
Enhancing the Effectiveness of Oligonucleotide Therapeutics Using Cell-Penetrating Peptide Conjugation, Chemical Modification, and Carrier-Based Delivery Strategies


Abstract
:1. Introduction
2. ON-Based Therapeutic Platforms
2.1. Gapmers
2.2. Aptamers

2.3. Steric-Blocking ONs
2.4. Immunostimulant ONs (ISOs)
2.5. Antagomirs, RNA Sponges, and Blockmirs
2.6. Agomirs
2.7. Small Interfering RNAs (siRNAs)
2.8. Choice of Antisense Oligonucleotide Platforms for Therapeutic Applications
3. Challenges Associated with the Therapeutic Use of ON Drugs
4. Gymnotic Uptake of ONs
5. Chemical Modifications to Enhance Stability, Cellular Uptake, and Safety
5.1. Backbone Modification
5.2. Nucleobase Modification
5.3. Sugar Modification
6. Bioconjugation
6.1. Peptide Conjugates
6.2. Lipid-Based Conjugates
6.3. Receptor–Ligand Conjugates
6.4. Antibody and Aptamer Conjugates
6.5. Polymer Conjugates
6.6. The Optimal Bioconjugation Strategy
7. ON Delivery Systems
7.1. Lipoplex, Liposomes, and Lipid Nanoparticles
7.2. Peptide-Based Delivery Systems
7.3. Polymer-Based Delivery Systems
7.4. Antibody Complexation Delivery Systems
7.5. Extracellular Vesicle-Mediated Delivery Systems
7.6. Spherical Nucleic Acids
7.7. DNA Nanostructures
7.8. Stimuli-Responsive Delivery Technologies
7.9. Multilayered Biopolymer Nanopolymer-Based Carriers
7.10. Advantages and Limitations of ON Delivery Systems
8. Challenges, Considerations, and Future Perspectives
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hawkins, J.W. A Brief History of Genetic Therapy: Gene Therapy, Antisense Technology, and Genomics. In Clinical Trials of Genetic Therapy with Antisense DNA and DNA Vectors; CRC Press: Boca Raton, FL, USA, 2020; pp. 1–38. ISBN 9781003064657. [Google Scholar]
- Wirth, T.; Parker, N.; Ylä-Herttuala, S. History of Gene Therapy. Gene 2013, 525, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.; Subramanian, G.; Silayeva, L.; Newkirk, I.; Doctor, D.; Chawla, K.; Chattopadhyay, S.; Chandra, D.; Chilukuri, N.; Betapudi, V. Gene Therapy Leaves a Vicious Cycle. Front. Oncol. 2019, 9, 297. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, L.K.; El-Khoury, R.; Thorpe, J.D.; Damha, M.J.; Hollenstein, M. Recent Progress in Non-Native Nucleic Acid Modifications. Chem. Soc. Rev. 2021, 50, 5126–5164. [Google Scholar] [CrossRef] [PubMed]
- Arechavala-Gomeza, V.; Garanto, A. Antisense RNA Therapeutics: A Brief Overview. Methods Mol. Biol. 2022, 2434, 33–49. [Google Scholar]
- Lee, J.J.A.; Yokota, T. Antisense Therapy in Neurology. J. Pers. Med. 2013, 3, 144–176. [Google Scholar] [CrossRef] [Green Version]
- Sasso, J.M.; Ambrose, B.J.B.; Tenchov, R.; Datta, R.S.; Basel, M.T.; DeLong, R.K.; Zhou, Q.A. The Progress and Promise of RNA Medicine–An Arsenal of Targeted Treatments. J. Med. Chem. 2022, 65, 6975–7015. [Google Scholar] [CrossRef]
- Khvorova, A.; Watts, J.K. The Chemical Evolution of Oligonucleotide Therapies of Clinical Utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Andrews, B.I.; Antia, F.D.; Brueggemeier, S.B.; Diorazio, L.J.; Koenig, S.G.; Kopach, M.E.; Lee, H.; Olbrich, M.; Watson, A.L. Sustainability Challenges and Opportunities in Oligonucleotide Manufacturing. J. Org. Chem. 2021, 86, 49–61. [Google Scholar] [CrossRef]
- Kuijper, E.C.; Bergsma, A.J.; Pijnappel, W.W.M.P.; Aartsma-Rus, A. Opportunities and Challenges for Antisense Oligonucleotide Therapies. J. Inherit. Metab. Dis. 2021, 44, 72–87. [Google Scholar] [CrossRef]
- Bost, J.P.; Barriga, H.; Holme, M.N.; Gallud, A.; Maugeri, M.; Gupta, D.; Lehto, T.; Valadi, H.; Esbjörner, E.K.; Stevens, M.M.; et al. Delivery of Oligonucleotide Therapeutics: Chemical Modifications, Lipid Nanoparticles, and Extracellular Vesicles. ACS Nano 2021, 15, 13993–14021. [Google Scholar] [CrossRef]
- Smith, C.I.E.; Zain, R. Therapeutic Oligonucleotides: State of the Art. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 605–630. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, Biodistribution and Cell Uptake of Antisense Oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obika, S.; Nanbu, D.; Hari, Y.; Andoh, J.; Morio, K.; Doi, T.; Imanishi, T. Stability and Structural Features of the Duplexes Containing Nucleoside Analogues with a Fixed N-Type Conformation, 2′-O,4′-C-Methyleneribonucleosides. Tetrahedron Lett. 1998, 39, 5401–5404. [Google Scholar] [CrossRef]
- Koller, E.; Vincent, T.M.; Chappell, A.; De, S.; Manoharan, M.; Bennett, C.F. Mechanisms of Single-Stranded Phosphorothioate Modified Antisense Oligonucleotide Accumulation in Hepatocytes. Nucleic Acids Res. 2011, 39, 4795–4807. [Google Scholar] [CrossRef] [Green Version]
- Fazil, M.H.U.T.; Ong, S.T.; Chalasani, M.L.S.; Low, J.H.; Kizhakeyil, A.; Mamidi, A.; Lim, C.F.H.; Wright, G.D.; Lakshminarayanan, R.; Kelleher, D.; et al. GapmeR Cellular Internalization by Macropinocytosis Induces Sequence-Specific Gene Silencing in Human Primary T-Cells. Sci. Rep. 2016, 6, 37721. [Google Scholar] [CrossRef] [Green Version]
- Pendergraff, H.M.; Krishnamurthy, P.M.; Debacker, A.J.; Moazami, M.P.; Sharma, V.K.; Niitsoo, L.; Yu, Y.; Tan, Y.N.; Haitchi, H.M.; Watts, J.K. Locked Nucleic Acid Gapmers and Conjugates Potently Silence ADAM33, an Asthma-Associated Metalloprotease with Nuclear-Localized MRNA. Mol. Ther. Nucleic Acids 2017, 8, 158–168. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.F.; Swayze, E.E. RNA Targeting Therapeutics: Molecular Mechanisms of Antisense Oligonucleotides as a Therapeutic Platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef]
