
Pulmonary Application of Novel Antigen-Loaded Chitosan Nano-Particles Co-Administered with the Mucosal Adjuvant C-Di-AMP Resulted in Enhanced Immune Stimulation and Dose Sparing Capacity
 ,
, 
Abstract
:1. Introduction
2. Material & Methods
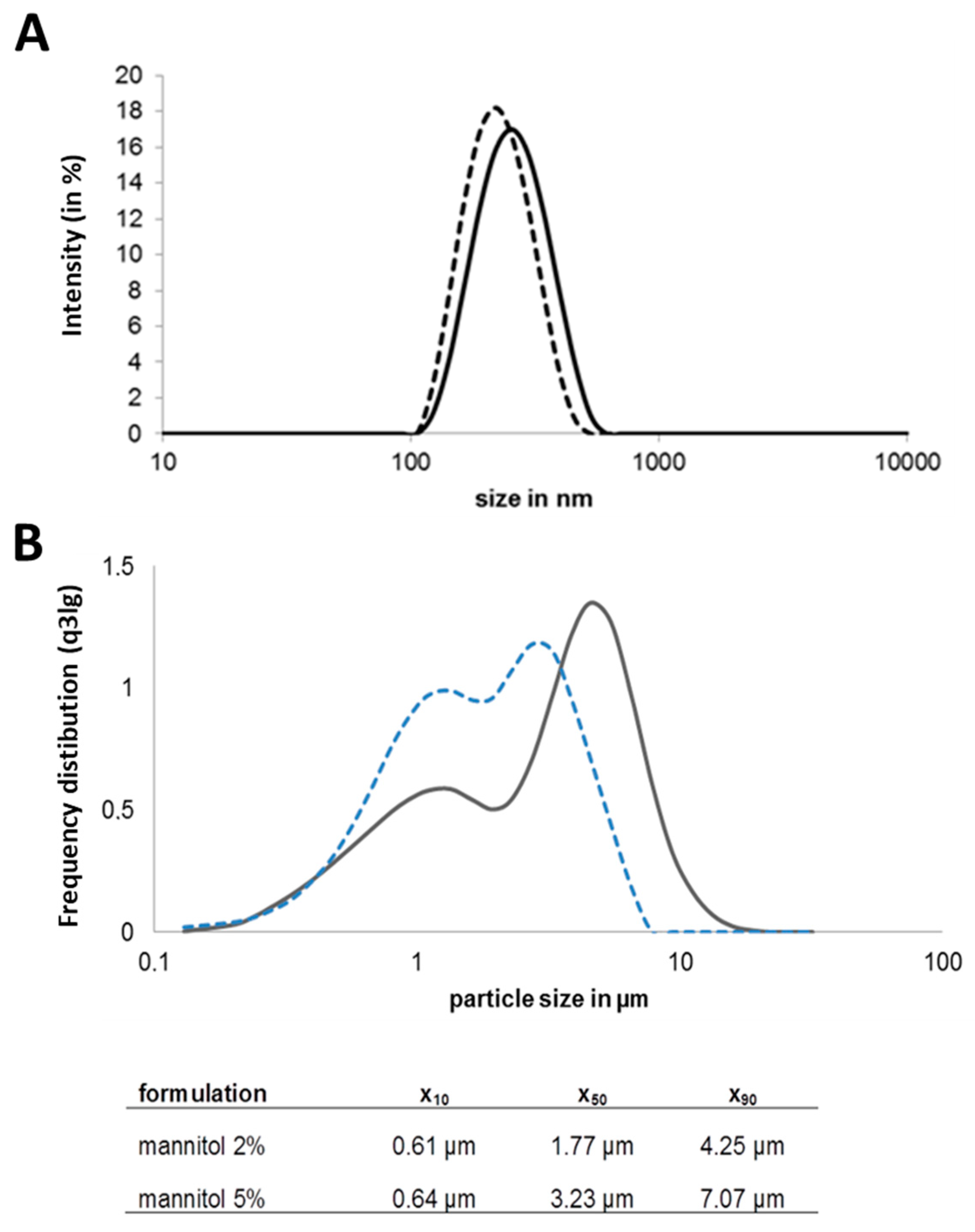
2.1. Nanoparticle Formation and Characterisation
2.2. Transfer to Dry Powder Nano-In-Microparticulate Formulation
2.3. Dispersion and Aerodynamic Assessment
2.4. Scanning Electron Microscopy (SEM) Images
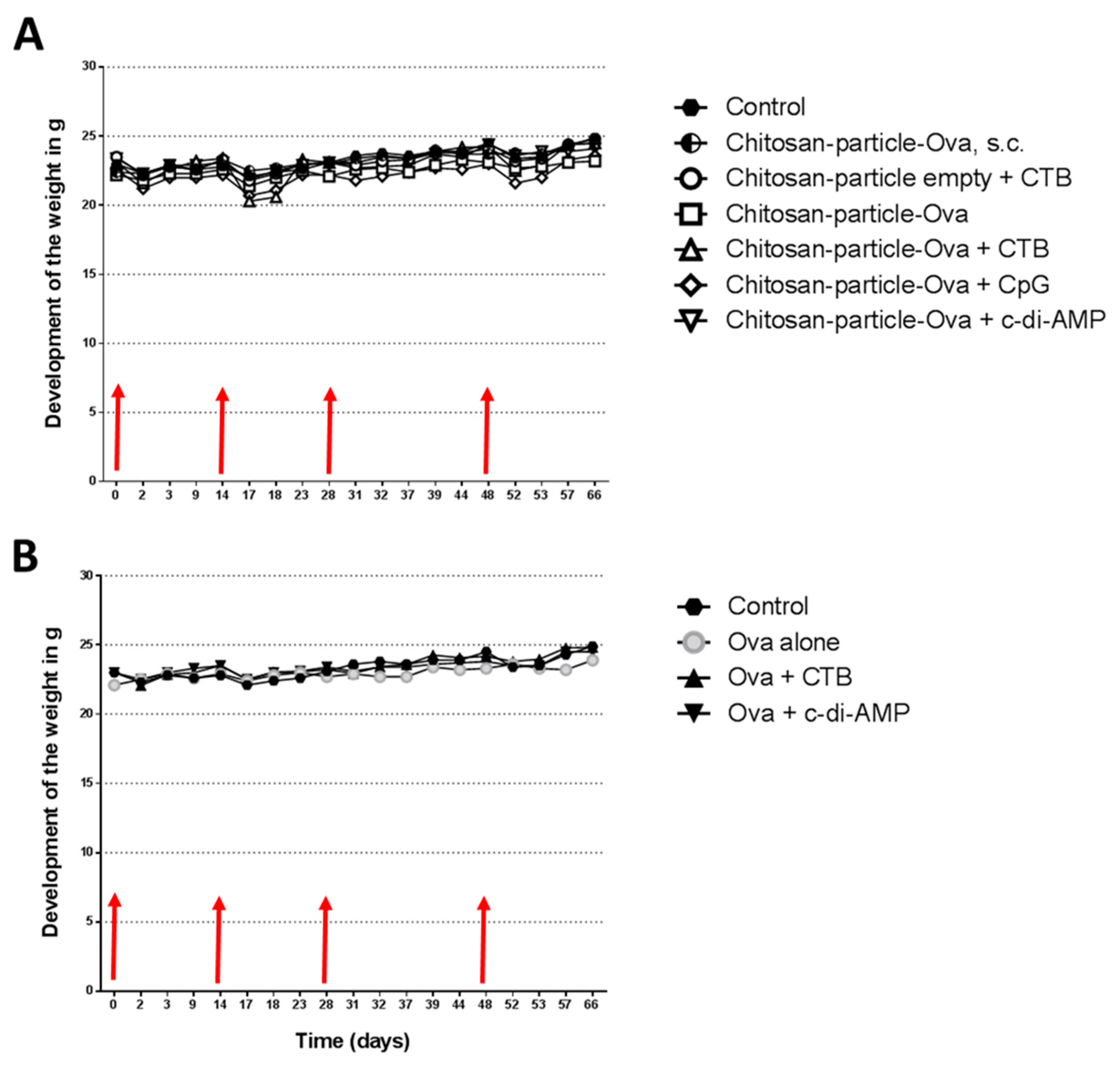
2.5. In Vivo Vaccination Studies
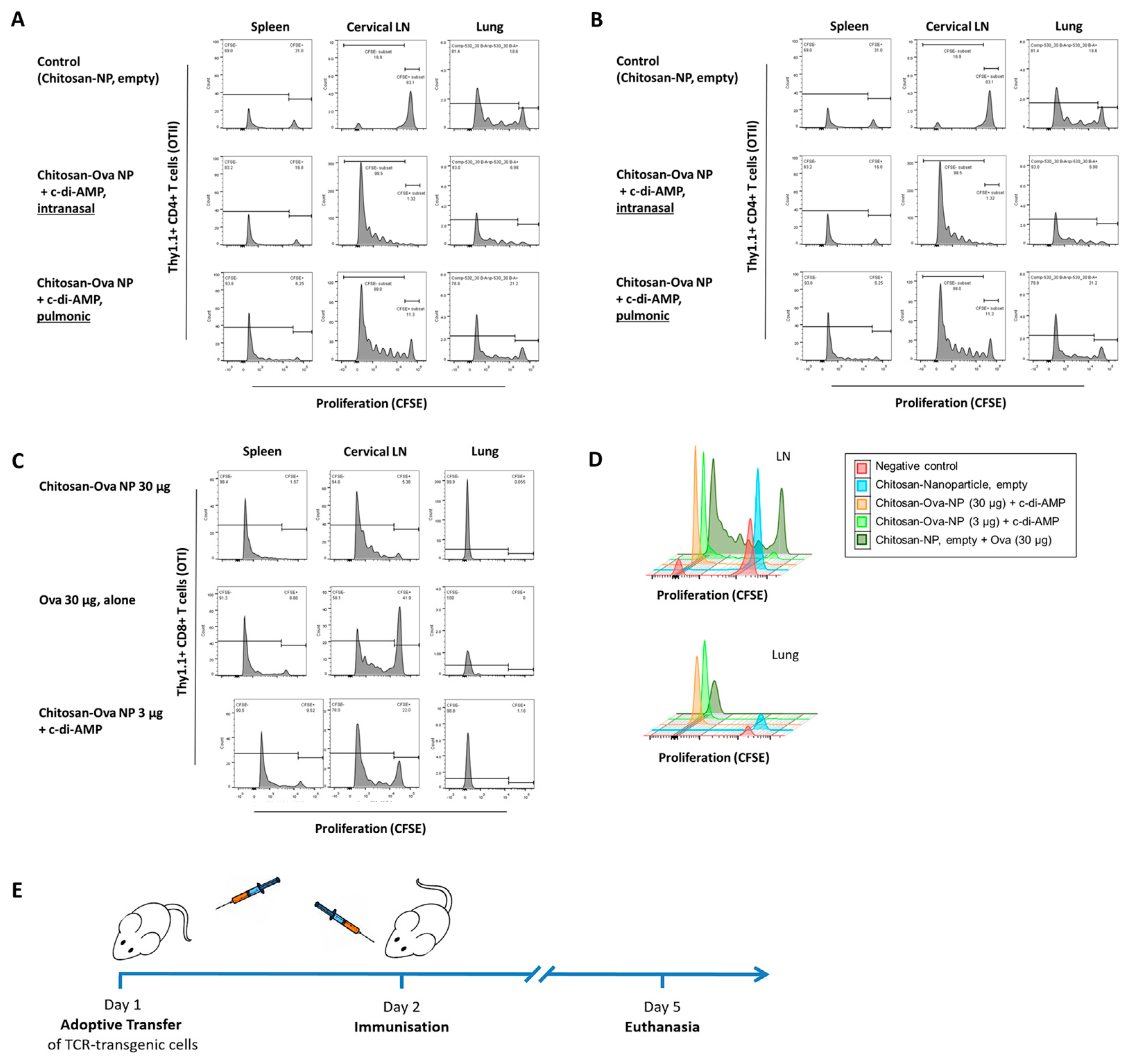
2.6. Adoptive Transfer
2.7. Enrichment of Naïve Non-Activated T Cells
2.8. Cell Labeling with Carboxyfluorescein Diacetate Succinimidyl Ester (CFDA-SE)
2.9. ELISA
2.10. ElISPOT
2.11. Proliferation
2.12. Statistical Analysis
3. Results
3.1. Preparation of Chitosan-OVA Nanoparticles and NiM Formulation Development
3.2. The Immunization with Chitosan-OVA Nanoparticle Co-Administered with c-di-AMP Is Well Tolerated
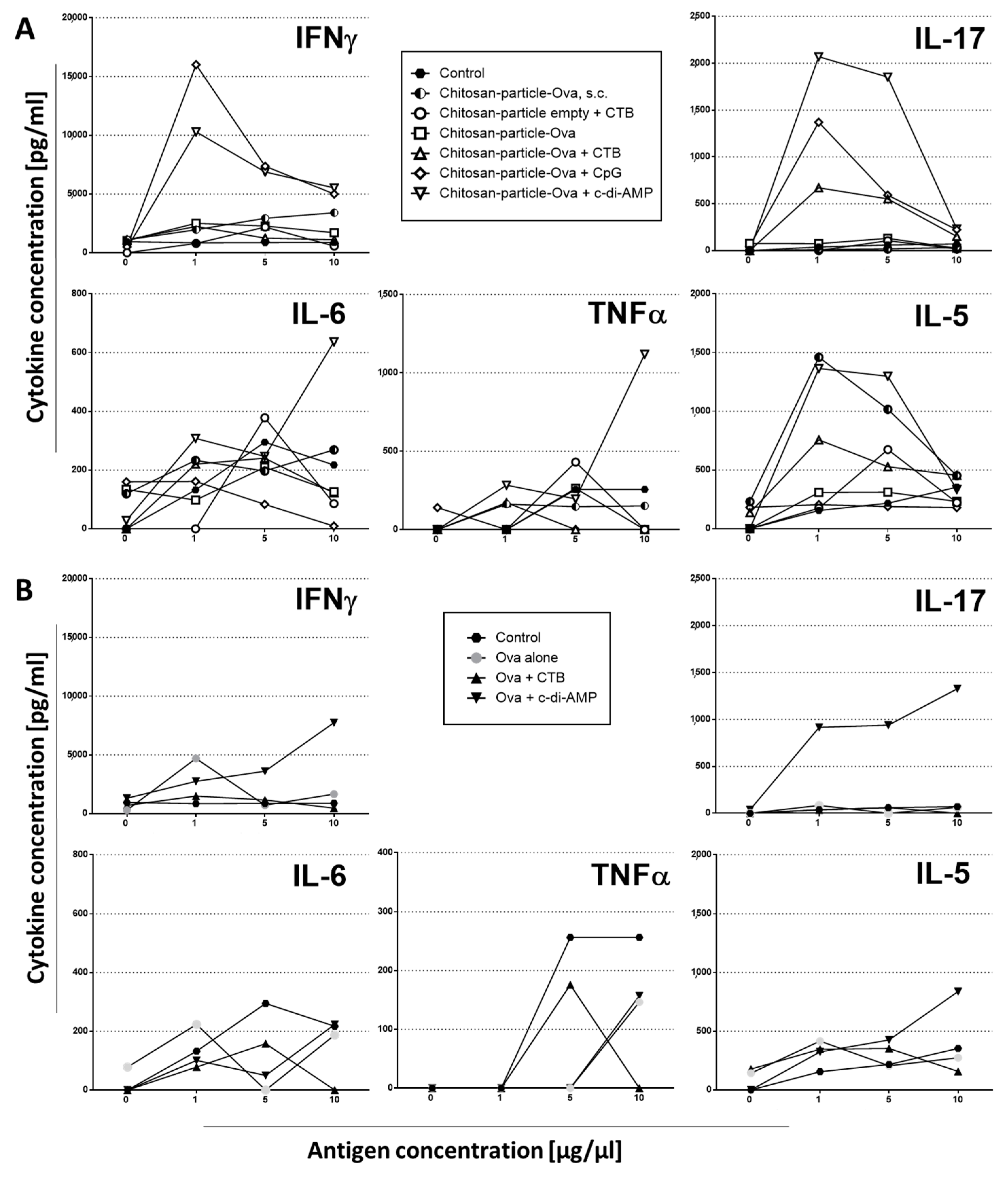
3.3. Mucosal Vaccination Strategies Using Chitosan-OVA Nanoparticles
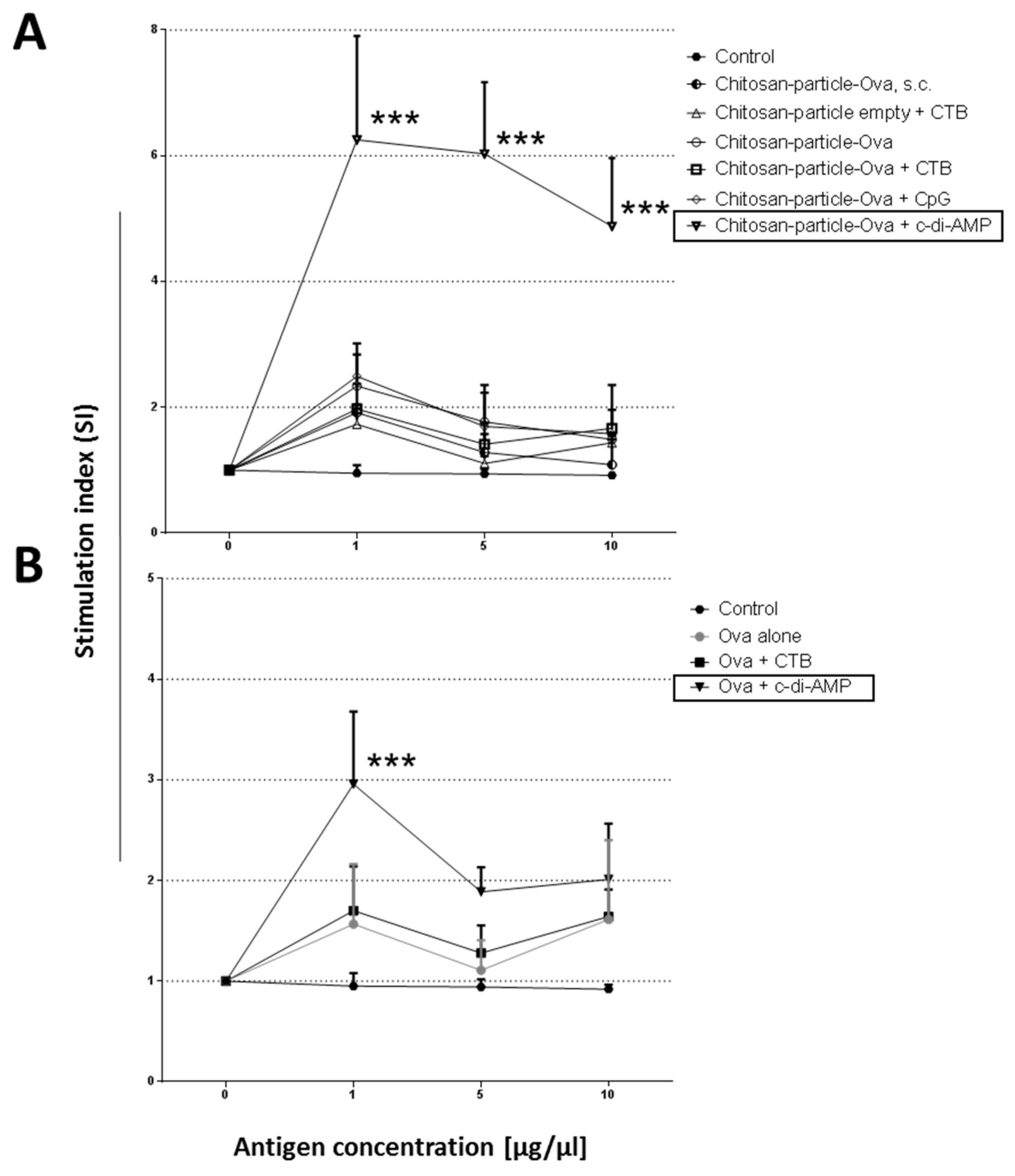
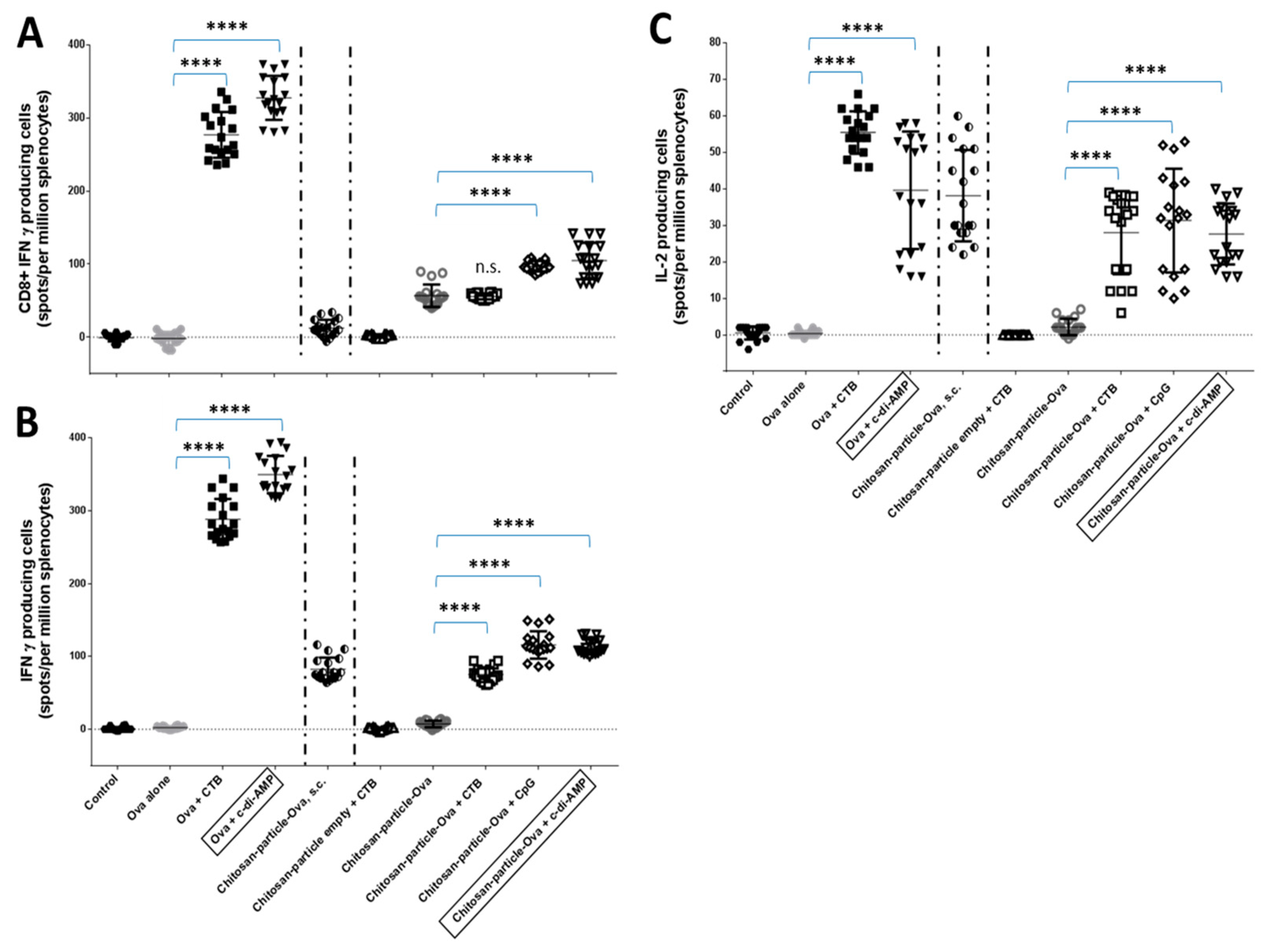
3.4. Pulmonary Administration of Chitosan-OVA Nanoparticles Co-Administered with c-di-AMP Induces Strong Cellular Immune Responses
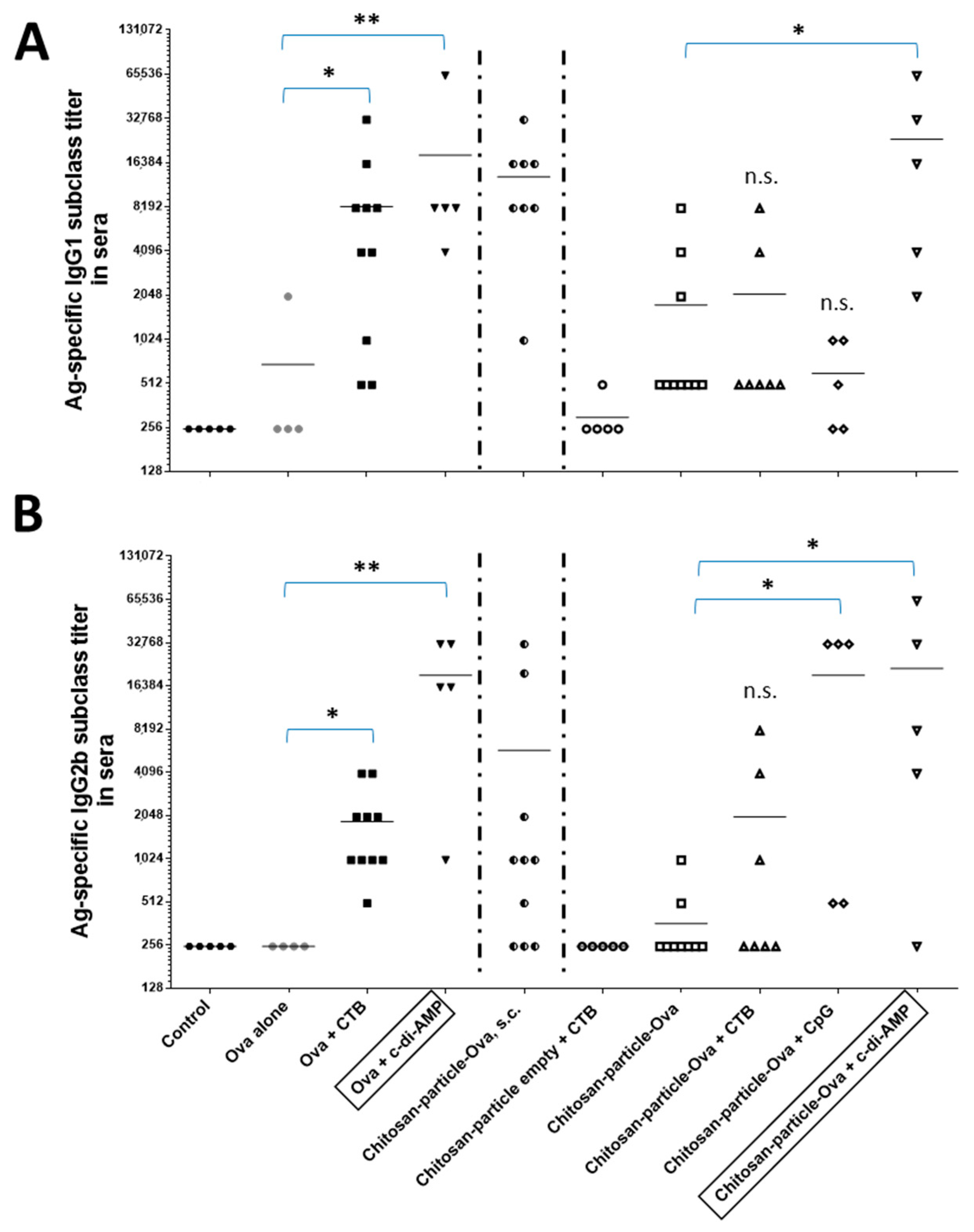
3.5. Chitosan-OVA Nanoparticles Co-Administered with c-di-AMP Elicit Strong Antigen-Specific Humoral Immune Responses
4. Discussion
4.1. Mucosal Administration of Chitosan Nanoparticles
4.2. Production and Preparation of Chitosan Nanoparticles and Transfer to a Dry Powder NiM
4.3. Selection of an Appropriate Mucosal Adjuvant
4.4. Dose Sparing Capacity of Chitosan-OVA Nanoparticles Co-Administered with c-di-AMP
4.5. Induction of OVA-Specific CD8+ T Cell Responses by Chitosan-OVA Nanoparticles Co-Administered with c-di-AMP
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | antigen-presenting cells |
| c-di-AMP | bis-(3′,5′)-cyclic dimeric adenosine monophosphate |
| CD | cluster of differentiation |
| CpG | CpG oligodeoxynucleotide. |
| CTB | cholera toxin B subunit |
| CTL | cytotoxic T lymphocytes |
| DPI | dry powder inhaler |
| ELISPOT | enzyme-linked immunospot |
| FPF | fine particle fraction |
| rHA | hemagglutinin |
| IgG | immune globulin |
| IL-2 | interleukin 2 |
| i.m. | intramuscular |
| IFNγ | interferon gamma |
| i.n. | intranasal |
| LN | lymph nodes |
| MMAD | mass median aerodynamic diameter |
| MHC | major histocompatibility complex |
| NiM | Nano-in-Microparticulate |
| NP | nanoparticles |
| OVA | Ovalbumin |
| s.c. | subcutaneous |
| Th1 | T helper cell 1 |
| TNFα | tumor necrosis factor alpha |
| TLR | Toll-like receptor |
| WHO | world health organisation |
References
- Rappuoli, R.; Miller, H.I.; Falkow, S. The intangible value of vaccination. Science 2002, 297, 937–939. [Google Scholar] [CrossRef] [PubMed]
