, M.; Seto, T.; Morita, S.; Yatabe, Y.; Okamoto, I.; Tsurutani, J.; Satouchi, M.; Hirashima, T.; Atagi, S.; et al. Final overall survival results of WJTOG3405, a randomized phase III trial comparing gefitinib versus cisplatin with docetaxel as the first-line treatment for patients with stage IIIB/IV or postoperative recurrent EGFR mutation-positive non-small-cell lung cancer. Ann. Oncol. 2019, 30, 1978–1984. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Final overall survival results from a randomised, phase III study of erlotinib versus chemotherapy as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer (OPTIMAL, CTONG-0802). Ann. Oncol. 2015, 26, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Zhou, C.; Liam, C.K.; Wu, G.; Liu, X.; Zhong, Z.; Lu, S.; Cheng, Y.; Han, B.; Chen, L.; et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: Analyses from the phase III, randomized, open-label, ENSURE study. Ann. Oncol. 2015, 26, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.K.; Wang, L.; Han, B.H.; Li, W.; Yu, P.; Liu, Y.P.; Ding, C.M.; Song, X.; Ma, Z.Y.; Ren, X.L.; et al. First-line icotinib versus cisplatin/pemetrexed plus pemetrexed maintenance therapy for patients with advanced EGFR mutation-positive lung adenocarcinoma (CONVINCE): A phase 3, open-label, randomized study. Ann. Oncol. 2017, 28, 2443–2450. [Google Scholar] [CrossRef]
- Yang, J.C.; Wu, Y.L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zhou, C.; Hu, C.P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): An open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Mok, T.S.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Chawla, A.; Rosell, R.; Corral, J.; Migliorino, M.R.; et al. Updated Overall Survival in a Randomized Study Comparing Dacomitinib with Gefitinib as First-Line Treatment in Patients with Advanced Non-Small-Cell Lung Cancer and EGFR-Activating Mutations. Drugs 2021, 81, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Tsuboi, M.; He, J.; John, T.; Grohe, C.; Majem, M.; Goldman, J.W.; Laktionov, K.; Kim, S.-W.; Kato, T.; et al. Osimertinib in Resected EGFR-Mutated Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Le, X.; Cornelissen, R.; Garassino, M.; Clarke, J.M.; Tchekmedyian, N.; Goldman, J.W.; Leu, S.Y.; Bhat, G.; Lebel, F.; Heymach, J.V.; et al. Poziotinib in Non-Small-Cell Lung Cancer Harboring HER2 Exon 20 Insertion Mutations after Prior Therapies: ZENITH20-2 Trial. J. Clin. Oncol. 2022, 40, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Ramalingam, S.S.; Kim, T.M.; Kim, S.W.; Yang, J.C.; Riely, G.J.; Mekhail, T.; Nguyen, D.; Garcia Campelo, M.R.; Felip, E.; et al. Treatment Outcomes and Safety of Mobocertinib in Platinum-Pretreated Patients with EGFR Exon 20 Insertion-Positive Metastatic Non-Small Cell Lung Cancer: A Phase 1/2 Open-label Nonrandomized Clinical Trial. JAMA Oncol. 2021, 7, e214761. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef] [PubMed]
- Cortot, A.B.; Madroszyk, A.; Giroux-Leprieur, E.; Molinier, O.; Quoix, E.; Bérard, H.; Otto, J.; Rault, I.; Moro-Sibilot, D.; Raimbourg, J.; et al. First-Line Afatinib plus Cetuximab for EGFR-Mutant Non-Small Cell Lung Cancer: Results from the Randomized Phase II IFCT-1503 ACE-Lung Study. Clin. Cancer Res. 2021, 27, 4168–4176. [Google Scholar] [CrossRef]
- Laface, C.; Fedele, P.; Maselli, F.M.; Ambrogio, F.; Foti, C.; Molinari, P.; Ammendola, M.; Lioce, M.; Ranieri, G. Targeted Therapy for Hepatocellular Carcinoma: Old and New Opportunities. Cancers 2022, 14, 4028. [Google Scholar] [CrossRef]
- Ranieri, G.; Laface, C.; Laforgia, M.; De Summa, S.; Porcelli, M.; Macina, F.; Ammendola, M.; Molinari, P.; Lauletta, G.; Di Palo, A.; et al. Bevacizumab Plus FOLFOX-4 Combined with Deep Electro-Hyperthermia as First-line Therapy in Metastatic Colon Cancer: A Pilot Study. Front. Oncol. 2020, 10, 590707. [Google Scholar] [CrossRef]
- Laface, C.; Laforgia, M.; Zito, A.F.; Loisi, D.; Zizzo, N.; Tamma, R.; Gadaleta, C.D.; Porcelli, M.; Currò, G.; Ammendola, M.; et al. Chymase-positive Mast cells correlate with tumor angiogenesis: First report in pancreatic cancer patients. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 6862–6873. [Google Scholar] [CrossRef]
- Laforgia, M.; Calabrò, C.; Scattone, A.; Laface, C.; Porcelli, M.; Gadaleta, C.D.; Nardulli, P.; Ranieri, G. Pharmacotherapy in Mast Cell Leukemia. Expert Opin. Pharmacother. 2020, 21, 1059–1069. [Google Scholar] [CrossRef]
- Rosell, R.; Dafni, U.; Felip, E.; Curioni-Fontecedro, A.; Gautschi, O.; Peters, S.; Massutí, B.; Palmero, R.; Aix, S.P.; Carcereny, E.; et al. Erlotinib and bevacizumab in patients with advanced non-small-cell lung cancer and activating EGFR mutations (BELIEF): An international, multicentre, single-arm, phase 2 trial. Lancet Respir. Med. 2017, 5, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Seto, T.; Nishio, M.; Goto, K.; Yamamoto, N.; Okamoto, I.; Yamanaka, T.; Tanaka, M.; Takahashi, K.; Fukuoka, M. Erlotinib plus bevacizumab vs erlotinib monotherapy as first-line treatment for advanced EGFR mutation-positive non-squamous non-small-cell lung cancer: Survival follow-up results of the randomized JO25567 study. Lung Cancer 2021, 151, 20–24. [Google Scholar] [CrossRef]
- Nakagawa, K.; Garon, E.B.; Seto, T.; Nishio, M.; Ponce Aix, S.; Paz-Ares, L.; Chiu, C.-H.; Park, K.; Novello, S.; Nadal, E.; et al. Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non-small-cell lung cancer (RELAY): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Luo, W.; Li, W.; Wang, T.; Huang, L.; Xu, F. First-Generation EGFR-TKI Plus Chemotherapy Versus EGFR-TKI Alone as First-Line Treatment in Advanced NSCLC with EGFR Activating Mutation: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Front. Oncol. 2021, 11, 598265. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Feng, P.H.; Karaseva, N.; Kim, S.W.; Kim, T.M.; Lee, C.K.; Poltoratskiy, A.; Yanagitani, N.; Marshall, R.; Huang, X.; et al. Osimertinib plus platinum-pemetrexed in newly diagnosed epidermal growth factor receptor mutation-positive advanced/metastatic non-small-cell lung cancer: Safety run-in results from the FLAURA2 study. ESMO Open 2021, 6, 100271. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, E.; Feld, E.; Horn, L. Driven by Mutations: The Predictive Value of Mutation Subtype in EGFR-Mutated Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 612–623. [Google Scholar] [CrossRef]
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef]
- Dong, R.F.; Zhu, M.L.; Liu, M.M.; Xu, Y.T.; Yuan, L.L.; Bian, J.; Xia, Y.Z.; Kong, L.Y. EGFR mutation mediates resistance to EGFR tyrosine kinase inhibitors in NSCLC: From molecular mechanisms to clinical research. Pharmacol. Res. 2021, 167, 105583. [Google Scholar] [CrossRef]
- Santoni-Rugiu, E.; Melchior, L.C.; Urbanska, E.M.; Jakobsen, J.N.; Stricker, K.; Grauslund, M.; Sørensen, J.B. Intrinsic resistance to EGFR-Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer: Differences and Similarities with Acquired Resistance. Cancers 2019, 11, 923. [Google Scholar] [CrossRef]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Shi, X.; Zhao, J.; He, Q.; Chen, M.; Yan, J.; Ou, Q.; Wu, X.; Shao, Y.W.; Yu, X. Mechanisms of primary resistance to EGFR targeted therapy in advanced lung adenocarcinomas. Lung Cancer 2018, 124, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Bareschino, M.A.; Bianco, R.; Tortora, G.; Ciardiello, F. Primary and acquired resistance to anti-EGFR targeted drugs in cancer therapy. Differ. Res. Biol. Divers. 2007, 75, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef]