- Hair, P.; Cameron, F.; McKeage, K. Mipomersen Sodium: First Global Approval. Drugs 2013, 73, 487–493. [Google Scholar] [CrossRef]
- Keam, S.J. Inotersen: First Global Approval. Drugs 2018, 78, 1371–1376. [Google Scholar] [CrossRef]
- Gold, L.; Janjic, N.; Jarvis, T.; Schneider, D.; Walker, J.J.; Wilcox, S.K.; Zichi, D. Aptamers and the RNA World, Past and Present. Cold Spring Harb. Perspect. Biol. 2012, 4, a003582. [Google Scholar] [CrossRef]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as Therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.H.; Lim, S.; Wong, W.F. Antisense Oligonucleotides: From Design to Therapeutic Application. Clin. Exp. Pharmacol. Physiol. 2006, 33, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Corey, D.R. Chemistry, Mechanism and Clinical Status of Antisense Oligonucleotides and Duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Aartsma-Rus, A.; Alves, S.; Borgos, S.E.; Buijsen, R.A.M.; Collin, R.W.J.; Covello, G.; Denti, M.A.; Desviat, L.R.; Echevarría, L.; et al. Delivery of Oligonucleotide-based Therapeutics: Challenges and Opportunities. EMBO Mol. Med. 2021, 13, e13243. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, S.; Vester, B.; Wengel, J. Locked Nucleic Acid (LNA): High Affinity Targeting of RNA for Diagnostics and Therapeutics. Drug Discov. Today Technol. 2005, 2, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Järver, P.; O’Donovan, L.; Gait, M.J. A Chemical View of Oligonucleotides for Exon Skipping and Related Drug Applications. Nucleic Acid Ther. 2014, 24, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Eckstein, F. Phosphorothioates, Essential Components of Therapeutic Oligonucleotides. Nucleic Acid Ther. 2014, 24, 374–387. [Google Scholar] [CrossRef]
- Sun, H.; Zhu, X.; Lu, P.Y.; Rosato, R.R.; Tan, W.; Zu, Y. Oligonucleotide Aptamers: New Tools for Targeted Cancer Therapy. Mol. Ther. Nucleic Acids 2014, 3, e182. [Google Scholar] [CrossRef]
- Yüce, M.; Kurt, H.; Hussain, B.; Budak, H. Systematic Evolution of Ligands by Exponential Enrichment for Aptamer Selection. In Biomedical Applications of Functionalized Nanomaterials; Elsevier: Amsterdam, The Netherlands, 2018; pp. 211–243. [Google Scholar]
- Ellington, A.D.; Szostak, J.W. In Vitro Selection of RNA Molecules That Bind Specific Ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Avci-Adali, M.; Steinle, H.; Michel, T.; Schlensak, C.; Wendel, H.P. Potential Capacity of Aptamers to Trigger Immune Activation in Human Blood. PLoS ONE 2013, 8, e68810. [Google Scholar] [CrossRef] [Green Version]
- Eyetech Study Group. Preclinical and Phase 1a Clinical Evaluation of an Anti-Vegf Pegylated Aptamer (eye001) for the Treatment of Exudative Age-Related Macular Degeneration. Retina 2002, 22, 143–152. [Google Scholar] [CrossRef]
- Klettner, A.; Roider, J. Comparison of Bevacizumab, Ranibizumab, and Pegaptanib In Vitro: Efficiency and Possible Additional Pathways. Investig. Opthalmology Vis. Sci. 2008, 49, 4523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, B.T.; Paul, S.; Jastrzebska, K.; Langer, H.; Caruthers, M.H.; Veedu, R.N. Thiomorpholino Oligonucleotides as a Robust Class of next Generation Platforms for Alternate MRNA Splicing. Proc. Natl. Acad. Sci. USA 2022, 119, e2207956119. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; van Ommen, G.-J.B. Antisense-Mediated Exon Skipping: A Versatile Tool with Therapeutic and Research Applications. RNA 2007, 13, 1609–1624. [Google Scholar] [CrossRef] [Green Version]
- Kole, R.; Krainer, A.R.; Altman, S. RNA Therapeutics: Beyond RNA Interference and Antisense Oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.C.; Aurelian, L.; Reddy, M.P.; Miller, P.S.; Ts’o, P.O. Antiviral Effect of an Oligo(Nucleoside Methylphosphonate) Complementary to the Splice Junction of Herpes Simplex Virus Type 1 Immediate Early Pre-MRNAs 4 and 5. Proc. Natl. Acad. Sci. USA 1986, 83, 2787–2791. [Google Scholar] [CrossRef] [Green Version]
- Lundin, K.E.; Gissberg, O.; Smith, C.I.E. Oligonucleotide Therapies: The Past and the Present. Hum. Gene Ther. 2015, 26, 475–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, H.; Xue, X.; Hou, Z.; Zhou, Y.; Meng, J.; Luo, X. Antisense Antibiotics: A Brief Review of Novel Target Discovery and Delivery. Curr. Drug Discov. Technol. 2010, 7, 76–85. [Google Scholar] [CrossRef]
- Deas, T.S.; Bennett, C.J.; Jones, S.A.; Tilgner, M.; Ren, P.; Behr, M.J.; Stein, D.A.; Iversen, P.L.; Kramer, L.D.; Bernard, K.A.; et al. In Vitro Resistance Selection and In Vivo Efficacy of Morpholino Oligomers against West Nile Virus. Antimicrob. Agents Chemother. 2007, 51, 2470–2482. [Google Scholar] [CrossRef] [Green Version]
- Syed, Y.Y. Eteplirsen: First Global Approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef]
- Dhillon, S. Viltolarsen: First Approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Nusinersen: First Global Approval. Drugs 2017, 77, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hu, C.; Moufawad El Achkar, C.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the Treatment of Duchenne Muscular Dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Anwar, S.; Yokota, T. Golodirsen for Duchenne Muscular Dystrophy. Drugs Today 2020, 56, 491–504. [Google Scholar] [CrossRef]