- Reed, S.G.; Bertholet, S.; Coler, R.N.; Friede, M. New horizons in adjuvants for vaccine development. Trends Immunol. 2009, 30, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Roush, S.W.; Murphy, T.V.; The Vaccine-Preventable Disease Table Working Group. Historical comparisons of morbidity and mortality for vaccine-preventable diseases in the United States. JAMA 2007, 298, 2155–2163. [Google Scholar] [CrossRef] [PubMed]
- Perrie, Y.; Mohammed, A.R.; Kirby, D.J.; McNeil, S.E.; Bramwell, V.W. Vaccine adjuvant systems: Enhancing the efficacy of sub-unit protein antigens. Int. J. Pharm. 2008, 364, 272–280. [Google Scholar] [CrossRef]
- Lamichhane, A.; Azegamia, T.; Kiyonoa, H. The mucosal immune system for vaccine development. Vaccine 2014, 32, 6711–6723. [Google Scholar] [CrossRef] [Green Version]
- Depreter, F.; Pilcer, G.; Amighi, K. Inhaled proteins: Challenges and perspectives. Int. J. Pharm. 2013, 447, 251–280. [Google Scholar] [CrossRef]
- Lawson, L.B.; Norton, E.B.; Clements, J.D. Defending the mucosa: Adjuvant and carrier formulations for mucosal immunity. Curr. Opin. Immunol. 2011, 23, 414–420. [Google Scholar] [CrossRef]
- Srivastava, A.; Gowda, D.; Madhunapantula, S.; Shinde, C.; Iyer, M. Mucosal vaccines: A paradigm shift in the development of mucosal adjuvants and delivery vehicles. APMIS 2015, 123, 275–288. [Google Scholar] [CrossRef]
- Woodrow, K.; Bennett, K.; Lo, D. Mucosal vaccine design and delivery. Annu. Rev. Biomed. Eng. 2012, 14, 17–46. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, K.; Koizumi, H.; Akashi, M.; Nakagawa, S.; Fujita, T.; Yamamoto, A.; Okada, N. Intranasal immunization with poly(gamma-glutamic acid) nanoparticles entrapping antigenic proteins can induce potent tumor immunity. J. Control. Release 2011, 152, 310–316. [Google Scholar] [CrossRef]
- Darrow, J.J.; Kesselheim, A.S. A New Wave of Vaccines for Non-Communicable Diseases: What Are the Regulatory Challenges? Food Drug Law. J. 2015, 70, 243–258. [Google Scholar]
- Cordeiro, A.S.; Alonso, M.J. Recent advances in vaccine delivery. Pharm. Pat. Anal. 2016, 5, 49–73. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.T.; Valiante, N.M. Recent advances in the discovery and delivery of vaccine adjuvants. Nat. Rev. Drug Discov. 2003, 2, 727–735. [Google Scholar] [CrossRef] [PubMed]
- De Temmerman, M.-L.; Rejman, J.; Demeester, J.; Irvine, D.J.; Gander, B.; De Smedt, S.C. Particulate vaccines: On the quest for optimal delivery and immune response. Drug Discov. Today 2011, 16, 569–582. [Google Scholar] [CrossRef]
- Babiuch, K.; Gottschaldt, M.; Werz, O.; Schubert, U.S. Particulate transepithelial drug carriers: Barriers and functional polymers. RSC Adv. 2012, 2, 10427–10465. [Google Scholar] [CrossRef]
- Vyas, S.P.; Gupta, P.N. Implication of nanoparticles/microparticles in mucosal vaccine delivery. Expert Rev. Vaccines 2007, 6, 401–418. [Google Scholar] [CrossRef]