- Reita, D.; Pabst, L.; Pencreach, E.; Guérin, E.; Dano, L.; Rimelen, V.; Voegeli, A.-C.; Vallat, L.; Mascaux, C.; Beau-Faller, M. Molecular Mechanism of EGFR-TKI Resistance in EGFR-Mutated Non-Small Cell Lung Cancer: Application to Biological Diagnostic and Monitoring. Cancers 2021, 13, 4926. [Google Scholar] [CrossRef]
- Attili, I.; Karachaliou, N.; Conte, P.; Bonanno, L.; Rosell, R. Therapeutic approaches for T790M mutation positive non-small-cell lung cancer. Expert Rev. Anticancer. Ther. 2018, 18, 1021–1030. [Google Scholar] [CrossRef]
- Chen, L.Y.; Molina-Vila, M.A.; Ruan, S.Y.; Su, K.Y.; Liao, W.Y.; Yu, K.L.; Ho, C.C.; Shih, J.Y.; Yu, C.J.; Yang, J.C.; et al. Coexistence of EGFR T790M mutation and common activating mutations in pretreatment non-small cell lung cancer: A systematic review and meta-analysis. Lung Cancer 2016, 94, 46–53. [Google Scholar] [CrossRef]
- Eck, M.J.; Yun, C.-H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2010, 1804, 559–566. [Google Scholar] [CrossRef]
- De Pas, T.; Toffalorio, F.; Manzotti, M.; Fumagalli, C.; Spitaleri, G.; Catania, C.; Delmonte, A.; Giovannini, M.; Spaggiari, L.; de Braud, F.; et al. Activity of Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors in Patients with Non-small Cell Lung Cancer Harboring Rare Epidermal Growth Factor Receptor Mutations. J. Thorac. Oncol. 2011, 6, 1895–1901. [Google Scholar] [CrossRef]
- Costa, D.B. More Than Just an Oncogene Translocation and a Kinase Inhibitor: Kevin’s Story. J. Clin. Oncol. 2011, 30, 110–112. [Google Scholar] [CrossRef]
- Pao, W.; Girard, N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011, 12, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Molina, M.A.; Drozdowskyj, A.; Giménez-Capitán, A.; Bertran-Alamillo, J.; Karachaliou, N.; Gervais, R.; Massuti, B.; Wei, J.; Moran, T.; et al. The Impact of EGFR T790M Mutations and BIM mRNA Expression on Outcome in Patients with EGFR-Mutant NSCLC Treated with Erlotinib or Chemotherapy in the Randomized Phase III EURTAC Trial. Clin. Cancer Res. 2014, 20, 2001–2010. [Google Scholar] [CrossRef]
- Soejima, K.; Yasuda, H.; Hirano, T. Osimertinib for EGFR T790M mutation-positive non-small cell lung cancer. Expert Rev. Clin. Pharmacol. 2017, 10, 31–38. [Google Scholar] [CrossRef]
- Ji, H.; Zhao, X.; Yuza, Y.; Shimamura, T.; Li, D.; Protopopov, A.; Jung, B.L.; McNamara, K.; Xia, H.; Glatt, K.A.; et al. Epidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 7817–7822. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, I.; Kenyon, L.C.; Emlet, D.R.; Mori, T.; Sasaki, J.; Hirosako, S.; Ichikawa, Y.; Kishi, H.; Godwin, A.K.; Yoshioka, M.; et al. Expression of constitutively activated EGFRvIII in non-small cell lung cancer. Cancer Sci. 2003, 94, 50–56. [Google Scholar] [CrossRef]
- Sasaki, H.; Kawano, O.; Endo, K.; Yukiue, H.; Yano, M.; Fujii, Y. EGFRvIII mutation in lung cancer correlates with increased EGFR copy number. Oncol. Rep. 2007, 17, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Faber, A.C.; Corcoran, R.B.; Ebi, H.; Sequist, L.V.; Waltman, B.A.; Chung, E.; Incio, J.; Digumarthy, S.R.; Pollack, S.F.; Song, Y.; et al. BIM Expression in Treatment-Naïve Cancers Predicts Responsiveness to Kinase Inhibitors. Cancer Discov. 2011, 1, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Nokihara, H.; Shimizu, T.; Seto, T.; Keating, A.; Krivoshik, A.; Uegaki, K.; Morita, S.; Nakagawa, K.; Fukuoka, M. 9LBA Antitumor activity of ASP8273, an irreversible mutant selective EGFR-TKI, in NSCLC patients with tumors harboring EGFR activating mutations and T790M resistance mutation. Eur. J. Cancer 2014, 50, 198. [Google Scholar] [CrossRef]
- de Bruin, E.C.; Cowell, C.; Warne, P.H.; Jiang, M.; Saunders, R.E.; Melnick, M.A.; Gettinger, S.; Walther, Z.; Wurtz, A.; Heynen, G.J.; et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 2014, 4, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Calvayrac, O.; Mazières, J.; Figarol, S.; Marty-Detraves, C.; Raymond-Letron, I.; Bousquet, E.; Farella, M.; Clermont-Taranchon, E.; Milia, J.; Rouquette, I.; et al. The RAS-related GTPase RHOB confers resistance to EGFR-tyrosine kinase inhibitors in non-small-cell lung cancer via an AKT-dependent mechanism. EMBO Mol. Med. 2017, 9, 238–250. [Google Scholar] [CrossRef]
- Tsui, D.W.Y.; Murtaza, M.; Wong, A.S.C.; Rueda, O.M.; Smith, C.G.; Chandrananda, D.; Soo, R.A.; Lim, H.L.; Goh, B.C.; Caldas, C.; et al. Dynamics of multiple resistance mechanisms in plasma DNA during EGFR-targeted therapies in non-small cell lung cancer. EMBO Mol. Med. 2018, 10, e7945. [Google Scholar] [CrossRef]
- Park, K.-S.; Raffeld, M.; Moon, Y.W.; Xi, L.; Bianco, C.; Pham, T.; Lee, L.C.; Mitsudomi, T.; Yatabe, Y.; Okamoto, I.; et al. CRIPTO1 expression in EGFR-mutant NSCLC elicits intrinsic EGFR-inhibitor resistance. J. Clin. Investig. 2014, 124, 3003–3015. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.; Woo, K.M.; Sima, C.S.; Plodkowski, A.; Hellmann, M.D.; Chaft, J.E.; Kris, M.G.; Arcila, M.E.; Ladanyi, M.; Drilon, A. Impact of Concurrent PIK3CA Mutations on Response to EGFR Tyrosine Kinase Inhibition in EGFR-Mutant Lung Cancers and on Prognosis in Oncogene-Driven Lung Adenocarcinomas. J. Thorac. Oncol. 2015, 10, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Canale, M.; Petracci, E.; Delmonte, A.; Chiadini, E.; Dazzi, C.; Papi, M.; Capelli, L.; Casanova, C.; De Luigi, N.; Mariotti, M.; et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated with First-Line Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2017, 23, 2195–2202. [Google Scholar] [CrossRef]
- Karachaliou, N.; Chaib, I.; Cardona, A.F.; Berenguer, J.; Bracht, J.W.P.; Yang, J.; Cai, X.; Wang, Z.; Hu, C.; Drozdowskyj, A.; et al. Common Co-activation of AXL and CDCP1 in EGFR-mutation-positive Non-smallcell Lung Cancer Associated with Poor Prognosis. EBioMedicine 2018, 29, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Michels, S.; Heydt, C.; van Veggel, B.; Deschler-Baier, B.; Pardo, N.; Monkhorst, K.; Rüsseler, V.; Stratmann, J.; Griesinger, F.; Steinhauser, S.; et al. Genomic Profiling Identifies Outcome-Relevant Mechanisms of Innate and Acquired Resistance to Third-Generation Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Therapy in Lung Cancer. JCO Precis. Oncol. 2019, 3, PO.18.00210. [Google Scholar] [CrossRef]
- Minari, R.; Bordi, P.; Del Re, M.; Facchinetti, F.; Mazzoni, F.; Barbieri, F.; Camerini, A.; Comin, C.E.; Gnetti, L.; Azzoni, C.; et al. Primary resistance to osimertinib due to SCLC transformation: Issue of T790M determination on liquid re-biopsy. Lung Cancer 2018, 115, 21–27. [Google Scholar] [CrossRef]
- Jackman, D.; Pao, W.; Riely, G.J.; Engelman, J.A.; Kris, M.G.; Jänne, P.A.; Lynch, T.; Johnson, B.E.; Miller, V.A. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 357–360. [Google Scholar] [CrossRef]
- Ortiz-Cuaran, S.; Scheffler, M.; Plenker, D.; Dahmen, I.; Scheel, A.H.; Fernandez-Cuesta, L.; Meder, L.; Lovly, C.M.; Persigehl, T.; Merkelbach-Bruse, S.; et al. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin. Cancer Res. 2016, 22, 4837–4847. [Google Scholar] [CrossRef]
- Gandara, D.R.; Li, T.; Lara, P.N.; Kelly, K.; Riess, J.W.; Redman, M.W.; Mack, P.C. Acquired Resistance to Targeted Therapies Against Oncogene-Driven Non–Small-Cell Lung Cancer: Approach to Subtyping Progressive Disease and Clinical Implications. Clin. Lung Cancer 2014, 15, 1–6. [Google Scholar] [CrossRef]