- Roshmi, R.R.; Yokota, T. Viltolarsen for the Treatment of Duchenne Muscular Dystrophy. Drugs Today 2019, 55, 627–639. [Google Scholar] [CrossRef]
- Roshmi, R.R.; Yokota, T. Pharmacological Profile of Viltolarsen for the Treatment of Duchenne Muscular Dystrophy: A Japanese Experience. Clin. Pharmacol. 2021, 13, 235–242. [Google Scholar] [CrossRef]
- Wilton-Clark, H.; Yokota, T. Toshifumi Yokota Casimersen for Duchenne Muscular Dystrophy. Drugs Today 2021, 57, 707. [Google Scholar] [CrossRef]
- Goodkey, K.; Aslesh, T.; Maruyama, R.; Yokota, T. Nusinersen in the Treatment of Spinal Muscular Atrophy. Methods Mol. Biol. 2018, 1828, 69–76. [Google Scholar] [CrossRef]
- Bauer, S.; Kirschning, C.J.; Häcker, H.; Redecke, V.; Hausmann, S.; Akira, S.; Wagner, H.; Lipford, G.B. Human TLR9 Confers Responsiveness to Bacterial DNA via Species-Specific CpG Motif Recognition. Proc. Natl. Acad. Sci. USA 2001, 98, 9237–9242. [Google Scholar] [CrossRef] [Green Version]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like Receptor Recognizes Bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Wagner, H. Bacterial CpG DNA Activates Immune Cells to Signal Infectious Danger. Adv. Immunol. 1999, 73, 329–368. [Google Scholar] [PubMed]
- Vollmer, J.; Krieg, A.M. Immunotherapeutic Applications of CpG Oligodeoxynucleotide TLR9 Agonists. Adv. Drug Deliv. Rev. 2009, 61, 195–204. [Google Scholar] [CrossRef]
- Klinman, D.M. Immunotherapeutic Uses of CpG Oligodeoxynucleotides. Nat. Rev. Immunol. 2004, 4, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; An, M.; Li, M.; Liu, H. Immunostimulatory Properties of Lipid Modified CpG Oligonucleotides. Mol. Pharm. 2017, 14, 2815–2823. [Google Scholar] [CrossRef] [PubMed]
- Bodera, P. Immunostimulatory Oligonucleotides. Recent Pat. Inflamm. Allergy Drug Discov. 2011, 5, 87–93. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C.A. Innate Immunity: The Virtues of a Nonclonal System of Recognition. Cell 1997, 91, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.D. Development of the CpG Adjuvant 1018: A Case Study. Methods Mol. Biol. 2016, 1494, 15–27. [Google Scholar]
- Scheiermann, J.; Klinman, D.M. Clinical Evaluation of CpG Oligonucleotides as Adjuvants for Vaccines Targeting Infectious Diseases and Cancer. Vaccine 2014, 32, 6377–6389. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Moynihan, K.D.; Zheng, Y.; Szeto, G.L.; Li, A.V.; Huang, B.; van Egeren, D.S.; Park, C.; Irvine, D.J. Structure-Based Programming of Lymph-Node Targeting in Molecular Vaccines. Nature 2014, 507, 519–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Titta, A.; Ballester, M.; Julier, Z.; Nembrini, C.; Jeanbart, L.; van der Vlies, A.J.; Swartz, M.A.; Hubbell, J.A. Nanoparticle Conjugation of CpG Enhances Adjuvancy for Cellular Immunity and Memory Recall at Low Dose. Proc. Natl. Acad. Sci. USA 2013, 110, 19902–19907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourquin, C.; Anz, D.; Zwiorek, K.; Lanz, A.-L.; Fuchs, S.; Weigel, S.; Wurzenberger, C.; von der Borch, P.; Golic, M.; Moder, S.; et al. Targeting CpG Oligonucleotides to the Lymph Node by Nanoparticles Elicits Efficient Antitumoral Immunity. J. Immunol. 2008, 181, 2990–2998. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, M.B. Pharmaceutical Approval Update. Pharm. Ther. 2018, 43, 83–84. [Google Scholar]
- Halperin, S.A.; Dobson, S.; McNeil, S.; Langley, J.M.; Smith, B.; McCall-Sani, R.; Levitt, D.; van Nest, G.; Gennevois, D.; Eiden, J.J. Comparison of the Safety and Immunogenicity of Hepatitis B Virus Surface Antigen Co-Administered with an Immunostimulatory Phosphorothioate Oligonucleotide and a Licensed Hepatitis B Vaccine in Healthy Young Adults. Vaccine 2006, 24, 20–26. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Evaluating the Safety and Immunogenicity of HIV-1 BG505 SOSIP.664 Gp140 with TLR Agonist and/or Alum Adjuvants in Healthy, HIV-Uninfected Adults. Available online: https://clinicaltrials.gov/ct2/show/NCT04177355 (accessed on 29 November 2022).
- Hsieh, S.-M.; Chang, S.-C.; Cheng, H.-Y.; Shih, S.-R.; Lien, C.E. Durability and Immunogenicity of Neutralizing Antibodies Response Against Omicron Variants After Three Doses of Subunit SARS-CoV-2 Vaccine MVC-COV1901: An Extension to an Open-Label, Dose-Escalation Phase 1 Study. Infect. Dis. Ther. 2022, 11, 1493–1504. [Google Scholar] [CrossRef]
- Hsieh, S.-M.; Liu, W.-D.; Huang, Y.-S.; Lin, Y.-J.; Hsieh, E.-F.; Lian, W.-C.; Chen, C.; Janssen, R.; Shih, S.-R.; Huang, C.-G.; et al. Safety and Immunogenicity of a Recombinant Stabilized Prefusion SARS-CoV-2 Spike Protein Vaccine (MVC COV1901) Adjuvanted with CpG 1018 and Aluminum Hydroxide in Healthy Adults: A Phase 1, Dose-Escalation Study. EClinicalMedicine 2021, 38, 100989. [Google Scholar] [CrossRef]
- Ward, B.J.; Gobeil, P.; Séguin, A.; Atkins, J.; Boulay, I.; Charbonneau, P.-Y.; Couture, M.; D’Aoust, M.-A.; Dhaliwall, J.; Finkle, C.; et al. Phase 1 Randomized Trial of a Plant-Derived Virus-like Particle Vaccine for COVID-19. Nat. Med. 2021, 27, 1071–1078. [Google Scholar] [CrossRef]