- Csaba, N.; Garcia-Fuentes, M.; Alonso, M.J. Nanoparticles for nasal vaccination. Adv. Drug Deliv. Rev. 2009, 61, 140–157. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Perelman, M.; Hinchcliffe, M. Chitosan: A promising safe and immune-enhancing adjuvant for intranasal vaccines. Hum. Vaccin. Immunother. 2014, 10, 797–807. [Google Scholar] [CrossRef]
- Andrade, F.; Antunes, F.; Nascimento, A.V.; Da Silva, S.B.; Das Neves, J.; Ferreira, D.; Sarmento, B. Chitosan formulations as carriers for therapeutic proteins. Curr. Drug Discov. Technol. 2011, 8, 157–172. [Google Scholar] [CrossRef]
- Scherliess, R.; Buske, S.; Young, K.; Weber, B.; Rades, T.; Hook, S. In vivo evaluation of chitosan as an adjuvant in subcutaneous vaccine formulations. Vaccine 2013, 31, 4812–4819. [Google Scholar] [CrossRef]
- Ohmes, J.; Saure, L.M.; Schütt, F.; Trenkel, M.; Seekamp, A.; Scherließ, R.; Adelung, R.; Fuchs, S. Injectable Thermosensitive Chitosan-Collagen Hydrogel as A Delivery System for Marine Polysaccharide Fucoidan. Mar. Drugs 2022, 20, 402. [Google Scholar] [CrossRef] [PubMed]
- Walter, F.; Winter, E.; Rahn, S.; Heidland, J.; Meier, S.; Struzek, A.-M.; Lettau, M.; Philipp, L.-M.; Beckinger, S.; Otto, L.; et al. Chitosan nanoparticles as antigen vehicles to induce effective tumor specific T cell responses. PLoS ONE 2020, 15, e0239369. [Google Scholar] [CrossRef]
- Scherliess, R.; Mönckedieck, M.; Young, K.; Trows, S.; Buske, S.; Hook, S. First in vivo evaluation of particulate nasal dry powder vaccine formulations containing ovalbumin in mice. Int. J. Pharm. 2015, 479, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Scherliess, R.; Ajmera, A.; Dennis, M.; Carroll, M.W.; Altrichter, J.; Silman, N.J.; Scholz, M.; Kemter, K.; Marriott, A.C. Induction of protective immunity against H1N1 influenza A(H1N1)pdm09 with spray-dried and electron-beam sterilised vaccines in non-human primates. Vaccine 2014, 32, 2231–2240. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Firdous, J.; Choi, Y.J.; Yun, C.H.; Cho, C.S. Design and application of chitosan microspheres as oral and nasal vaccine carriers: An updated review. Int. J. Nanomed. 2012, 7, 6077–6093. [Google Scholar] [CrossRef] [Green Version]
- Baleeiro, R.B.; Schweinlin, M.; Rietscher, R.; Diedrich, A.; Czaplewska, J.A.; Metzger, M.; Lehr, C.M.; Scherließ, R.; Hanefeld, A.; Gottschaldt, M.; et al. Nanoparticle-Based Mucosal Vaccines Targeting Tumor-Associated Antigens to Human Dendritic Cells. J. Biomed. Nanotechnol. 2016, 12, 1527–1543. [Google Scholar] [CrossRef] [PubMed]
- Hanefeld, A.; Weigandt, M.; Wolf, M.; Knolle, P.; Schröder, M.; Scherließ, R.; Walden, P.; Diedrich, A.; Steckel, H.; Baleeiro, R.B. Antigen-Loaded Chitosan Nanoparticles for Immunotherapy; WO2015/185180A1; Merck Patent Gesellschaft: New York, NY, USA, 2014. [Google Scholar]
- Savelkoul, H.F.; Ferro, V.A.; Strioga, M.M.; Schijns, V.E. Choice and Design of Adjuvants for Parenteral and Mucosal Vaccines. Vaccines 2015, 3, 148–171. [Google Scholar] [CrossRef] [Green Version]