- Passaro, A.; Jänne, P.A.; Mok, T.; Peters, S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Arcila, M.E.; Sima, C.S.; Riely, G.J.; Chmielecki, J.; Kris, M.G.; Pao, W.; Ladanyi, M.; Miller, V.A. Acquired Resistance to EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Lung Cancer: Distinct Natural History of Patients with Tumors Harboring the T790M Mutation. Clin. Cancer Res. 2011, 17, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.-e.C.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Chmielecki, J.; Mok, T.; Wu, Y.L.; Han, J.Y.; Ahn, M.J.; Ramalingam, S.S.; John, T.; Okamoto, I.; Yang, J.C.; Shepherd, F.A.; et al. Analysis of acquired resistance mechanisms to osimertinib in patients with EGFR-mutated advanced non-small cell lung cancer from the AURA3 trial. Nat. Commun. 2023, 14, 1071. [Google Scholar] [CrossRef]
- Fang, W.; Gan, J.; Huang, Y.; Zhou, H.; Zhang, L. Acquired EGFR L718V Mutation and Loss of T790M-Mediated Resistance to Osimertinib in a Patient with NSCLC Who Responded to Afatinib. J. Thorac. Oncol. 2019, 14, e274–e275. [Google Scholar] [CrossRef]
- Lin, C.C.; Shih, J.Y.; Yu, C.J.; Ho, C.C.; Liao, W.Y.; Lee, J.H.; Tsai, T.H.; Su, K.Y.; Hsieh, M.S.; Chang, Y.L.; et al. Outcomes in patients with non-small-cell lung cancer and acquired Thr790Met mutation treated with osimertinib: A genomic study. Lancet Respir. Med. 2018, 6, 107–116. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.A.; Han, J.Y.; Ahn, M.J.; Ramalingam, S.S.; Delmonte, A.; Hsia, T.C.; Laskin, J.; Kim, S.W.; He, Y.; Tsai, C.M.; et al. Epidermal growth factor receptor mutation analysis in tissue and plasma from the AURA3 trial: Osimertinib versus platinum-pemetrexed for T790M mutation-positive advanced non-small cell lung cancer. Cancer 2020, 126, 373–380. [Google Scholar] [CrossRef]
- Lu, X.; Yu, L.; Zhang, Z.; Ren, X.; Smaill, J.B.; Ding, K. Targeting EGFR(L858R/T790M) and EGFR(L858R/T790M/C797S) resistance mutations in NSCLC: Current developments in medicinal chemistry. Med. Res. Rev. 2018, 38, 1550–1581. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Chan, J.M.; Kubota, D.; Sato, H.; Rizvi, H.; Daneshbod, Y.; Chang, J.C.; Paik, P.K.; Offin, M.; Arcila, M.E.; et al. Tumor Analyses Reveal Squamous Transformation and Off-Target Alterations As Early Resistance Mechanisms to First-line Osimertinib in EGFR-Mutant Lung Cancer. Clin. Cancer Res. 2020, 26, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.-H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yang, N.; Ou, Q.; Xiang, Y.; Jiang, T.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Shao, Y.W.; et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients. Clin. Cancer Res. 2018, 24, 3097–3107. [Google Scholar] [CrossRef] [PubMed]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef]
- Arulananda, S.; Do, H.; Musafer, A.; Mitchell, P.; Dobrovic, A.; John, T. Combination Osimertinib and Gefitinib in C797S and T790M EGFR-Mutated Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1728–1732. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, J.J.; Huang, J.; Ye, J.Y.; Zhang, X.C.; Tu, H.Y.; Han-Zhang, H.; Wu, Y.L. Lung Adenocarcinoma Harboring EGFR T790M and In Trans C797S Responds to Combination Therapy of First- and Third-Generation EGFR TKIs and Shifts Allelic Configuration at Resistance. J. Thorac. Oncol. 2017, 12, 1723–1727. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mao, T.; Wang, J.; Zheng, H.; Hu, Z.; Cao, P.; Yang, S.; Zhu, L.; Guo, S.; Zhao, X.; et al. Toward the next generation EGFR inhibitors: An overview of osimertinib resistance mediated by EGFR mutations in non-small cell lung cancer. Cell Commun. Signal. 2023, 21, 71. [Google Scholar] [CrossRef]
- Ou, S.I.; Cui, J.; Schrock, A.B.; Goldberg, M.E.; Zhu, V.W.; Albacker, L.; Stephens, P.J.; Miller, V.A.; Ali, S.M. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/R and L792F/H mutations in one EGFR (L858R/T790M) NSCLC patient who progressed on osimertinib. Lung Cancer 2017, 108, 228–231. [Google Scholar] [CrossRef]
- Zheng, D.; Hu, M.; Bai, Y.; Zhu, X.; Lu, X.; Wu, C.; Wang, J.; Liu, L.; Wang, Z.; Ni, J.; et al. EGFR G796D mutation mediates resistance to osimertinib. Oncotarget 2017, 8, 49671–49679. [Google Scholar] [CrossRef]
- Lim, S.M.; Syn, N.L.; Cho, B.C.; Soo, R.A. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat. Rev. 2018, 65, 1–10. [Google Scholar] [CrossRef]
- Ercan, D.; Choi, H.G.; Yun, C.H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Jänne, P.A. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. [Google Scholar] [CrossRef]
- Zhang, Y.; He, B.; Zhou, D.; Li, M.; Hu, C. Newly emergent acquired EGFR exon 18 G724S mutation after resistance of a T790M specific EGFR inhibitor osimertinib in non-small-cell lung cancer: A case report. OncoTargets Ther. 2019, 12, 51–56. [Google Scholar] [CrossRef]
- Oztan, A.; Fischer, S.; Schrock, A.B.; Erlich, R.L.; Lovly, C.M.; Stephens, P.J.; Ross, J.S.; Miller, V.; Ali, S.M.; Ou, S.I.; et al. Emergence of EGFR G724S mutation in EGFR-mutant lung adenocarcinoma post progression on osimertinib. Lung Cancer 2017, 111, 84–87. [Google Scholar] [CrossRef]
- Brown, B.P.; Zhang, Y.K.; Westover, D.; Yan, Y.; Qiao, H.; Huang, V.; Du, Z.; Smith, J.A.; Ross, J.S.; Miller, V.A.; et al. On-target Resistance to the Mutant-Selective EGFR Inhibitor Osimertinib Can Develop in an Allele-Specific Manner Dependent on the Original EGFR-Activating Mutation. Clin. Cancer Res. 2019, 25, 3341–3351. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.E.; Okamoto, I.; Sriuranpong, V.; Vansteenkiste, J.; Imamura, F.; Lee, J.S.; Pang, Y.K.; Cobo, M.; Kasahara, K.; Cheng, Y.; et al. Tissue and Plasma EGFR Mutation Analysis in the FLAURA Trial: Osimertinib versus Comparator EGFR Tyrosine Kinase Inhibitor as First-Line Treatment in Patients with EGFR-Mutated Advanced Non-Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 6644–6652. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Hoyo, A.; Moliner, L.; Arriola, E. Acquired Mechanisms of Resistance to Osimertinib—The Next Challenge. Cancers 2022, 14, 1931. [Google Scholar] [CrossRef]
- Kohsaka, S.; Nagano, M.; Ueno, T.; Suehara, Y.; Hayashi, T.; Shimada, N.; Takahashi, K.; Suzuki, K.; Takamochi, K.; Takahashi, F.; et al. A method of high-throughput functional evaluation of EGFR gene variants of unknown significance in cancer. Sci. Transl. Med. 2017, 9, eaan6566. [Google Scholar] [CrossRef] [PubMed]
- Chmielecki, J.; Foo, J.; Oxnard, G.R.; Hutchinson, K.; Ohashi, K.; Somwar, R.; Wang, L.; Amato, K.R.; Arcila, M.; Sos, M.L.; et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci. Transl. Med. 2011, 3, 90ra59. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Jänne, P.A. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin. Cancer Res. 2008, 14, 2895–2899. [Google Scholar] [CrossRef]
- Weingertner, N.; Meyer, N.; Voegeli, A.C.; Guenot, D.; Renaud, S.; Massard, G.; Falcoz, P.E.; Olland, A.; Mennecier, B.; Gaub, M.P.; et al. Correlation between MET protein expression and MET gene copy number in a Caucasian cohort of non-small cell lung cancers according to the new IASLC/ATS/ERS classification. Pathology 2015, 47, 320–328. [Google Scholar] [CrossRef]
- Drilon, A.; Cappuzzo, F.; Ou, S.I.; Camidge, D.R. Targeting MET in Lung Cancer: Will Expectations Finally Be MET? J. Thorac. Oncol. 2017, 12, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.; Hong, L.; Zhang, J.; Heymach, J.; Hong, D.; Le, X. Beyond epidermal growth factor receptor: MET amplification as a general resistance driver to targeted therapy in oncogene-driven non-small-cell lung cancer. ESMO Open 2021, 6, 100319. [Google Scholar] [CrossRef]
- Schildhaus, H.U.; Schultheis, A.M.; Rüschoff, J.; Binot, E.; Merkelbach-Bruse, S.; Fassunke, J.; Schulte, W.; Ko, Y.D.; Schlesinger, A.; Bos, M.; et al. MET amplification status in therapy-naïve adeno- and squamous cell carcinomas of the lung. Clin. Cancer Res. 2015, 21, 907–915. [Google Scholar] [CrossRef]