- Czech, M.P. MicroRNAs as Therapeutic Targets. N. Engl. J. Med. 2006, 354, 1194–1195. [Google Scholar] [CrossRef]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of MicroRNAs In Vivo with ‘Antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Sharp, P.A. MicroRNA Sponges: Progress and Possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupaimoole, R.; Slack, F.J. MicroRNA Therapeutics: Towards a New Era for the Management of Cancer and Other Diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overby, S.J.; Cerro-Herreros, E.; González-Martínez, I.; Varela, M.A.; Seoane-Miraz, D.; Jad, Y.; Raz, R.; Møller, T.; Pérez-Alonso, M.; Wood, M.J.; et al. Proof of Concept of Peptide-Linked BlockmiR-Induced MBNL Functional Rescue in Myotonic Dystrophy Type 1 Mouse Model. Mol. Ther. Nucleic Acids 2022, 27, 1146–1155. [Google Scholar] [CrossRef]
- Young, J.A.; Ting, K.K.; Li, J.; Moller, T.; Dunn, L.; Lu, Y.; Lay, A.J.; Moses, J.; Prado-Lourenço, L.; Khachigian, L.M.; et al. Regulation of Vascular Leak and Recovery from Ischemic Injury by General and VE-Cadherin–Restricted MiRNA Antagonists of MiR-27. Blood 2013, 122, 2911–2919. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z. The Guideline of the Design and Validation of MiRNA Mimics. Methods Mol. Biol. 2011, 676, 211–223. [Google Scholar]
- Fu, Y.; Chen, J.; Huang, Z. Recent Progress in MicroRNA-Based Delivery Systems for the Treatment of Human Disease. ExRNA 2019, 1, 24. [Google Scholar] [CrossRef] [Green Version]
- Bader, A.G.; Brown, D.; Winkler, M. The Promise of MicroRNA Replacement Therapy. Cancer Res. 2010, 70, 7027–7030. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.-J. Therapeutic SiRNA: State of the Art. Signal Transduct. Target. Ther. 2020, 5, 101. [Google Scholar] [CrossRef]
- Vickers, T.A. Fully Modified 2’ MOE Oligonucleotides Redirect Polyadenylation. Nucleic Acids Res. 2001, 29, 1293–1299. [Google Scholar] [CrossRef] [Green Version]
- Weng, Y.; Xiao, H.; Zhang, J.; Liang, X.-J.; Huang, Y. RNAi Therapeutic and Its Innovative Biotechnological Evolution. Biotechnol. Adv. 2019, 37, 801–825. [Google Scholar] [CrossRef]
- Guo, D.; Ji, X.; Peng, F.; Zhong, Y.; Chu, B.; Su, Y.; He, Y. Photostable and Biocompatible Fluorescent Silicon Nanoparticles for Imaging-Guided Co-Delivery of SiRNA and Doxorubicin to Drug-Resistant Cancer Cells. Nanomicro. Lett. 2019, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, M.; Liu, Y.; Wang, Y.; Zhang, D.; Zou, Y.; Ruan, W.; Yin, J.; Tao, W.; Park, J.B.; Shi, B. ROS-Responsive Polymeric SiRNA Nanomedicine Stabilized by Triple Interactions for the Robust Glioblastoma Combinational RNAi Therapy. Adv. Mater. 2019, 31, 1903277. [Google Scholar] [CrossRef]
- Liu, J.; Guo, N.; Gao, C.; Liu, N.; Zheng, X.; Tan, Y.; Lei, J.; Hao, Y.; Chen, L.; Zhang, X. Effective Gene Silencing Mediated by Polypeptide Nanoparticles LAH4-L1-SiMDR1 in Multi-Drug Resistant Human Breast Cancer. J. Biomed. Nanotechnol. 2019, 15, 531–543. [Google Scholar] [CrossRef]
- Kawamata, T.; Tomari, Y. Making RISC. Trends Biochem. Sci. 2010, 35, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M.; Furset, G.; Cekaite, L. Suppression of Immunostimulatory SiRNA-Driven Innate Immune Activation by 2′-Modified RNAs. Biochem. Biophys. Res. Commun. 2007, 361, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, X.; Ma, Y.; Liang, Z.; Yang, Z.; Cao, H. Site-Specific Modification Using the 2′-Methoxyethyl Group Improves the Specificity and Activity of SiRNAs. Mol. Ther. Nucleic Acids 2017, 9, 242–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fluiter, K.; Mook, O.R.F.; Baas, F. The Therapeutic Potential of LNA-Modified SiRNAs: Reduction of Off-Target Effects by Chemical Modification of the SiRNA Sequence. Methods Mol. Biol. 2009, 487, 189–203. [Google Scholar]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis. N. Engl. J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef]
- Mehta, A.; Michler, T.; Merkel, O.M. SiRNA Therapeutics against Respiratory Viral Infections—What Have We Learned for Potential COVID-19 Therapies? Adv. Healthc. Mater. 2021, 10, 2001650. [Google Scholar] [CrossRef]
- Bramsen, J.B.; Laursen, M.B.; Damgaard, C.K.; Lena, S.W.; Ravindra Babu, B.; Wengel, J.; Kjems, J. Improved Silencing Properties Using Small Internally Segmented Interfering RNAs. Nucleic Acids Res. 2007, 35, 5886–5897. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Behlke, M.A.; Rose, S.D.; Chang, M.-S.; Choi, S.; Rossi, J.J. Synthetic DsRNA Dicer Substrates Enhance RNAi Potency and Efficacy. Nat. Biotechnol. 2005, 23, 222–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullard, A. FDA Approves Fifth RNAi Drug—Alnylam’s next-Gen HATTR Treatment. Nat. Rev. Drug Discov. 2022, 21, 548–549. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Inclisiran: First Approval. Drugs 2021, 81, 389–395. [Google Scholar] [CrossRef]
- Keam, S.J. Vutrisiran: First Approval. Drugs 2022, 82, 1419–1425. [Google Scholar] [CrossRef]
- Syed, Y.Y. Givosiran: A Review in Acute Hepatic Porphyria. Drugs 2021, 81, 841–848. [Google Scholar] [CrossRef]
- Scott, L.J.; Keam, S.J. Lumasiran: First Approval. Drugs 2021, 81, 277–282. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Corey, D.R. The 10th Oligonucleotide Therapy Approved: Golodirsen for Duchenne Muscular Dystrophy. Nucleic Acid Ther. 2020, 30, 67–70. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, C.; Wood, M.J.A. Antisense Oligonucleotides: The next Frontier for Treatment of Neurological Disorders. Nat. Rev. Neurol. 2018, 14, 9–21. [Google Scholar] [CrossRef]