- Riese, P.; Schulze, K.; Ebensen, T.; Prochnow, B.; Guzman, C.A. Vaccine adjuvants: Key tools for innovative vaccine design. Curr. Top. Med. Chem. 2013, 13, 2562–2580. [Google Scholar] [CrossRef]
- Heegaard, P.H.; Fang, Y.; Jungersen, G. Vaccine Technologies for Veterinary Viral Diseases; Methods in Molecular Biology; Brun, A., Ed.; Springer: New York, NY, USA, 2016; Volume 1349, pp. 63–82. [Google Scholar]
- Del Giudice, G.; Rappuoli, R.; Didierlaurent, A.M. Correlates of adjuvanticity: A review on adjuvants in licensed vaccines. Semin. Immunol. 2018, 39, 14–21. [Google Scholar] [CrossRef]
- Ebensen, T.; Guzman, C.A. Immune modulators with defined molecular targets: Cornerstone to optimize rational vaccine design. Adv. Exp. Med. Biol. 2009, 655, 171–188. [Google Scholar] [CrossRef] [Green Version]
- Schaap, P. Cyclic di-nucleotide signaling enters the eukaryote domain. IUBMB Life 2013, 65, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Kim, J.-Y.; Lee, Y.; Kim, J.M.; Kim, Y.B.; Chun, T.; Oh, Y.-K. Enhanced humoral and cellular immune responses after sublingual immunization against human papillomavirus 16 L1 protein with adjuvants. Vaccine 2010, 28, 2598–2606. [Google Scholar] [CrossRef]
- Lycke, N. From toxin to adjuvant: Basic mechanisms for the control of mucosal IgA immunity and tolerance. Immunol. Lett. 2005, 97, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Pizza, M.; Giuliani, M.; Fontana, M.; Monaci, E.; Douce, G.; Dougan, G.; Mills, K.; Rappuoli, R.; Del Giudice, G. Mucosal vaccines: Non toxic derivatives of LT and CT as mucosal adjuvants. Vaccine 2001, 19, 2534–2541. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Yin, Y.; Yu, Q.; Huang, L.; Wang, X.; Lin, J.; Yang, Q. CpG Oligodeoxynucleotides Facilitate Delivery of Whole Inactivated H9N2 Influenza Virus via Transepithelial Dendrites of Dendritic Cells in Nasal Mucosa. J. Virol. 2015, 89, 5904–5918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iho, S.; Maeyama, J.; Suzuki, F. CpG oligodeoxynucleotides as mucosal adjuvants. Hum. Vaccines Immunother. 2015, 11, 755–760. [Google Scholar] [CrossRef] [Green Version]
- Nierkens, S.; Brok, M.H.D.; Roelofsen, T.; Wagenaars, J.A.L.; Figdor, C.G.; Ruers, T.J.; Adema, G.J. Route of administration of the TLR9 agonist CpG critically determines the efficacy of cancer immunotherapy in mice. PLoS ONE 2009, 4, e8368. [Google Scholar] [CrossRef] [Green Version]
- Palma, C.; Iona, E.; Ebensen, T.; Guzman, C.A.; Cassone, A. The toll-like receptor 2/6 ligand MALP-2 reduces the viability of Mycobacterium tuberculosis in murine macrophages. Open Microbiol. J. 2009, 3, 47–52. [Google Scholar] [CrossRef]
- Borsutzky, S.; Ebensen, T.; Link, C.; Becker, P.D.; Fiorelli, V.; Cafaro, A.; Ensoli, B.; Guzmán, C.A. Efficient systemic and mucosal responses against the HIV-1 Tat protein by prime/boost vaccination using the lipopeptide MALP-2 as adjuvant. Vaccine 2006, 24, 2049–2056. [Google Scholar] [CrossRef]