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Jänne, P.A.; Verma, S.; et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated with Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J. Clin. Oncol. 2016, 34, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zou, Q.; Liu, H.; Qiu, B.; Li, Q.; Lin, Y.; Liang, Y. Management of Non-small Cell Lung Cancer Patients with MET Exon 14 Skipping Mutations. Curr. Treat. Options Oncol. 2020, 21, 33. [Google Scholar] [CrossRef] [PubMed]
- Sankar, K.; Gadgeel, S.M.; Qin, A. Molecular therapeutic targets in non-small cell lung cancer. Expert Rev. Anticancer. Ther. 2020, 20, 647–661. [Google Scholar] [CrossRef]
- Ren, S.; Wang, J.; Ying, J.; Mitsudomi, T.; Lee, D.H.; Wang, Z.; Chu, Q.; Mack, P.C.; Cheng, Y.; Duan, J.; et al. Consensus for HER2 alterations testing in non-small-cell lung cancer. ESMO Open 2022, 7, 100395. [Google Scholar] [CrossRef]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012, 2, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Loriot, Y.; André, F.; Gobert, A.; Auger, N.; Lacroix, L.; Soria, J.C. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann. Oncol. 2015, 26, 2073–2078. [Google Scholar] [CrossRef]
- Fan, W.; Tang, Z.; Yin, L.; Morrison, B.; Hafez-Khayyata, S.; Fu, P.; Huang, H.; Bagai, R.; Jiang, S.; Kresak, A.; et al. MET-independent lung cancer cells evading EGFR kinase inhibitors are therapeutically susceptible to BH3 mimetic agents. Cancer Res. 2011, 71, 4494–4505. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, C.A.; Stetson, D.; Markovets, A.A.; Al-Kadhimi, K.J.; Lai, Z.; Fisher, P.R.; Meador, C.B.; Spitzler, P.; Ichihara, E.; Ross, S.J.; et al. Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015, 75, 2489–2500. [Google Scholar] [CrossRef]
- Chen, W.; Yu, D.; Sun, S.Y.; Li, F. Nanoparticles for co-delivery of osimertinib and selumetinib to overcome osimertinib-acquired resistance in non-small cell lung cancer. Acta Biomater. 2021, 129, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Yang, J.C.; Yu, H.; Kim, S.W.; Saka, H.; Horn, L.; Goto, K.; Ohe, Y.; Mann, H.; Thress, K.S.; et al. TATTON: A multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann. Oncol. 2020, 31, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Ohe, Y.; Chiu, C.H.; Ou, X.; Cantarini, M.; Jänne, P.A.; Hartmaier, R.J.; Ahn, M.J. Osimertinib plus Selumetinib in EGFR-Mutated Non-Small Cell Lung Cancer after Progression on EGFR-TKIs: A Phase Ib, Open-Label, Multicenter Trial (TATTON Part B). Clin. Cancer Res. 2022, 28, 4222–4231. [Google Scholar] [CrossRef]
- Ho, C.C.; Liao, W.Y.; Lin, C.A.; Shih, J.Y.; Yu, C.J.; Yang, J.C. Acquired BRAF V600E Mutation as Resistant Mechanism after Treatment with Osimertinib. J. Thorac. Oncol. 2017, 12, 567–572. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, M.; Liu, H.; Yin, W. AZD9291 promotes autophagy and inhibits PI3K/Akt pathway in NSCLC cancer cells. J. Cell. Biochem. 2019, 120, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, H.S.; Lee, B.; Kim, H.K.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K.; Lee, S.H. Genomic landscape of acquired resistance to third-generation EGFR tyrosine kinase inhibitors in EGFR T790M-mutant non-small cell lung cancer. Cancer 2020, 126, 2704–2712. [Google Scholar] [CrossRef]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Vojnic, M.; Kubota, D.; Kurzatkowski, C.; Offin, M.; Suzawa, K.; Benayed, R.; Schoenfeld, A.J.; Plodkowski, A.J.; Poirier, J.T.; Rudin, C.M.; et al. Acquired BRAF Rearrangements Induce Secondary Resistance to EGFR therapy in EGFR-Mutated Lung Cancers. J. Thorac. Oncol. 2019, 14, 802–815. [Google Scholar] [CrossRef]
- Aldea, M.; Andre, F.; Marabelle, A.; Dogan, S.; Barlesi, F.; Soria, J.C. Overcoming Resistance to Tumor-Targeted and Immune-Targeted Therapies. Cancer Discov. 2021, 11, 874–899. [Google Scholar] [CrossRef]
- Offin, M.; Somwar, R.; Rekhtman, N.; Benayed, R.; Chang, J.C.; Plodkowski, A.; Lui, A.J.W.; Eng, J.; Rosenblum, M.; Li, B.T.; et al. Acquired ALK and RET Gene Fusions as Mechanisms of Resistance to Osimertinib in EGFR-Mutant Lung Cancers. JCO Precis. Oncol. 2018, 2. [Google Scholar] [CrossRef]
- Schrock, A.B.; Zhu, V.W.; Hsieh, W.S.; Madison, R.; Creelan, B.; Silberberg, J.; Costin, D.; Bharne, A.; Bonta, I.; Bosemani, T.; et al. Receptor Tyrosine Kinase Fusions and BRAF Kinase Fusions are Rare but Actionable Resistance Mechanisms to EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2018, 13, 1312–1323. [Google Scholar] [CrossRef]
- Nelson, A.W.; Schrock, A.B.; Pavlick, D.C.; Ali, S.M.; Atkinson, E.C.; Chachoua, A. Novel SPECC1L-MET Fusion Detected in Circulating Tumor DNA in a Patient with Lung Adenocarcinoma following Treatment with Erlotinib and Osimertinib. J. Thorac. Oncol. 2019, 14, e27–e29. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, Z.; Isozaki, H.; Lennerz, J.K.; Gainor, J.F.; Lennes, I.T.; Zhu, V.W.; Marcoux, N.; Banwait, M.K.; Digumarthy, S.R.; Su, W.; et al. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov. 2018, 8, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Woo, J.K.; Kim, E.S.; Hong, W.K.; Lee, H.Y. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res. 2006, 66, 10100–10111. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Rho, J.K.; Jeon, B.S.; Choi, S.J.; Park, S.C.; Lee, S.S.; Kim, H.R.; Kim, C.H.; Lee, J.C. Combined inhibition of IGFR enhances the effects of gefitinib in H1650: A lung cancer cell line with EGFR mutation and primary resistance to EGFR-TK inhibitors. Cancer Chemother. Pharmacol. 2010, 66, 381–388. [Google Scholar] [CrossRef]
- Ware, K.E.; Marshall, M.E.; Heasley, L.R.; Marek, L.; Hinz, T.K.; Hercule, P.; Helfrich, B.A.; Doebele, R.C.; Heasley, L.E. Rapidly Acquired Resistance to EGFR Tyrosine Kinase Inhibitors in NSCLC Cell Lines through De-Repression of FGFR2 and FGFR3 Expression. PLoS ONE 2010, 5, e14117. [Google Scholar] [CrossRef]
- Li, L.; Wang, H.; Li, C.; Wang, Z.; Zhang, P.; Yan, X. Transformation to small-cell carcinoma as an acquired resistance mechanism to AZD9291: A case report. Oncotarget 2017, 8, 18609–18614. [Google Scholar] [CrossRef]
- Sutherland, K.D.; Proost, N.; Brouns, I.; Adriaensen, D.; Song, J.Y.; Berns, A. Cell of origin of small cell lung cancer: Inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 2011, 19, 754–764. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 2003, 15, 740–746. [Google Scholar] [CrossRef]
- Kang, Y.; Massagué, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef]
- Yauch, R.L.; Januario, T.; Eberhard, D.A.; Cavet, G.; Zhu, W.; Fu, L.; Pham, T.Q.; Soriano, R.; Stinson, J.; Seshagiri, S.; et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin. Cancer Res. 2005, 11, 8686–8698. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, C.M.; Bellevicine, C.; Vicidomini, G.; Vitagliano, D.; Malapelle, U.; Accardo, M.; Fabozzi, A.; Fiorelli, A.; Fasano, M.; Papaccio, F.; et al. SMO Gene Amplification and Activation of the Hedgehog Pathway as Novel Mechanisms of Resistance to Anti-Epidermal Growth Factor Receptor Drugs in Human Lung Cancer. Clin. Cancer Res. 2015, 21, 4686–4697. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.Y.; Zhang, X.C.; Yang, S.Q.; An, S.J.; Chen, Z.H.; Su, J.; Xie, Z.; Gou, L.Y.; Wu, Y.L. Blockade of Hedgehog Signaling Synergistically Increases Sensitivity to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small-Cell Lung Cancer Cell Lines. PLoS ONE 2016, 11, e0149370. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Han, J.Y.; Ahn, M.J.; Cho, B.C.; Yu, H.; Kim, S.W.; Yang, J.C.; Lee, J.S.; Su, W.C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet Oncology 2020, 21, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Zhang, L.; Kim, D.W.; Liu, X.; Lee, D.H.; Yang, J.C.; Ahn, M.J.; Vansteenkiste, J.F.; Su, W.C.; Felip, E.; et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib After Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients with EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 3101–3109. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, J.; Lu, S.; Zhang, Y.; Zhao, J.; Kim, D.W.; Soo, R.A.; Kim, S.W.; Pan, H.; et al. Tepotinib plus gefitinib in patients with EGFR-mutant non-small-cell lung cancer with MET overexpression or MET amplification and acquired resistance to previous EGFR inhibitor (INSIGHT study): An open-label, phase 1b/2, multicentre, randomised trial. Lancet Respir. Med. 2020, 8, 1132–1143. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.C.; Felip, E.; Hayashi, H.; Thomas, M.; Lu, S.; Besse, B.; Sun, T.; Martinez, M.; Sethi, S.N.; Shreeve, S.M.; et al. MARIPOSA: Phase 3 study of first-line amivantamab + lazertinib versus osimertinib in EGFR-mutant non-small-cell lung cancer. Future Oncol. 2022, 18, 639–647. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Smit, E.F.; Groen, H.J.; Horn, L.; Gettinger, S.; Camidge, D.R.; Riely, G.J.; Wang, B.; Fu, Y.; Chand, V.K.; et al. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov. 2014, 4, 1036–1045. [Google Scholar] [CrossRef]
- Lee, J.Y.; Sun, J.M.; Lim, S.H.; Kim, H.S.; Yoo, K.H.; Jung, K.S.; Song, H.N.; Ku, B.M.; Koh, J.; Bae, Y.H.; et al. A Phase Ib/II Study of Afatinib in Combination with Nimotuzumab in Non-Small Cell Lung Cancer Patients with Acquired Resistance to Gefitinib or Erlotinib. Clin. Cancer Res. 2016, 22, 2139–2145. [Google Scholar] [CrossRef]
- Thatcher, N.; Hirsch, F.R.; Luft, A.V.; Szczesna, A.; Ciuleanu, T.E.; Dediu, M.; Ramlau, R.; Galiulin, R.K.; Bálint, B.; Losonczy, G.; et al. Necitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first-line therapy in patients with stage IV squamous non-small-cell lung cancer (SQUIRE): An open-label, randomised, controlled phase 3 trial. Lancet Oncol. 2015, 16, 763–774. [Google Scholar] [CrossRef]
- Bertino, E.M.; Gentzler, R.D.; Clifford, S.; Kolesar, J.; Muzikansky, A.; Haura, E.B.; Piotrowska, Z.; Camidge, D.R.; Stinchcombe, T.E.; Hann, C.; et al. Phase IB Study of Osimertinib in Combination with Navitoclax in EGFR-mutant NSCLC Following Resistance to Initial EGFR Therapy (ETCTN 9903). Clin. Cancer Res. 2021, 27, 1604–1611. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Kim, S.-W.; Wu, Y.-L.; Nakagawa, K.; Yang, J.-J.; Ahn, M.-J.; Wang, J.; Yang, J.C.-H.; Lu, Y.; Atagi, S.; et al. Gefitinib Plus Chemotherapy Versus Chemotherapy in Epidermal Growth Factor Receptor Mutation–Positive Non–Small-Cell Lung Cancer Resistant to First-Line Gefitinib (IMPRESS): Overall Survival and Biomarker Analyses. J. Clin. Oncol. 2017, 35, 4027–4034. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, E.; Morita, S.; Nakamura, A.; Hosomi, Y.; Watanabe, K.; Ikeda, S.; Seike, M.; Fujita, Y.; Minato, K.; Ko, R.; et al. Updated Analysis of NEJ009: Gefitinib-Alone Versus Gefitinib Plus Chemotherapy for Non-Small-Cell Lung Cancer with Mutated EGFR. J. Clin. Oncol. 2022, 40, 3587–3592. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Asahina, H.; Kishimoto, J.; Miyata, Y.; Uchida, T.; Watanabe, K.; Hamai, K.; Harada, T.; Tsubata, Y.; Sugawara, S.; et al. Osimertinib versus osimertinib plus chemotherapy for non-small cell lung cancer with EGFR (T790M)-associated resistance to initial EGFR inhibitor treatment: An open-label, randomised phase 2 clinical trial. Eur. J. Cancer 2021, 149, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.; Hellmann, M.D.; Chow, L.Q.M.; Borghaei, H.; Antonia, S.; Brahmer, J.R.; Goldman, J.W.; Gerber, D.E.; Juergens, R.A.; Shepherd, F.A.; et al. Nivolumab Plus Erlotinib in Patients with EGFR-Mutant Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Shepherd, F.A.; Kim, D.W.; Lee, G.W.; Lee, J.S.; Chang, G.C.; Lee, S.S.; Wei, Y.F.; Lee, Y.G.; Laus, G.; et al. Osimertinib Plus Durvalumab versus Osimertinib Monotherapy in EGFR T790M-Positive NSCLC following Previous EGFR TKI Therapy: CAURAL Brief Report. J. Thorac. Oncol. 2019, 14, 933–939. [Google Scholar] [CrossRef]
- Reck, M.; Mok, T.S.K.; Nishio, M.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): Key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir. Med. 2019, 7, 387–401. [Google Scholar] [CrossRef]
- Lu, S.; Wu, L.; Jian, H.; Chen, Y.; Wang, Q.; Fang, J.; Wang, Z.; Hu, Y.; Sun, M.; Han, L.; et al. Sintilimab plus bevacizumab biosimilar IBI305 and chemotherapy for patients with EGFR-mutated non-squamous non-small-cell lung cancer who progressed on EGFR tyrosine-kinase inhibitor therapy (ORIENT-31): First interim results from a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2022, 23, 1167–1179. [Google Scholar] [CrossRef]
- Lu, S.; Wang, Q.; Zhang, G.; Dong, X.; Yang, C.-T.; Song, Y.; Chang, G.-C.; Lu, Y.; Pan, H.; Chiu, C.-H.; et al. Abstract CT190: A multicenter, open-label, single-arm, phase II study: The third generation EGFR tyrosine kinase inhibitor almonertinib for pretreated EGFR T790M-positive locally advanced or metastatic non-small cell lung cancer (APOLLO). Cancer Res. 2020, 80, CT190. [Google Scholar] [CrossRef]
- Shi, Y.; Hu, X.; Zhang, S.; Lv, D.; Zhang, Y.; Yu, Q.; Wu, L.; Liu, L.; Wang, X.; Ma, Z.; et al. Efficacy and safety of alflutinib (AST2818) in patients with T790M mutation-positive NSCLC: A phase IIb multicenter single-arm study. J. Clin. Oncol. 2020, 38, 9602. [Google Scholar] [CrossRef]
- Ahn, M.-J.; Han, J.-Y.; Lee, K.H.; Kim, S.-W.; Kim, D.-W.; Lee, Y.-G.; Cho, E.K.; Kim, J.-H.; Lee, G.-W.; Lee, J.-S.; et al. Lazertinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: Results from the dose escalation and dose expansion parts of a first-in-human, open-label, multicentre, phase 1–2 study. Lancet Oncol. 2019, 20, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zheng, X.; Zhao, H.; Fang, W.; Zhang, Y.; Ge, J.; Wang, L.; Wang, W.; Jiang, J.; Chuai, S.; et al. First-in-Human Phase I Study of AC0010, a Mutant-Selective EGFR Inhibitor in Non–Small Cell Lung Cancer: Safety, Efficacy, and Potential Mechanism of Resistance. J. Thorac. Oncol. 2018, 13, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Zhou, Q.; Liu, X.; Zhang, L.; Zhou, J.; Wu, L.; An, T.; Cheng, Y.; Zheng, X.; Hu, B.; et al. MA16.06 Phase I/II Study of AC0010, Mutant-Selective EGFR Inhibitor, in Non-Small Cell Lung Cancer (NSCLC) Patients with EGFR T790M Mutation. J. Thorac. Oncol. 2017, 12, S437–S438. [Google Scholar] [CrossRef]
- Tan, D.S.W.; Kim, S.W.; Ponce Aix, S.; Sequist, L.V.; Smit, E.F.; Yang, J.C.H.; Hida, T.; Toyozawa, R.; Felip, E.; Wolf, J.; et al. Nazartinib for treatment-naive EGFR-mutant non-small cell lung cancer: Results of a phase 2, single-arm, open-label study. Eur. J. Cancer 2022, 172, 276–286. [Google Scholar] [CrossRef]
- Nagasaka, M.; Zhu, V.W.; Lim, S.M.; Greco, M.; Wu, F.; Ou, S.I. Beyond Osimertinib: The Development of Third-Generation EGFR Tyrosine Kinase Inhibitors For Advanced EGFR+ NSCLC. J. Thorac. Oncol. 2021, 16, 740–763. [Google Scholar] [CrossRef]
- Park, K.; Jӓnne, P.A.; Kim, D.W.; Han, J.Y.; Wu, M.F.; Lee, J.S.; Kang, J.H.; Lee, D.H.; Cho, B.C.; Yu, C.J.; et al. Olmutinib in T790M-positive non-small cell lung cancer after failure of first-line epidermal growth factor receptor-tyrosine kinase inhibitor therapy: A global, phase 2 study. Cancer 2021, 127, 1407–1416. [Google Scholar] [CrossRef]
- Murakami, H.; Nokihara, H.; Hayashi, H.; Seto, T.; Park, K.; Azuma, K.; Tsai, C.M.; Yang, J.C.; Nishio, M.; Kim, S.W.; et al. Clinical activity of ASP8273 in Asian patients with non-small-cell lung cancer with EGFR activating and T790M mutations. Cancer Sci. 2018, 109, 2852–2862. [Google Scholar] [CrossRef]