- Klabenkova, K.; Fokina, A.; Stetsenko, D. Chemistry of Peptide-Oligonucleotide Conjugates: A Review. Molecules 2021, 26, 5420. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Takeda, S.; Lu, Q.-L.; Partridge, T.A.; Nakamura, A.; Hoffman, E.P. A Renaissance for Antisense Oligonucleotide Drugs in Neurology. Arch. Neurol. 2009, 66, 32–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, S.; Sinhari, A.; Jain, P.; Jadhav, H.R. A Perspective on Oligonucleotide Therapy: Approaches to Patient Customization. Front. Pharmacol. 2022, 13, 1006304. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F. Overcoming Cellular Barriers for RNA Therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Juliano, R.; Bauman, J.; Kang, H.; Ming, X. Biological Barriers to Therapy with Antisense and SiRNA Oligonucleotides. Mol. Pharm. 2009, 6, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Pandit, R.; Chen, L.; Götz, J. The Blood-Brain Barrier: Physiology and Strategies for Drug Delivery. Adv. Drug Deliv. Rev. 2020, 165–166, 1–14. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Weng, Y.; Huang, Q.; Li, C.; Yang, Y.; Wang, X.; Yu, J.; Huang, Y.; Liang, X.-J. Improved Nucleic Acid Therapy with Advanced Nanoscale Biotechnology. Mol. Ther. Nucleic Acids 2020, 19, 581–601. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, M.C.P.; Kont, A.; Aburto, M.R.; Cryan, J.F.; O’Driscoll, C.M. Advances in the Design of (Nano)Formulations for Delivery of Antisense Oligonucleotides and Small Interfering RNA: Focus on the Central Nervous System. Mol. Pharm. 2021, 18, 1491–1506. [Google Scholar] [CrossRef]
- Souleimanian, N.; Deleavey, G.F.; Soifer, H.; Wang, S.; Tiemann, K.; Damha, M.J.; Stein, C.A. Antisense 2′-Deoxy, 2′-Fluoroarabino Nucleic Acid (2′F-ANA) Oligonucleotides: In Vitro Gymnotic Silencers of Gene Expression Whose Potency Is Enhanced by Fatty Acids. Mol. Ther. Nucleic Acids 2012, 1, e43. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Hansen, J.B.; Lai, J.; Wu, S.; Voskresenskiy, A.; H⊘g, A.; Worm, J.; Hedtjärn, M.; Souleimanian, N.; Miller, P.; et al. Efficient Gene Silencing by Delivery of Locked Nucleic Acid Antisense Oligonucleotides, Unassisted by Transfection Reagents. Nucleic Acids Res. 2010, 38, e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soifer, H.S.; Koch, T.; Lai, J.; Hansen, B.; Hoeg, A.; Oerum, H.; Stein, C.A. Silencing of Gene Expression by Gymnotic Delivery of Antisense Oligonucleotides. Methods Mol. Biol. 2012, 815, 333–346. [Google Scholar]
- Deprey, K.; Batistatou, N.; Kritzer, J.A. A Critical Analysis of Methods Used to Investigate the Cellular Uptake and Subcellular Localization of RNA Therapeutics. Nucleic Acids Res. 2020, 48, 7623–7639. [Google Scholar] [CrossRef]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X. Cellular Uptake and Trafficking of Antisense Oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ming, X.; Nakagawa, O. Cellular Uptake and Intracellular Trafficking of Antisense and SiRNA Oligonucleotides. Bioconjug. Chem. 2012, 23, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyenvalle, A.; Leumann, C.; Garcia, L. Therapeutic Potential of Tricyclo-DNA Antisense Oligonucleotides. J. Neuromuscul. Dis. 2016, 3, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Wang, X.-Q.; Holmes, T.R.; Bonkowski, M.; Roth, E.W.; Ponedal, A.; Mirkin, C.; Paller, A.S. Epidermal SR-A Complexes Are Lipid Raft Based and Promote Nucleic Acid Nanoparticle Uptake. J. Investig. Dermatol. 2021, 141, 1428–1437.e8. [Google Scholar] [CrossRef] [PubMed]
- DeWitte-Orr, S.J.; Collins, S.E.; Bauer, C.M.T.; Bowdish, D.M.; Mossman, K.L. An Accessory to the ‘Trinity’: SR-As Are Essential Pathogen Sensors of Extracellular DsRNA, Mediating Entry and Leading to Subsequent Type I IFN Responses. PLoS Pathog. 2010, 6, e1000829. [Google Scholar] [CrossRef] [Green Version]
- Miyatake, S.; Mizobe, Y.; Tsoumpra, M.K.; Lim, K.R.Q.; Hara, Y.; Shabanpoor, F.; Yokota, T.; Takeda, S.; Aoki, Y. Scavenger Receptor Class A1 Mediates Uptake of Morpholino Antisense Oligonucleotide into Dystrophic Skeletal Muscle. Mol. Ther. Nucleic Acids 2019, 14, 520–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.M.; Donner, A.J.; Blank, E.E.; Egger, A.W.; Kellar, B.M.; Østergaard, M.E.; Seth, P.P.; Harris, E.N. Stabilin-1 and Stabilin-2 Are Specific Receptors for the Cellular Internalization of Phosphorothioate-Modified Antisense Oligonucleotides (ASOs) in the Liver. Nucleic Acids Res. 2016, 44, 2782–2794. [Google Scholar] [CrossRef]
- Miller, C.M.; Wan, W.B.; Seth, P.P.; Harris, E.N. Endosomal Escape of Antisense Oligonucleotides Internalized by Stabilin Receptors Is Regulated by Rab5C and EEA1 During Endosomal Maturation. Nucleic Acid Ther. 2018, 28, 86–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Allen, N.; Vickers, T.A.; Revenko, A.S.; Sun, H.; Liang, X.; Crooke, S.T. Cellular Uptake Mediated by Epidermal Growth Factor Receptor Facilitates the Intracellular Activity of Phosphorothioate-Modified Antisense Oligonucleotides. Nucleic Acids Res. 2018, 46, 3579–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Contu, V.R.; Kabuta, C.; Hase, K.; Fujiwara, Y.; Wada, K.; Kabuta, T. SIDT2 Mediates Gymnosis, the Uptake of Naked Single-Stranded Oligonucleotides into Living Cells. RNA Biol. 2017, 14, 1534–1543. [Google Scholar] [CrossRef]
- Pearse, B.M. Clathrin: A Unique Protein Associated with Intracellular Transfer of Membrane by Coated Vesicles. Proc. Natl. Acad. Sci. USA 1976, 73, 1255–1259. [Google Scholar] [CrossRef] [Green Version]
- Pearse, B.M.F. Coated Vesicles from Pig Brain: Purification and Biochemical Characterization. J. Mol. Biol. 1975, 97, 93–98. [Google Scholar] [CrossRef]