- Rharbaoui, F.; Drabner, B.; Borsutzky, S.; Winckler, U.; Morr, M.; Ensoli, B.; Mühlradt, P.F.; Guzmán, C.A. The Mycoplasma-derived lipopeptide MALP-2 is a potent mucosal adjuvant. Eur. J. Immunol. 2002, 32, 2857–2865. [Google Scholar] [CrossRef]
- Skrnjug, I.; Rueckert, C.; Libanova, R.; Lienenklaus, S.; Weiss, S.; Guzmán, C. The mucosal adjuvant cyclic di-AMP exerts immune stimulatory effects on dendritic cells and macrophages. PLoS ONE 2014, 9, e95728. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.V.; Ebensen, T.; Schulze, K.; Cargnelutti, D.E.; Blazejewska, P.; Scodeller, E.A.; Guzmán, C.A. Intranasal delivery of influenza rNP adjuvanted with c-di-AMP induces strong humoral and cellular immune responses and provides protection against virus challenge. PLoS ONE 2014, 9, e104824. [Google Scholar] [CrossRef]
- Ebensen, T.; Libanova, R.; Schulze, K.; Yevsa, T.; Morr, M.; Guzmán, C.A. Bis-(3′,5′)-cyclic dimeric adenosine monophosphate: Strong Th1/Th2/Th17 promoting mucosal adjuvant. Vaccine 2011, 29, 5210–5220. [Google Scholar] [CrossRef]
- Libanova, R.; Ebensen, T.; Schulze, K.; Bruhn, D.; Nörder, M.; Yevsa, T.; Morr, M.; Guzmán, C.A. The member of the cyclic di-nucleotide family bis-(3′,5′)-cyclic dimeric inosine monophosphate exerts potent activity as mucosal adjuvant. Vaccine 2010, 28, 2249–2258. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Sater, A.A.; Grajkowski, A.; Erdjument-Bromage, H.; Plumlee, C.; Levi, A.; Schreiber, M.T.; Lee, C.; Shuman, H.; Beaucage, S.L.; Schindler, C. The overlapping host responses to bacterial cyclic dinucleotides. Microbes Infect. 2012, 14, 188–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosolini, M.; Pont, F.; Verhoeyen, E.; Fournie, J.J. Cyclic dinucleotides modulate human T-cell response through monocyte cell death. Eur. J. Immunol. 2015, 45, 3313–3323. [Google Scholar] [CrossRef]
- Sanchez Alberti, A.; Bivona, A.E.; Cerny, N.; Schulze, K.; Weißmann, S.; Ebensen, T.; Morales, C.; Padilla, A.M.; Cazorla, S.I.; Tarleton, R.L.; et al. Engineered trivalent immunogen adjuvanted with a STING agonist confers protection against Trypanosoma cruzi infection. NPJ Vaccines 2017, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Matos, M.N.; Cazorla, S.I.; Schulze, K.; Ebensen, T.; Guzmán, C.A.; Malchiodi, E.L. Immunization with Tc52 or its amino terminal domain adjuvanted with c-di-AMP induces Th17+Th1 specific immune responses and confers protection against Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2017, 11, e0005300. [Google Scholar] [CrossRef] [Green Version]