- Cho, B.C.; Goldberg, S.B.; Kim, D.W.; Socinski, M.A.; Burns, T.F.; Lwin, Z.; Pathan, N.; Ma, W.D.; Masters, J.C.; Cossons, N.; et al. A phase 1b/2 study of PF-06747775 as monotherapy or in combination with Palbociclib in patients with epidermal growth factor receptor mutant advanced non-small cell lung cancer. Expert Opin. Investig. Drugs 2022, 31, 747–757. [Google Scholar] [CrossRef]
- Wang, S.; Cang, S.; Liu, D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 34. [Google Scholar] [CrossRef]
- Rangachari, D.; To, C.; Shpilsky, J.E.; VanderLaan, P.A.; Kobayashi, S.S.; Mushajiang, M.; Lau, C.J.; Paweletz, C.P.; Oxnard, G.R.; Jänne, P.A.; et al. EGFR-Mutated Lung Cancers Resistant to Osimertinib through EGFR C797S Respond to First-Generation Reversible EGFR Inhibitors but Eventually Acquire EGFR T790M/C797S in Preclinical Models and Clinical Samples. J. Thorac. Oncol. 2019, 14, 1995–2002. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, L.; Yin, J.C.; Wu, X.; Shao, Y.W.; Gao, B. Lung Adenocarcinoma Harboring EGFR 19del/C797S/T790M Triple Mutations Responds to Brigatinib and Anti-EGFR Antibody Combination Therapy. J. Thorac. Oncol. 2019, 14, e85–e88. [Google Scholar] [CrossRef] [PubMed]
- Uchibori, K.; Inase, N.; Araki, M.; Kamada, M.; Sato, S.; Okuno, Y.; Fujita, N.; Katayama, R. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat. Commun. 2017, 8, 14768. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xu, H.; Ma, L.; Yang, L.; Yang, G.; Zhang, S.; Ai, X.; Zhang, S.; Wang, Y. Possibility of brigatinib-based therapy, or chemotherapy plus anti-angiogenic treatment after resistance of osimertinib harboring EGFR T790M-cis-C797S mutations in lung adenocarcinoma patients. Cancer Med. 2021, 10, 8328–8337. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, N.; Zhang, Y.; Li, L.; Han, R.; Zhu, M.; Feng, M.; Chen, H.; Lizaso, A.; Qin, T.; et al. Effective Treatment of Lung Adenocarcinoma Harboring EGFR Activating Mutation, T790M, and cis-C797S Triple Mutations by Brigatinib and Cetuximab Combination Therapy. J. Thorac. Oncol. 2020, 15, 1369–1375. [Google Scholar] [CrossRef]
- Song, Z.; Ren, G.; Hu, L.; Wang, X.; Song, J.; Jia, Y.; Zhao, G.; Zang, A.; Du, H.; Sun, Y.; et al. Two case reports of non-small cell lung cancer patients harboring acquired EGFR T790M-cis-C797S benefit from immune checkpoint inhibitor combined with platinum-based doublet chemotherapy. Ann. Transl. Med. 2022, 10, 719. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, Y.; Liu, D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef]
- To, C.; Jang, J.; Chen, T.; Park, E.; Mushajiang, M.; De Clercq, D.J.H.; Xu, M.; Wang, S.; Cameron, M.D.; Heppner, D.E.; et al. Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discov. 2019, 9, 926–943. [Google Scholar] [CrossRef] [PubMed]
- Schalm, S.S.; Dineen, T.; Lim, S.M.; Park, C.W.; Hsieh, J.; Woessner, R.; Zhang, Z.; Wilson, K.; Eno, M.; Wilson, D.; et al. 1296P BLU-945, a highly potent and selective 4th generation EGFR TKI for the treatment of EGFR T790M/C797S resistant NSCLC. Ann. Oncol. 2020, 31, S839. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.-H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef]
- To, C.; Beyett, T.S.; Jang, J.; Feng, W.W.; Bahcall, M.; Haikala, H.M.; Shin, B.H.; Heppner, D.E.; Rana, J.K.; Leeper, B.A.; et al. An allosteric inhibitor against the therapy-resistant mutant forms of EGFR in non-small cell lung cancer. Nat. Cancer 2022, 3, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Kashima, K.; Kawauchi, H.; Tanimura, H.; Tachibana, Y.; Chiba, T.; Torizawa, T.; Sakamoto, H. CH7233163 Overcomes Osimertinib-Resistant EGFR-Del19/T790M/C797S Mutation. Mol. Cancer Ther. 2020, 19, 2288–2297. [Google Scholar] [CrossRef]
- Conti, C.; Campbell, J.; Woessner, R.; Guo, J.; Timsit, Y.; Iliou, M.; Wardwell, S.; Davis, A.; Chicklas, S.; Hsieh, J.; et al. Abstract 1262: BLU-701 is a highly potent, brain-penetrant and WT-sparing next-generation EGFR TKI for the treatment of sensitizing (ex19del, L858R) and C797S resistance mutations in metastatic NSCLC. Cancer Res. 2021, 81, 1262. [Google Scholar] [CrossRef]
- Tavera, L.; Schalm, S.; Campbell, J.; Guo, J.; Medendorp, C.; Chen, M.; Albayya, F.; Dineen, T.; Zhang, Z.; Iliou, M.; et al. Abstract 3328: Antitumor activity of BLU-945 and BLU-701 as single agents and in combination in EGFR L858R-driven models of NSCLC. Cancer Res. 2022, 82, 3328. [Google Scholar] [CrossRef]
- Eno, M.S.; Brubaker, J.D.; Campbell, J.E.; De Savi, C.; Guzi, T.J.; Williams, B.D.; Wilson, D.; Wilson, K.; Brooijmans, N.; Kim, J.; et al. Discovery of BLU-945, a Reversible, Potent, and Wild-Type-Sparing Next-Generation EGFR Mutant Inhibitor for Treatment-Resistant Non-Small-Cell Lung Cancer. J. Med. Chem. 2022, 65, 9662–9677. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.; Goto, K.; Camidge, D.R.; Elamin, Y.; de Langen, A.J.; Leighl, N.B.; Minchom, A.; Piotrowska, Z.; Planchard, D.; Reckamp, K.; et al. Abstract P230: A phase 1/2 study of BLU-945, a highly potent and selective inhibitor of epidermal growth factor receptor (EGFR) resistance mutations, in patients with EGFR-mutant non-small cell lung cancer (NSCLC). Mol. Cancer Ther. 2021, 20, P230. [Google Scholar] [CrossRef]
- Lim, S.M.; Park, C.W.; Zhang, Z.; Woessner, R.; Dineen, T.; Stevison, F.; Hsieh, J.; Eno, M.; Wilson, D.; Campbell, J.; et al. Abstract 1467: BLU-945, a fourth-generation, potent and highly selective epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) with intracranial activity, demonstrates robust in vivo antitumor activity in models of osimertinib-resistant non-small cell lung cancer (NSCLC). Cancer Res. 2021, 81, 1467. [Google Scholar] [CrossRef]
- Shum, E.; Elamin, Y.; Reckamp, K.L.; Piotrowska, Z.; Rotow, J.; Tan, D.S.; Goto, K.; Parepally, J.; Albayya, F.; Louie-Gao, M.; et al. Abstract CT184: Emerging evidence of activity of BLU-945 in patients with advanced EGFR-mutant NSCLC utilizing circulating tumor DNA (ctDNA) in the phase 1/2 SYMPHONY study. Cancer Res. 2022, 82, CT184. [Google Scholar] [CrossRef]
- Le, X.; Puri, S.; Negrao, M.V.; Nilsson, M.B.; Robichaux, J.; Boyle, T.; Hicks, J.K.; Lovinger, K.L.; Roarty, E.; Rinsurongkawong, W.; et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin. Cancer Res. 2018, 24, 6195–6203. [Google Scholar] [CrossRef]
- Piper-Vallillo, A.J.; Sequist, L.V.; Piotrowska, Z. Emerging Treatment Paradigms for EGFR-Mutant Lung Cancers Progressing on Osimertinib: A Review. J. Clin. Oncol. 2020, 38, 2926–2936. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Han, J.-Y.; Kato, T.; Barlesi, F.; Garon, E.B.; Cappuzzo, F.; Shibata, Y.; Smith, N.; Khanna, S.; Belli, R.; et al. Capmatinib plus osimertinib versus platinum-pemetrexed doublet chemotherapy as second-line therapy in patients with stage IIIb/IIIc or IV EGFR-mutant, T790M-negative NSCLC harboring MET amplification. J. Clin. Oncol. 2022, 40, TPS9153. [Google Scholar] [CrossRef]
- Deng, L.; Kiedrowski, L.A.; Ravera, E.; Cheng, H.; Halmos, B. Response to Dual Crizotinib and Osimertinib Treatment in a Lung Cancer Patient with MET Amplification Detected by Liquid Biopsy Who Acquired Secondary Resistance to EGFR Tyrosine Kinase Inhibition. J. Thorac. Oncol. 2018, 13, e169–e172. [Google Scholar] [CrossRef] [PubMed]
- York, E.R.; Varella-Garcia, M.; Bang, T.J.; Aisner, D.L.; Camidge, D.R. Tolerable and Effective Combination of Full-Dose Crizotinib and Osimertinib Targeting MET Amplification Sequentially Emerging after T790M Positivity in EGFR-Mutant Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, e85–e88. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tian, P.; Xia, L.; Li, L.; Han, R.; Zhu, M.; Lizaso, A.; Qin, T.; Li, M.; Yu, B.; et al. The clinical efficacy of combinatorial therapy of EGFR-TKI and crizotinib in overcoming MET amplification-mediated resistance from prior EGFR-TKI therapy. Lung Cancer 2020, 146, 165–173. [Google Scholar] [CrossRef] [PubMed]