- Kirkham, M.; Parton, R.G. Clathrin-Independent Endocytosis: New Insights into Caveolae and Non-Caveolar Lipid Raft Carriers. Biochim. Biophys. Acta 2005, 1745, 273–286. [Google Scholar] [CrossRef] [Green Version]
- Keren, K. Membrane Tension Leads the Way. Proc. Natl. Acad. Sci. USA 2011, 108, 14379–14380. [Google Scholar] [CrossRef] [Green Version]
- Bonifacino, J.S.; Neefjes, J. Moving and Positioning the Endolysosomal System. Curr. Opin. Cell Biol. 2017, 47, 1–8. [Google Scholar] [CrossRef]
- Hu, Y.-B.; Dammer, E.B.; Ren, R.-J.; Wang, G. The Endosomal-Lysosomal System: From Acidification and Cargo Sorting to Neurodegeneration. Transl. Neurodegener. 2015, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowdy, S.F.; Setten, R.L.; Cui, X.-S.; Jadhav, S.G. Delivery of RNA Therapeutics: The Great Endosomal Escape! Nucleic Acid Ther. 2022, 32, 361–368. [Google Scholar] [CrossRef]
- Vermeulen, L.M.P.; de Smedt, S.C.; Remaut, K.; Braeckmans, K. The Proton Sponge Hypothesis: Fable or Fact? Eur. J. Pharm. Biopharm. 2018, 129, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.-Y.; Chang, C.C.Y.; Ohgami, N.; Yamauchi, Y. Cholesterol Sensing, Trafficking, and Esterification. Annu. Rev. Cell Dev. Biol. 2006, 22, 129–157. [Google Scholar] [CrossRef]
- Wang, S.; Allen, N.; Liang, X.; Crooke, S.T. Membrane Destabilization Induced by Lipid Species Increases Activity of Phosphorothioate-Antisense Oligonucleotides. Mol. Ther. Nucleic Acids 2018, 13, 686–698. [Google Scholar] [CrossRef] [Green Version]
- Hullin-Matsuda, F.; Taguchi, T.; Greimel, P.; Kobayashi, T. Lipid Compartmentalization in the Endosome System. Semin. Cell Dev. Biol. 2014, 31, 48–56. [Google Scholar] [CrossRef]
- Wang, S.; Sun, H.; Tanowitz, M.; Liang, X.; Crooke, S.T. Intra-Endosomal Trafficking Mediated by Lysobisphosphatidic Acid Contributes to Intracellular Release of Phosphorothioate-Modified Antisense Oligonucleotides. Nucleic Acids Res. 2017, 45, 5309–5322. [Google Scholar] [CrossRef] [Green Version]
- Chevallier, J.; Chamoun, Z.; Jiang, G.; Prestwich, G.; Sakai, N.; Matile, S.; Parton, R.G.; Gruenberg, J. Lysobisphosphatidic Acid Controls Endosomal Cholesterol Levels. J. Biol. Chem. 2008, 283, 27871–27880. [Google Scholar] [CrossRef] [Green Version]
- Falguières, T.; Luyet, P.-P.; Gruenberg, J. Molecular Assemblies and Membrane Domains in Multivesicular Endosome Dynamics. Exp. Cell Res. 2009, 315, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Bizot, F.; Fayssoil, A.; Gastaldi, C.; Irawan, T.; Phongsavanh, X.; Mansart, A.; Tensorer, T.; Brisebard, E.; Garcia, L.; Juliano, R.L.; et al. Oligonucleotide Enhancing Compound Increases Tricyclo-DNA Mediated Exon-Skipping Efficacy in the Mdx Mouse Model. Cells 2023, 12, 702. [Google Scholar] [CrossRef]
- Dang, Y.; van Heusden, C.; Nickerson, V.; Chung, F.; Wang, Y.; Quinney, N.L.; Gentzsch, M.; Randell, S.H.; Moulton, H.M.; Kole, R.; et al. Enhanced delivery of peptide-morpholino oligonucleotides with a small molecule to correct splicing defects in the lung. Nucleic Acids Res. 2021, 49, 6100–6113. [Google Scholar] [CrossRef]
- Bissig, C.; Gruenberg, J. ALIX and the Multivesicular Endosome: ALIX in Wonderland. Trends Cell Biol. 2014, 24, 19–25. [Google Scholar] [CrossRef]
- Pei, D.; Buyanova, M. Overcoming Endosomal Entrapment in Drug Delivery. Bioconjug. Chem. 2019, 30, 273–283. [Google Scholar] [CrossRef]
- Wan, W.B.; Seth, P.P. The Medicinal Chemistry of Therapeutic Oligonucleotides. J. Med. Chem. 2016, 59, 9645–9667. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.H.S. RNA Interference Using Boranophosphate SiRNAs: Structure-Activity Relationships. Nucleic Acids Res. 2004, 32, 5991–6000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Chen, S.; Xu, T.; Taghizadeh, K.; Wishnok, J.S.; Zhou, X.; You, D.; Deng, Z.; Dedon, P.C. Phosphorothioation of DNA in Bacteria by Dnd Genes. Nat. Chem. Biol. 2007, 3, 709–710. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M.; Yi, A.-K.; Matson, S.; Waldschmidt, T.J.; Bishop, G.A.; Teasdale, R.; Koretzky, G.A.; Klinman, D.M. CpG Motifs in Bacterial DNA Trigger Direct B-Cell Activation. Nature 1995, 374, 546–549. [Google Scholar] [CrossRef]
- Krieg, A.M. CpG Still Rocks! Update on an Accidental Drug. Nucleic Acid Ther. 2012, 22, 77–89. [Google Scholar] [CrossRef]
- Braasch, D.A.; Jensen, S.; Liu, Y.; Kaur, K.; Arar, K.; White, M.A.; Corey, D.R. RNA Interference in Mammalian Cells by Chemically-Modified RNA. Biochemistry 2003, 42, 7967–7975. [Google Scholar] [CrossRef]
- Roberts, T.C.; Ezzat, K.; El Andaloussi, S.; Weinberg, M.S. Synthetic SiRNA Delivery: Progress and Prospects. Methods Mol. Biol. 2016, 1364, 291–310. [Google Scholar] [CrossRef]
- Ezzat, K.; Aoki, Y.; Koo, T.; McClorey, G.; Benner, L.; Coenen-Stass, A.; O’Donovan, L.; Lehto, T.; Garcia-Guerra, A.; Nordin, J.; et al. Self-Assembly into Nanoparticles Is Essential for Receptor Mediated Uptake of Therapeutic Antisense Oligonucleotides. Nano Lett. 2015, 15, 4364–4373. [Google Scholar] [CrossRef] [Green Version]