- Lirussi, D.; Ebensen, T.; Schulze, K.; Trittel, S.; Duran, V.; Liebich, I.; Kalinke, U.; Guzmán, C.A. Type I IFN and not TNF, is Essential for Cyclic Di-nucleotide-elicited CTL by a Cytosolic Cross-presentation Pathway. eBioMedicine 2017, 22, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Landi, A.; Law, J.; Hockman, D.; Logan, M.; Crawford, K.; Chen, C.; Kundu, J.; Ebensen, T.; Guzman, C.; Deschatelets, L.; et al. Superior immunogenicity of HCV envelope glycoproteins when adjuvanted with cyclic-di-AMP, a STING activator or archaeosomes. Vaccine 2017, 35, 6949–6956. [Google Scholar] [CrossRef]
- Ebensen, T.; DeBarry, J.; Pedersen, G.; Blazejewska, P.; Weissmann, S.; Schulze, K.; McCullough, K.C.; Cox, R.J.; Guzmán, C.A. Mucosal Administration of Cycle-Di-Nucleotide-Adjuvanted Virosomes Efficiently Induces Protection against Influenza H5N1 in Mice. Front. Immunol. 2017, 8, 1223. [Google Scholar] [CrossRef] [PubMed]
- Lirussi, D.; Weissmann, S.; Ebensen, T.; Nitsche-Gloy, U.; Franz, H.; Guzmán, C. Cyclic Di-Adenosine Monophosphate: A Promising Adjuvant Candidate for the Development of Neonatal Vaccines. Pharmaceutics 2021, 13, 188. [Google Scholar] [CrossRef]
- Lanfermann, C.; Wintgens, S.; Ebensen, T.; Kohn, M.; Laudeley, R.; Schulze, K.; Rheinheimer, C.; Hegemann, J.; Guzmán, C.; Klos, A. Prophylactic Multi-Subunit Vaccine against Chlamydia trachomatis: In Vivo Evaluation in Mice. Vaccines 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Demoulins, T.; Ruggli, N.; Gerber, M.; Thomann-Harwood, L.J.; Ebensen, T.; Schulze, K.; Guzmán, C.A.; McCullough, K.C. Self-Amplifying Pestivirus Replicon RNA Encoding Influenza Virus Nucleoprotein and Hemagglutinin Promote Humoral and Cellular Immune Responses in Pigs. Front. Immunol. 2020, 11, 622385. [Google Scholar] [CrossRef] [PubMed]
- Sanchez Alberti, A.; Bivona, A.E.; Matos, M.N.; Cerny, N.; Schulze, K.; Weißmann, S.; Ebensen, T.; González, G.; Morales, C.; Cardoso, A.C.; et al. Mucosal Heterologous Prime/Boost Vaccination Induces Polyfunctional Systemic Immunity, Improving Protection Against Trypanosoma cruzi. Front. Immunol. 2020, 11, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volckmar, J.; Knop, L.; Stegemann-Koniszewski, S.; Schulze, K.; Ebensen, T.; Guzmán, C.A.; Bruder, D. The STING activator c-di-AMP exerts superior adjuvant properties than the formulation poly(I:C)/CpG after subcutaneous vaccination with soluble protein antigen or DEC-205-mediated antigen targeting to dendritic cells. Vaccine 2019, 37, 4963–4974. [Google Scholar] [CrossRef]
- Ebensen, T.; Delandre, S.; Prochnow, B.; Guzman, C.A.; Schulze, K. The Combination Vaccine Adjuvant System Alum/c-di-AMP Results in Quantitative and Qualitative Enhanced Immune Responses Post Immunization. Front. Cell. Infect. Microbiol. 2019, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, D.; Chen, D.; Cummings, R. Vaccine stabilization: Research, commercialization, and potential impact. Vaccine 2011, 29, 7122–7124. [Google Scholar] [CrossRef]
- Lal, M. Technologies for the Development of Thermostable Vaccines. 1-24 (PATH, 2013). Available online: https://www.path.org/media-center/new-technology-for-producing-thermostable-vaccines/ (accessed on 28 November 2022).