- La Monica, S.; Fumarola, C.; Cretella, D.; Bonelli, M.; Minari, R.; Cavazzoni, A.; Digiacomo, G.; Galetti, M.; Volta, F.; Mancini, M.; et al. Efficacy of the CDK4/6 Dual Inhibitor Abemaciclib in EGFR-Mutated NSCLC Cell Lines with Different Resistance Mechanisms to Osimertinib. Cancers 2021, 13, 6. [Google Scholar] [CrossRef]
- Qin, Q.; Li, X.; Liang, X.; Zeng, L.; Wang, J.; Sun, L.; Zhong, D. CDK4/6 inhibitor palbociclib overcomes acquired resistance to third-generation EGFR inhibitor osimertinib in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 2389–2397. [Google Scholar] [CrossRef]
- Perrone, M.; Giuliani, F.; Sanna, V.; Bruno, S.; Melaccio, A.; Santoro, A.N.; Laface, C.; Maselli, F.M.; Iaia, M.L.; Guarini, C.; et al. Advances in pharmacotherapies that target the cell cycle for treatment of breast cancer: Where are we at today? Expert Opin. Pharmacother. 2023, 24, 887–900. [Google Scholar] [CrossRef]
- Ji, W.; Choi, Y.J.; Kang, M.-H.; Sung, K.J.; Kim, D.H.; Jung, S.; Choi, C.-M.; Lee, J.C.; Rho, J.K. Efficacy of the CDK7 Inhibitor on EMT-Associated Resistance to 3rd Generation EGFR-TKIs in Non-Small Cell Lung Cancer Cell Lines. Cells 2020, 9, 2596. [Google Scholar] [CrossRef]
- Regales, L.; Gong, Y.; Shen, R.; de Stanchina, E.; Vivanco, I.; Goel, A.; Koutcher, J.A.; Spassova, M.; Ouerfelli, O.; Mellinghoff, I.K.; et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J. Clin. Investig. 2009, 119, 3000–3010. [Google Scholar] [CrossRef]
- van Veggel, B.; de Langen, A.J.; Hashemi, S.M.S.; Monkhorst, K.; Heideman, D.A.M.; Thunnissen, E.; Smit, E.F. Afatinib and Cetuximab in Four Patients with EGFR Exon 20 Insertion–Positive Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1222–1226. [Google Scholar] [CrossRef]
- Fang, W.; Huang, Y.; Gan, J.; Shao, Y.W.; Zhang, L. Durable Response of Low-Dose Afatinib plus Cetuximab in an Adenocarcinoma Patient with a Novel EGFR Exon 20 Insertion Mutation. J. Thorac. Oncol. 2019, 14, e220–e221. [Google Scholar] [CrossRef]
- Marcoux, N.; Gettinger, S.N.; O’Kane, G.; Arbour, K.C.; Neal, J.W.; Husain, H.; Evans, T.L.; Brahmer, J.R.; Muzikansky, A.; Bonomi, P.D.; et al. EGFR-Mutant Adenocarcinomas That Transform to Small-Cell Lung Cancer and Other Neuroendocrine Carcinomas: Clinical Outcomes. J. Clin. Oncol. 2018, 37, 278–285. [Google Scholar] [CrossRef]
- Mu, Y.; Hao, X.; Yang, K.; Ma, D.; Wang, S.; Xu, Z.; Li, J.; Xing, P. Clinical Modality of Resistance and Subsequent Management of Patients with Advanced Non-small Cell Lung Cancer Failing Treatment with Osimertinib. Target. Oncol. 2019, 14, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Wu, Y.-L.; Nakagawa, K.; Kim, S.-W.; Yang, J.-J.; Ahn, M.-J.; Wang, J.; Yang, J.C.-H.; Lu, Y.; Atagi, S.; et al. Gefitinib plus chemotherapy versus placebo plus chemotherapy in EGFR-mutation-positive non-small-cell lung cancer after progression on first-line gefitinib (IMPRESS): A phase 3 randomised trial. Lancet Oncol. 2015, 16, 990–998. [Google Scholar] [CrossRef]
- Hosomi, Y.; Morita, S.; Sugawara, S.; Kato, T.; Fukuhara, T.; Gemma, A.; Takahashi, K.; Fujita, Y.; Harada, T.; Minato, K.; et al. Gefitinib alone versus Gefitinib Plus Chemotherapy for Non–Small-Cell Lung Cancer with Mutated Epidermal Growth Factor Receptor: NEJ009 Study. J. Clin. Oncol. 2019, 38, 115–123. [Google Scholar] [CrossRef]
- Laface, C.; Ranieri, G.; Maselli, F.M.; Ambrogio, F.; Foti, C.; Ammendola, M.; Laterza, M.; Cazzato, G.; Memeo, R.; Mastrandrea, G.; et al. Immunotherapy and the Combination with Targeted Therapies for Advanced Hepatocellular Carcinoma. Cancers 2023, 15, 654. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Lisberg, A.; Cummings, A.; Goldman, J.W.; Bornazyan, K.; Reese, N.; Wang, T.; Coluzzi, P.; Ledezma, B.; Mendenhall, M.; Hunt, J.; et al. A Phase II Study of Pembrolizumab in EGFR-Mutant, PD-L1+, Tyrosine Kinase Inhibitor Naïve Patients with Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, F.; Zhong, M.; Yarden, Y.; Fu, L. The biomarkers of hyperprogressive disease in PD-1/PD-L1 blockage therapy. Mol. Cancer 2020, 19, 81. [Google Scholar] [CrossRef]
- Kim, C.G.; Kim, K.H.; Pyo, K.H.; Xin, C.F.; Hong, M.H.; Ahn, B.C.; Kim, Y.; Choi, S.J.; Yoon, H.I.; Lee, J.G.; et al. Hyperprogressive disease during PD-1/PD-L1 blockade in patients with non-small-cell lung cancer. Ann. Oncol. 2019, 30, 1104–1113. [Google Scholar] [CrossRef]
- To, K.K.W.; Fong, W.; Cho, W.C.S. Immunotherapy in Treating EGFR-Mutant Lung Cancer: Current Challenges and New Strategies. Front. Oncol. 2021, 11, 635007. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.C.M.; Fares, A.F.; Le, L.W.; Mackay, K.M.; Soberano, S.; Chan, S.W.; Smith, E.; Ryan, M.; Tsao, M.S.; Bradbury, P.A.; et al. Subtypes of EGFR- and HER2-Mutant Metastatic NSCLC Influence Response to Immune Checkpoint Inhibitors. Clin. Lung Cancer 2021, 22, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Haratani, K.; Hayashi, H.; Tanaka, T.; Kaneda, H.; Togashi, Y.; Sakai, K.; Hayashi, K.; Tomida, S.; Chiba, Y.; Yonesaka, K.; et al. Tumor immune microenvironment and nivolumab efficacy in EGFR mutation-positive non-small-cell lung cancer based on T790M status after disease progression during EGFR-TKI treatment. Ann. Oncol. 2017, 28, 1532–1539. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Chow, L.Q.M.; Borghaei, H.; Shen, Y.; Harbison, C.; Alaparthy, S.; Chen, A.C.; Gettinger, S.N. Safety and response with nivolumab (anti-PD-1; BMS-936558, ONO-4538) plus erlotinib in patients (pts) with epidermal growth factor receptor mutant (EGFR MT) advanced NSCLC. J. Clin. Oncol. 2014, 32, 8022. [Google Scholar] [CrossRef]
- Oshima, Y.; Tanimoto, T.; Yuji, K.; Tojo, A. EGFR–TKI-Associated Interstitial Pneumonitis in Nivolumab-Treated Patients with Non–Small Cell Lung Cancer. JAMA Oncol. 2018, 4, 1112–1115. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Chow, L.Q.; Kim, D.W.; Kim, S.W.; Yeh, T.; Song, X.; Jiang, H.; Taylor, R.; Karakunnel, J.; Creelan, B. 57O Efficacy, safety and tolerability of MEDI4736 (durvalumab [D]), a human IgG1 anti-programmed cell death-ligand-1 (PD-L1) antibody, combined with gefitinib (G): A phase I expansion in TKI-naïve patients (pts) with EGFR mutant NSCLC. J. Thorac. Oncol. 2016, 11, S79. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Arbour, K.C.; Rizvi, H.; Iqbal, A.N.; Gadgeel, S.M.; Girshman, J.; Kris, M.G.; Riely, G.J.; Yu, H.A.; Hellmann, M.D. Severe immune-related adverse events are common with sequential PD-(L)1 blockade and osimertinib. Ann. Oncol. 2019, 30, 839–844. [Google Scholar] [CrossRef]
- Hsu, P.-C.; Jablons, D.M.; Yang, C.-T.; You, L. Epidermal Growth Factor Receptor (EGFR) Pathway, Yes-Associated Protein (YAP) and the Regulation of Programmed Death-Ligand 1 (PD-L1) in Non-Small Cell Lung Cancer (NSCLC). Int. J. Mol. Sci. 2019, 20, 3821. [Google Scholar] [CrossRef]
- Kaira, K.; Kagamu, H. Drastic Response of Re-challenge of EGFR-TKIs Immediately After Nivolumab Therapy in EGFR-TKI–Resistant Patients. J. Thorac. Oncol. 2019, 14, e135–e136. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Kobayashi, K.; Shiono, A.; Yamaguchi, O.; Hashimoto, K.; Mouri, A.; Shinomiya, S.; Miura, Y.; Imai, H.; Kagamu, H. Effectiveness of EGFR-TKI rechallenge immediately after PD-1 blockade failure. Thorac. Cancer 2021, 12, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.C.; Tsang, K.C.; Choi, H.C.; Lee, V.H.; Lam, K.O.; Chiang, C.L.; So, T.H.; Chan, W.W.; Nyaw, S.F.; Lim, F.; et al. Combination atezolizumab, bevacizumab, pemetrexed and carboplatin for metastatic EGFR mutated NSCLC after TKI failure. Lung Cancer 2021, 159, 18–26. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| EGFR Mutations | Specific Alteration | Frequency | Clinical Characteristics | Response to EGFR-TKIs | |||