- Gaus, H.J.; Gupta, R.; Chappell, A.E.; Østergaard, M.E.; Swayze, E.E.; Seth, P.P. Characterization of the Interactions of Chemically-Modified Therapeutic Nucleic Acids with Plasma Proteins Using a Fluorescence Polarization Assay. Nucleic Acids Res. 2019, 47, 1110–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shemesh, C.S.; Yu, R.Z.; Gaus, H.J.; Seth, P.P.; Swayze, E.E.; Bennett, F.C.; Geary, R.S.; Henry, S.P.; Wang, Y. Pharmacokinetic and Pharmacodynamic Investigations of ION-353382, a Model Antisense Oligonucleotide: Using Alpha-2-Macroglobulin and Murinoglobulin Double-Knockout Mice. Nucleic Acid Ther. 2016, 26, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Weidner, D.A.; Valdez, B.C.; Henning, D.; Greenberg, S.; Busch, H. Phosphorothioate Oligonucleotides Bind in a Non Sequence-Specific Manner to the Nucleolar Protein C23/Nucleolin. FEBS Lett. 1995, 366, 146–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Shen, W.; Sun, H.; Prakash, T.P.; Crooke, S.T. TCP1 Complex Proteins Interact with Phosphorothioate Oligonucleotides and Can Co-Localize in Oligonucleotide-Induced Nuclear Bodies in Mammalian Cells. Nucleic Acids Res. 2014, 42, 7819–7832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monia, B.P.; Johnston, J.F.; Sasmor, H.; Cummins, L.L. Nuclease Resistance and Antisense Activity of Modified Oligonucleotides Targeted to Ha-. J. Biol. Chem. 1996, 271, 14533–14540. [Google Scholar] [CrossRef] [Green Version]
- Moulton, J.D. Using Morpholinos to Control Gene Expression. Curr. Protoc. Nucleic Acid Chem. 2006, 27, 4301–43024. [Google Scholar] [CrossRef] [PubMed]
- SUMMERTON, J.; WELLER, D. Morpholino Antisense Oligomers: Design, Preparation, and Properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukashyaka, M.C.; Wu, C.-L.; Ha, K.; Zhang, J.; Wood, J.; Foley, S.; Mastis, B.; Jungels, N.; Sun, H.; Shadid, M.; et al. Pharmacokinetic/Pharmacodynamic Modeling of a Cell-Penetrating Peptide Phosphorodiamidate Morpholino Oligomer in Mdx Mice. Pharm. Res. 2021, 38, 1731–1745. [Google Scholar] [CrossRef]
- Liu, Y.; Dodart, J.-C.; Tran, H.; Berkovitch, S.; Braun, M.; Byrne, M.; Durbin, A.F.; Hu, X.S.; Iwamoto, N.; Jang, H.G.; et al. Variant-Selective Stereopure Oligonucleotides Protect against Pathologies Associated with C9orf72-Repeat Expansion in Preclinical Models. Nat. Commun. 2021, 12, 847. [Google Scholar] [CrossRef]
- Byrne, M.; Vathipadiekal, V.; Apponi, L.; Iwamoto, N.; Kandasamy, P.; Longo, K.; Liu, F.; Looby, R.; Norwood, L.; Shah, A.; et al. Stereochemistry Enhances Potency, Efficacy, and Durability of Malat1 Antisense Oligonucleotides In Vitro and In Vivo in Multiple Species. Transl. Vis. Sci. Technol. 2021, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, N.; Butler, D.C.D.; Svrzikapa, N.; Mohapatra, S.; Zlatev, I.; Sah, D.W.Y.; Meena; Standley, S.M.; Lu, G.; Apponi, L.H.; et al. Control of Phosphorothioate Stereochemistry Substantially Increases the Efficacy of Antisense Oligonucleotides. Nat. Biotechnol. 2017, 35, 845–851. [Google Scholar] [CrossRef]
- Kandasamy, P.; McClorey, G.; Shimizu, M.; Kothari, N.; Alam, R.; Iwamoto, N.; Kumarasamy, J.; Bommineni, G.R.; Bezigian, A.; Chivatakarn, O.; et al. Control of Backbone Chemistry and Chirality Boost Oligonucleotide Splice Switching Activity. Nucleic Acids Res. 2022, 50, 5443–5466. [Google Scholar] [CrossRef] [PubMed]
- Eulberg, D.; Klussmann, S. Spiegelmers: Biostable Aptamers. ChemBioChem 2003, 4, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Oukacine, F.; Ravelet, C.; Peyrin, E. Enantiomeric Sensing and Separation by Nucleic Acids. TrAC Trends Anal. Chem. 2020, 122, 115733. [Google Scholar] [CrossRef]
- Deleavey, G.F.; Damha, M.J. Designing Chemically Modified Oligonucleotides for Targeted Gene Silencing. Chem. Biol. 2012, 19, 937–954. [Google Scholar] [CrossRef] [Green Version]
- Herdewijn, P. Heterocyclic Modifications of Oligonucleotides and Antisense Technology. Antisense Nucleic Acid Drug Dev. 2000, 10, 297–310. [Google Scholar] [CrossRef]
- Terrazas, M.; Kool, E.T. RNA Major Groove Modifications Improve SiRNA Stability and Biological Activity. Nucleic Acids Res. 2009, 37, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Pendergraff, H.; Narayanannair, K.J.; Lackey, J.G.; Kuchimanchi, S.; Rajeev, K.G.; Manoharan, M.; Hu, J.; Corey, D.R. RNA Duplexes with Abasic Substitutions Are Potent and Allele-Selective Inhibitors of Huntingtin and Ataxin-3 Expression. Nucleic Acids Res. 2013, 41, 8788–8801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-S.; Seok, H.; Lee, D.H.; Ham, J.; Lee, W.; Youm, E.M.; Yoo, J.S.; Lee, Y.-S.; Jang, E.-S.; Chi, S.W. Abasic Pivot Substitution Harnesses Target Specificity of RNA Interference. Nat. Commun. 2015, 6, 10154. [Google Scholar] [CrossRef] [Green Version]
- Prakash, T.P. An Overview of Sugar-Modified Oligonucleotides for Antisense Therapeutics. Chem. Biodivers. 2011, 8, 1616–1641. [Google Scholar] [CrossRef] [PubMed]
- Monia, B.P.; Lesnik, E.A.; Gonzalez, C.; Lima, W.F.; McGee, D.; Guinosso, C.J.; Kawasaki, A.M.; Cook, P.D.; Freier, S.M. Evaluation of 2′-Modified Oligonucleotides Containing 2′-Deoxy Gaps as Antisense Inhibitors of Gene Expression. J. Biol. Chem. 1993, 268, 14514–14522. [Google Scholar] [CrossRef] [PubMed]
- Southwell, A.L.; Skotte, N.H.; Bennett, C.F.; Hayden, M.R. Antisense Oligonucleotide Therapeutics for Inherited Neurodegenerative Diseases. Trends Mol. Med. 2012, 18, 634–643. [Google Scholar] [CrossRef]