- Diedrich, A.; Scherließ, R. In Proceedings of the DDL 25, Edinburgh, Scotland, 10–12 December 2014.
- Schulze, K.; Ebensen, T.; Riese, P.; Prochnow, B.; Lehr, C.-M.; Guzmán, C.A. New Horizons in the Development of Novel Needle-Free Immunization Strategies to Increase Vaccination Efficacy. Curr. Top. Microbiol. Immunol. 2016, 398, 207–234. [Google Scholar] [CrossRef]
- Wang, J.J.; Xiao, R.Z.; Xie, T.; Zhou, G.L.; Zhan, X.R.; Wang, S.L. Recent advances of chitosan nanoparticles as drug carriers. Int. J. Nanomed. 2011, 6, 765–774. [Google Scholar]
- Topham, D.J.; Castrucci, M.R.; Wingo, F.S.; Belz, G.T.; Doherty, P.C. The role of antigen in the localization of naive, acutely activated, and memory CD8(+) T cells to the lung during influenza pneumonia. J. Immunol. 2001, 167, 6983–6990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garulli, B.; Di Mario, G.; Sciaraffia, E.; Kawaoka, Y.; Castrucci, M.R. Immunogenicity of a recombinant influenza virus bearing both the CD4+ and CD8+ T cell epitopes of ovalbumin. J. Biomed. Biotechnol. 2011, 2011, 497364. [Google Scholar] [CrossRef] [Green Version]
- Chapman, T.J.; Castrucci, M.R.; Padrick, R.C.; Bradley, L.M.; Topham, D.J. Antigen-specific and non-specific CD4+ T cell recruitment and proliferation during influenza infection. Virology 2005, 340, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Lyons, A.B.; Blake, S.J.; Doherty, K.V. Flow cytometric analysis of cell division by dilution of CFSE and related dyes. Curr. Protoc. Cytom. 2013, 9, 11–19. [Google Scholar] [CrossRef]
- Blank, F.; Stumbles, P.; von Garnier, C. Opportunities and challenges of the pulmonary route for vaccination. Expert Opin. Drug Deliv. 2011, 8, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Pape, K.A.; Kearney, E.R.; Khoruts, A.; Mondino, A.; Merica, R.; Chen, Z.-M.; Ingulli, E.; White, J.; Johnson, J.G.; Jenkins, M. Use of adoptive transfer of T-cell-antigen-receptor-transgenic T cell for the study of T-cell activation in vivo. Immunol. Rev. 1997, 156, 67–78. [Google Scholar] [CrossRef]
- Ma, P.; Liu, H.-T.; Wei, P.; Xu, Q.-S.; Bai, X.-F.; Du, Y.-G.; Yu, C. Chitosan oligosaccharides inhibit LPS-induced over-expression of IL-6 and TNF-α in RAW264.7 macrophage cells through blockade of mitogen-activated protein kinase (MAPK) and PI3K/Akt signaling pathways. Carbohydr. Polym. 2011, 84, 1391–1398. [Google Scholar] [CrossRef]
- Li, D.; Fu, D.; Kang, H.; Rong, G.; Jin, Z.; Wang, X.; Zhao, K. Advances and Potential Applications of Chitosan Nanoparticles as a Delivery Carrier for the Mucosal Immunity of Vaccine. Curr. Drug Deliv. 2017, 14, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Sun, B.; Jin, Z.; Zhao, K. Enhanced Immune Responses to Mucosa by Functionalized Chitosan-Based Composite Nanoparticles as a Vaccine Adjuvant for Intranasal Delivery. ACS Appl. Mater. Interfaces 2022, 14, 52691–52701. [Google Scholar] [CrossRef]
- Forbes, E.K.; Sander, C.; Ronan, E.O.; McShane, H.; Hill, A.V.S.; Beverley, P.C.L.; Tchilian, E.Z. Multifunctional, high-level cytokine-producing Th1 cells in the lung, but not spleen, correlate with protection against Mycobacterium tuberculosis aerosol challenge in mice. J. Immunol. 2008, 181, 4955–4964. [Google Scholar] [CrossRef] [Green Version]
- Mody, N.; Sharma, R.; Agrawal, U.; Vyas, S.P. Nanocarriers: A versatile approach for mucosal vaccine delivery. Ther. Deliv. 2015, 6, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Jabbal-Gill, I. Nasal vaccine innovation. J. Drug Target. 2010, 18, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Frank, L.A.; Sandri, G.; D’Autilia, F.; Contri, R.V.; Bonferoni, M.C.; Caramella, C.; Frank, A.G.; Pohlmann, A. Chitosan gel containing polymeric nanocapsules: A new formulation for vaginal drug delivery. Int. J. Nanomed. 2014, 9, 3151–3161. [Google Scholar] [CrossRef] [Green Version]
- Riese, P.; Sakthivel, P.; Trittel, S.; Guzman, C.A. Intranasal formulations: Promising strategy to deliver vaccines. Expert Opin. Drug Deliv. 2014, 11, 1619–1634. [Google Scholar] [CrossRef] [PubMed]
- van der Lubben, I.M.; Verhoef, J.C.; Borchard, G.; Junginger, H.E. Chitosan for mucosal vaccination. Adv. Drug Deliv. Rev. 2001, 52, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.J.; Lee, J.-S.; Talactac, M.R.; Chowdhury, M.Y.; Kim, J.-H.; Park, M.-E.; Choi, Y.-K.; Sung, M.-H.; Kim, C.-J. Mucosal immunization with recombinant influenza hemagglutinin protein and poly gamma-glutamate/chitosan nanoparticles induces protection against highly pathogenic influenza A virus. Vet. Microbiol. 2012, 160, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Jabbal-Gill, I.; Watts, P.; Smith, A. Chitosan-based delivery systems for mucosal vaccines. Expert Opin. Drug Deliv. 2012, 9, 1051–1067. [Google Scholar] [CrossRef]
- Carvalho, T.C.; Peters, J.I.; Williams III, R.O. Influence of particle size on regional lung deposition—What evidence is there? Int. J. Pharm. 2011, 406, 1–10. [Google Scholar] [CrossRef]
- Vehring, R. Pharmaceutical Particle Engineering via Spray Drying. Pharm. Res. 2008, 25, 999–1022. [Google Scholar] [CrossRef] [Green Version]
- Littringer, E.M.; Mescher, A.; Eckhard, S.; Schröttner, H.; Langes, C.; Fries, M.; Griesser, U.; Walzel, P.; Urbanetz, N.A. Spray Drying of Mannitol as a Drug Carrier—The Impact of Process Parameters on Product Properties. Dry. Technol. 2012, 30, 114–124. [Google Scholar] [CrossRef]