|---|---|---|---|---|---|---|---|
| Gefitinib/Erlotinib | Afatinib | Osimertininb | |||||
| Common | Exon 19 deletion | 45–50% | Adenocarcinoma Female gender Never smoker status Asian ethnicity | Sensitive | Sensitive | Sensitive | |
| Exon 21 L858R point mutation | 37–40% | Sensitive | Sensitive | Sensitive | |||
| Uncommon or rare | Exon 18 | G719X | 3% | Adenocarcinoma Male gender Smoker history Asian ethnicity | Intermediate | Sensitive | Inter./sensitive |
| E709X | 0.3% | E709K Intermediate | Sensitive | Sensitive | |||
| Del18 | 0.3% | Intermediate | Sensitive | Sensitive | |||
| Exon 19 | Ins19 | <0.6% | Intermediate | Inter./Sens. | Inter./Sens. | ||
| Exon 20 | Ins20 | >5.8% | Resistant | Resistant/Sens. | Inter./Sens. | ||
| S768I | <1.5% | Intermediate | Inter./Sens. | Sensitive | |||
| Exon 21 | L861Q | 0.9–3% | Intermediate | Inter./Sens. | Inter./Sens. | ||
| Other EGFR alterations |
| 5–15% | Depend on the specific combination of mutations | Depend on the specific combination of mutations | |||
| 3–6% | ||||||
| Variable | ||||||
| Generation EGFR-TKI | Clinical Trail | EGFR Status | Comparison | Results |
|---|---|---|---|---|
| First-generation | NEJ002 [28,29] | Common mutations | Gefitinib vs. carboplatin plus paclitaxel | * PFS: 10.8 vs. 5.4 mo. ** OS: 27.7 vs. 26.6 mo. |
| IPASS [30,31] | Common mutations | Gefitinib vs. carboplatin plus paclitaxel | * PFS: 9.5 vs. 6.3 mo. ** OS: 18.8 vs. 17.4 mo. | |
| WJTOG3405 [32,33] | Common mutations | Gefitinib vs. cisplatin plus docetaxel | * PFS: 9.2 vs. 6.3 mo. ** OS: 34.9 vs. 37.3 mo. | |
| OPTIMAL [34,35] | Common mutations | Erlotinib vs. gemcitabine plus carboplatin | * PFS: 13.1 vs. 4.6 mo. ** OS: 22.8 vs. 27.2 mo. | |
| ENSURE [36] | Common mutations | Erlotinib vs. gemcitabine plus cisplatin | * PFS: 11 vs. 5.5 mo. ** OS: 26.3 vs. 25.5 mo. | |
| EUTARC [37] | Common mutations | Erlotinib vs. cisplatin plus docetaxel or gemcitabine | * PFS: 9.7 vs. 5.2 mo. ** OS: 22.8 vs. 27.2 mo. | |
| CONVINCE [38] | Common mutations | Icotinib vs. cisplatin plus pemetrexed | * PFS: 11.2 vs. 7.9 mo. ** OS: 30.5 vs. 32.1 mo. | |
| Second-generation | LUX-Lung 3 [39] | Common mutations | Afatinib vs. cisplatin plus gemcitabine or pemetrexed | * PFS: 13.6 vs. 6.9 mo. ** OS: 28.2 vs. 28.2 mo. |
| LUX-Lung 6 [40] | Common mutations | Afatinib vs. cisplatin plus gemcitabine or pemetrexed | * PFS: 11 vs. 5.6 mo. ** OS: 23.1 vs. 23.5 mo. | |
| ARCHER 1050 [40,41] | Common mutations +/− T790M | Dacotinib vs. gefitinib | * PFS: 14.7 vs. 9.2 mo. ** OS: 34.1 vs. 27 mo. | |
| Third-generation | AURA3 [42] | T790M mutation | Osimertinib vs. cis/carboplatin plus pemetrexed after first-line EGFR-TKI therapy | * PFS: 10.1 vs. 4.4 mo. ** OS: 26.8 vs. 22.5 mo. |
| FLAURA [26] | Common mutations | Osimertinib vs. gefitinib or erlotinib | * PFS: 18.9 vs. 10.2 mo. ** OS: 38.6 vs. 31.8 mo. | |
| ADAURA [43] | Common mutations | Osimertinib for 3 years as adjuvant therapy | * 4ys-DFS: 73% vs. 38% | |
| EGFR-TKIs specific for Ins20 | ZENITH20-2 [44] | Exon 20 insertions | Poziotininb | ORR: 27.8% DCR: 70% PFS: 5.5 mo. |
| EXCLAIM [45] | Exon 20 insertions | Mobocertinib | ORR: 43% PFS: 7.3 mo. |
| Clinical Trail | EGFR Status | Comparison | Results |
|---|---|---|---|
| IFCT-1503 ACE-Lung [47] | Exon 19 deletions, L858R, G719X, L861Q, and S768I mutations or exon 19 insertions | Afatinib plus cetuximab vs. afatinib | Ended for futility |
| BELIEF [52] | Common mutations +/− T790M | Erlotinib plus bevacizumab | PFS in ITT: 13.2 mo. PFS in T790M-positive group: 16 mo. PFS in the T790M-negative group: 10.5 mo. |
| JO25567 [53] | Common mutations | Erlotinib single agent or with bevacizumab | * PFS: 16 vs. 9.7 ** OS: 47.0 vs. 47.4 mo. |
| RELAY [54] | Common mutations | Erlotinib plus ramucirumab vs. erlotinib | * PFS: 19.4 vs. 12.4 mo. OS: immature data |
| FLAURA2 [56] | Common mutations +/− T790M | Chemotherapy plus osimertinib | Ongoing |
| Clinical Trail | Phase | Arm(s) | Endpoint |
|---|---|---|---|
| NCT02143466 [155] | 1b | Osimertinib + savolitinib | ORR: 33% PFS: 5.4 mo. |
| SAVANNAH | 2 | Osimertinib + savolitinib | ORR (ongoing) |
| SACHI | 3 | Osimertinib + savolitinib vs. pemetrexed + platinum | PFS (ongoing) |
| TATTON [133] | 1b | Osimertinib plus: selumetinib (MEK1/2 inhibitor), savolitinib (MET-TKI), or durvalumab (anti-PD-L1). | Safety and tollerability |
| INC280 [156] | 1b/2 | Capmatinib plus gefitinib | ORR: 27% |
| GEOMETRY-E | 3 | Capmatinib + osimertinib vs. pemetrexed + platinum | DLT, PFS (ongoing) |
| INSIGHT [157] | 1b/2 | Tepotinib plus gefitinib vs. standard platinum chemotherapy | PFS: 4.9 vs. 4.4 mo. OS: 17.3 vs. 18.7 mo. |
| MARIPOSA [158] | 3 | Amivantanab + lazertinib vs. osimertinib | PFS (ongoing) |
| NCT04545710 | 2 | Osimertinib + abemaciclib | PFS |
| NCT03455829 | 1/2 | Osimertinib + lerociclib | DLT, RP2D, AEs, PFS |
| NCT01090011 [159] | 1b | Afatinib + cetuximab | ORR: 30% PFR: 5 mo. |
| NCT01861223 [160] | 1b/2 | Afatinib + nimotuzumab | ORR: 23% PFS: 4.3 mo. OS: 11.7 mo. |
| SQUIRE [161] | 3 | Necitumumab + gemcitabine + cisplatin vs. gemcitabine + cisplatin | OS: 11.5 vs. 9.9 mo. |
| NCT02520778 [162] | 1b | Osimertinib plus navitoclax | ORR: 100% PFS: 16.8 mo. |
| IMPRESS [163] | 3 | Gefitinib + cisplatin + pemetrexed vs. cisplatin + pemetrexed | OS: 13.4 vs. 19.5 mo. PFS: 5.4 vs. 5.4 mo. |
| NEJ009 [164] | 3 | Gefitinib vs. gefitinib + carboplatin+ pemetrexed | OS: 49 vs. 38.5 mo. PFS: 20.9 vs. 11.9 mo. |
| jRCTs071180062 [165] | 2 | Osimertinib + carboplatin + pemetrexed vs. osimertinib | PFS: 15.8 vs. 14.6 mo. ORR: 71.4% vs. 53.6% |
| FLAME | 2 | Osimertinib plus chemotherapy vs. osimertinib | PFS (ongoing) |
| COMPEL | 2 | Osimertinib plus chemotherapy vs. osimertinib | PFS (ongoing) |
| FLAURA 2 [56] | 3 | Osimertinib + platinum–pemetrexed vs. platinum–pemetrexed | PFS (ongoing) OS (ongoing) |
| CheckMate 012 [166] | 1 | Nivolumab + erlotinib | ORR:15% |
| CAURAL [167] | 3 | Osimertinib + durvalumab vs. osimertinib | AEs |
| IMpower150 [168] | 3 | Atezolizumab plus bevacizumab plus carboplatin plus paclitaxel vs. bevacizumab plus carboplatin plus paclitaxel | OS: 19.5 vs. 14.7 mo. |
| ORIENT-31 [169] | 3 | Sintilimab + bevacizumab biosimilar IBI305 + chemotherapy vs. chemotherapy alone | PFS: 6.9 vs. 4.3 mo. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laface, C.; Maselli, F.M.; Santoro, A.N.; Iaia, M.L.; Ambrogio, F.; Laterza, M.; Guarini, C.; De Santis, P.; Perrone, M.; Fedele, P. The Resistance to EGFR-TKIs in Non-Small Cell Lung Cancer: From Molecular Mechanisms to Clinical Application of New Therapeutic Strategies. Pharmaceutics 2023, 15, 1604. https://doi.org/10.3390/pharmaceutics15061604
Laface C, Maselli FM, Santoro AN, Iaia ML, Ambrogio F, Laterza M, Guarini C, De Santis P, Perrone M, Fedele P. The Resistance to EGFR-TKIs in Non-Small Cell Lung Cancer: From Molecular Mechanisms to Clinical Application of New Therapeutic Strategies. Pharmaceutics. 2023; 15(6):1604. https://doi.org/10.3390/pharmaceutics15061604
Chicago/Turabian StyleLaface, Carmelo, Felicia Maria Maselli, Anna Natalizia Santoro, Maria Laura Iaia, Francesca Ambrogio, Marigia Laterza, Chiara Guarini, Pierluigi De Santis, Martina Perrone, and Palma Fedele. 2023. "The Resistance to EGFR-TKIs in Non-Small Cell Lung Cancer: From Molecular Mechanisms to Clinical Application of New Therapeutic Strategies" Pharmaceutics 15, no. 6: 1604. https://doi.org/10.3390/pharmaceutics15061604