- Diedrich, A. Entwicklung einer nanopartikulären Formulierung zur Vakzinierung über den Respirationstrakt. Ph.D. Thesis, Kiel University, Kiel, Germany, 2015. [Google Scholar]
- Norton, E.B.; Bauer, D.L.; Weldon, W.C.; Oberste, M.S.; Lawson, L.B.; Clements, J.D. The novel adjuvant dmLT promotes dose sparing, mucosal immunity and longevity of antibody responses to the inactivated polio vaccine in a murine model. Vaccine 2015, 33, 1909–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wee, J.L.; Scheerlinck, J.-P.Y.; Snibson, K.J.; Edwards, S.; Pearse, M.; Quinn, C.; Sutton, P. Pulmonary delivery of ISCOMATRIX influenza vaccine induces both systemic and mucosal immunity with antigen dose sparing. Mucosal Immunol. 2008, 1, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svindland, S.C.; Pedersen, G.K.; Pathirana, R.D.; Bredholt, G.; Nøstbakken, J.K.; Jul-Larsen, Å.; Guzmán, C.A.; Montomoli, E.; Lapini, G.; Piccirella, S.; et al. A study of Chitosan and c-di-GMP as mucosal adjuvants for intranasal influenza H5N1 vaccine. Influenza Other Respir. Viruses 2013, 7, 1181–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svindland, S.C.; Jul-Larsen, .; Pathirana, R.; Andersen, S.; Madhun, A.; Montomoli, E.; Jabbal-Gill, I.; Cox, R.J. The mucosal and systemic immune responses elicited by a chitosan-adjuvanted intranasal influenza H5N1 vaccine. Influenza Other Respir. Viruses 2012, 6, 90–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutsch, M.; Zhou, W.; Rhodes, P.; Bopp, M.; Chen, R.T.; Linder, T.; Spyr, C.; Steffen, R. Use of the inactivated intranasal influenza vaccine and the risk of Bell’s palsy in Switzerland. N. Engl. J. Med. 2004, 350, 896–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, K.; Ebensen, T.; Babiuk, L.A.; Gerdts, V.; Guzman, C.A. Intranasal vaccination with an adjuvanted polyphosphazenes nanoparticle-based vaccine formulation stimulates protective immune responses in mice. Nanomedicine 2017, 13, 2169–2178. [Google Scholar] [CrossRef]
- Schulze, K.; Ebensen, T.; Chandrudu, S.; Skwarczynski, M.; Toth, I.; Olive, C.; Guzman, C.A. Bivalent mucosal peptide vaccines administered using the LCP carrier system stimulate protective immune responses against Streptococcus pyogenes infection. Nanomedicine 2017, 13, 2463–2474. [Google Scholar] [CrossRef]
- Mittal, A.; Schulze, K.; Ebensen, T.; Weissmann, S.; Hansen, S.; Guzmán, C.A.; Lehr, C.-M. Inverse micellar sugar glass (IMSG) nanoparticles for transfollicular vaccination. J. Control. Release 2015, 206, 140–152. [Google Scholar] [CrossRef] [Green Version]
- Van Dis, E.; Sogi, K.M.; Rae, C.S.; Sivick, K.E.; Surh, N.H.; Leong, M.L.; Kanne, D.B.; Metchette, K.; Leong, J.J.; Bruml, J.R.; et al. STING-Activating Adjuvants Elicit a Th17 Immune Response and Protect against Mycobacterium tuberculosis Infection. Cell. Rep. 2018, 23, 1435–1447. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Vaccine Formulation | Administration Route | C57BL/6 | Dose per Animal |
|---|---|---|---|---|
| 1 | Control | pulmonary | 5 | - |
| 2 | OVA alone | pulmonary | 5 | 30 µg/- |
| 3 | OVA + CTB | pulmonary | 10 | 30 µg/10 µg |
| 4 | OVA + c-di-AMP | pulmonary | 5 | 30 µg/10 µg |
| 5 | Chitosan-particle-OVA | Subcutan (s.c.) | 10 | 30 µg |
| 6 | Chitosan-particle empty + CTB | pulmonary | 5 | 0 µg/10 µg |
| 7 | Chitosan-particle-OVA | pulmonary | 10 | 30 µg |
| 8 | Chitosan-particle-OVA + CTB | pulmonary | 10 | 30 µg/10 µg |
| 9 | Chitosan-particle-OVA + CpG | pulmonary | 5 | 30 µg/10 µg |
| 10 | Chitosan-particle-OVA + c-di-AMP | pulmonary | 5 | 30 µg/10 µg |
| Group | Vaccine Formulation | Route | C57BL/6 | Number of Animals |
|---|---|---|---|---|
| 1 | Control | i.n./pulmonary | OTII | 1 |
| 2 | Chitosan NP, empty | pulmonary | OTII | 3 * |
| 3 | OVA 30 µg | i.n./pulmonary | OTII | 5 |
| 4 | Chitosan-OVA NP 30 µg + c-di-AMP | i.n./pulmonary | OTII | 5 |
| 5 | Chitosan-OVA NP 30 µg | i.n./pulmonary | OTI | 5 |
| 6 | Chitosan-OVA NP 30 µg | i.n./pulmonary | OTI | 3 * |
| 7 | Chitosan-OVA NP 3 µg + c-di-AMP | i.n./pulmonary | OTI | 3 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebensen, T.; Arntz, A.; Schulze, K.; Hanefeld, A.; Guzmán, C.A.; Scherließ, R. Pulmonary Application of Novel Antigen-Loaded Chitosan Nano-Particles Co-Administered with the Mucosal Adjuvant C-Di-AMP Resulted in Enhanced Immune Stimulation and Dose Sparing Capacity. Pharmaceutics 2023, 15, 1238. https://doi.org/10.3390/pharmaceutics15041238
Ebensen T, Arntz A, Schulze K, Hanefeld A, Guzmán CA, Scherließ R. Pulmonary Application of Novel Antigen-Loaded Chitosan Nano-Particles Co-Administered with the Mucosal Adjuvant C-Di-AMP Resulted in Enhanced Immune Stimulation and Dose Sparing Capacity. Pharmaceutics. 2023; 15(4):1238. https://doi.org/10.3390/pharmaceutics15041238
Chicago/Turabian StyleEbensen, Thomas, Andrea Arntz, Kai Schulze, Andrea Hanefeld, Carlos A. Guzmán, and Regina Scherließ. 2023. "Pulmonary Application of Novel Antigen-Loaded Chitosan Nano-Particles Co-Administered with the Mucosal Adjuvant C-Di-AMP Resulted in Enhanced Immune Stimulation and Dose Sparing Capacity" Pharmaceutics 15, no. 4: 1238. https://doi.org/10.3390/pharmaceutics15041238