- Manoharan, M. 2′-Carbohydrate Modifications in Antisense Oligonucleotide Therapy: Importance of Conformation, Configuration and Conjugation. Biochim. Et Biophys. Acta 1999, 1489, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Allerson, C.R.; Sioufi, N.; Jarres, R.; Prakash, T.P.; Naik, N.; Berdeja, A.; Wanders, L.; Griffey, R.H.; Swayze, E.E.; Bhat, B. Fully 2′-Modified Oligonucleotide Duplexes with Improved in Vitro Potency and Stability Compared to Unmodified Small Interfering RNA. J. Med. Chem. 2005, 48, 901–904. [Google Scholar] [CrossRef]
- Hassler, M.R.; Turanov, A.A.; Alterman, J.F.; Haraszti, R.A.; Coles, A.H.; Osborn, M.F.; Echeverria, D.; Nikan, M.; Salomon, W.E.; Roux, L.; et al. Comparison of Partially and Fully Chemically-Modified SiRNA in Conjugate-Mediated Delivery In Vivo. Nucleic Acids Res. 2018, 46, 2185–2196. [Google Scholar] [CrossRef] [Green Version]
- Jackson, A.L.; Burchard, J.; Leake, D.; Reynolds, A.; Schelter, J.; Guo, J.; Johnson, J.M.; Lim, L.; Karpilow, J.; Nichols, K.; et al. Position-Specific Chemical Modification of SiRNAs Reduces “off-Target” Transcript Silencing. RNA 2006, 12, 1197–1205. [Google Scholar] [CrossRef] [Green Version]
- Springer, A.D.; Dowdy, S.F. GalNAc-SiRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Garber, K. Alnylam Terminates Revusiran Program, Stock Plunges. Nat. Biotechnol. 2016, 34, 1213–1214. [Google Scholar] [CrossRef]
- Nair, J.K.; Attarwala, H.; Sehgal, A.; Wang, Q.; Aluri, K.; Zhang, X.; Gao, M.; Liu, J.; Indrakanti, R.; Schofield, S.; et al. Impact of Enhanced Metabolic Stability on Pharmacokinetics and Pharmacodynamics of GalNAc–SiRNA Conjugates. Nucleic Acids Res. 2017, 45, 10969–10977. [Google Scholar] [CrossRef] [Green Version]
- Obika, S.; Nanbu, D.; Hari, Y.; Morio, K.; In, Y.; Ishida, T.; Imanishi, T. Synthesis of 2′-O,4′-C-Methyleneuridine and -Cytidine. Novel Bicyclic Nucleosides Having a Fixed C3, -Endo Sugar Puckering. Tetrahedron Lett. 1997, 38, 8735–8738. [Google Scholar] [CrossRef]
- Jepsen, J.S.; Sørensen, M.D.; Wengel, J. Locked Nucleic Acid: A Potent Nucleic Acid Analog in Therapeutics and Biotechnology. Oligonucleotides 2004, 14, 130–146. [Google Scholar] [CrossRef]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-Generation Antisense Oligonucleotide Inhibitor of STAT3 with Early Evidence of Clinical Activity in Lymphoma and Lung Cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, K.; Hasegawa, C.; Kaneko, M.; Tsutsumi, S.; Sone, J.; Ishikawa, T.; Imanishi, T.; Koizumi, M. 2′-O,4′-C-Ethylene-Bridged Nucleic Acids (ENA): Highly Nuclease-Resistant and Thermodynamically Stable Oligonucleotides for Antisense Drug. Bioorg. Med. Chem. Lett. 2002, 12, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Vester, B.; Wengel, J. LNA (Locked Nucleic Acid): High-Affinity Targeting of Complementary RNA and DNA. Biochemistry 2004, 43, 13233–13241. [Google Scholar] [CrossRef] [PubMed]
- Veedu, R.N.; Wengel, J. Locked Nucleic Acid as a Novel Class of Therapeutic Agents. RNA Biol. 2009, 6, 321–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshkin, A.A.; Singh, S.K.; Nielsen, P.; Rajwanshi, V.K.; Kumar, R.; Meldgaard, M.; Olsen, C.E.; Wengel, J. LNA (Locked Nucleic Acids): Synthesis of the Adenine, Cytosine, Guanine, 5-Methylcytosine, Thymine and Uracil Bicyclonucleoside Monomers, Oligomerisation, and Unprecedented Nucleic Acid Recognition. Tetrahedron 1998, 54, 3607–3630. [Google Scholar] [CrossRef]
- Hung, G.; Xiao, X.; Peralta, R.; Bhattacharjee, G.; Murray, S.; Norris, D.; Guo, S.; Monia, B.P. Characterization of Target MRNA Reduction Through In Situ RNA Hybridization in Multiple Organ Systems Following Systemic Antisense Treatment in Animals. Nucleic Acid Ther. 2013, 23, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Obad, S.; dos Santos, C.O.; Petri, A.; Heidenblad, M.; Broom, O.; Ruse, C.; Fu, C.; Lindow, M.; Stenvang, J.; Straarup, E.M.; et al. Silencing of MicroRNA Families by Seed-Targeting Tiny LNAs. Nat. Genet. 2011, 43, 371–378. [Google Scholar] [CrossRef]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; El Andaloussi, S.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T.; et al. Functional Correction in Mouse Models of Muscular Dystrophy Using Exon-Skipping Tricyclo-DNA Oligomers. Nat. Med. 2015, 21, 270–275. [Google Scholar] [CrossRef]
- Imbert, M.; Blandel, F.; Leumann, C.; Garcia, L.; Goyenvalle, A. Lowering Mutant Huntingtin Using Tricyclo-DNA Antisense Oligonucleotides As a Therapeutic Approach for Huntington’s Disease. Nucleic Acid Ther. 2019, 29, 256–265. [Google Scholar] [CrossRef]
- Benizri, S.; Gissot, A.; Martin, A.; Vialet, B.; Grinstaff, M.W.; Barthélémy, P. Bioconjugated Oligonucleotides: Recent Developments and Therapeutic Applications. Bioconjug. Chem. 2019, 30, 366–383. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.; Saleh, A.F.; Arzumanov, A.A.; Hammond, S.M.; Godfrey, C.; Coursindel, T.; Gait, M.J.; Wood, M.J. Pip6-PMO, A New Generation of Peptide-Oligonucleotide Conjugates with Improved Cardiac Exon Skipping Activity for DMD Treatment. Mol. Ther. Nucleic Acids 2012, 1, e38. [Google Scholar] [CrossRef] [PubMed]
- Ämmälä, C.; Drury, W.J.; Knerr, L.; Ahlstedt, I.; Stillemark-Billton, P.; Wennberg-Huldt, C.; Andersson, E.-M.; Valeur, E.; Jansson-Löfmark, R.; Janzén, D.; et al. Targeted Delivery of Antisense Oligonucleotides to Pancreatic β-Cells. Sci. Adv. 2018, 4, eaat3386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, W.; Samarsky, D.; Liu, L.; Xu, Q.; Zhang, W.; Zhu, G.; Wu, P.; Zuo, X.; Deng, H.; et al. Tumor-Targeted in Vivo Gene Silencing via Systemic Delivery of CRGD-Conjugated SiRNA. Nucleic Acids Res. 2014, 42, 11805–11817. [Google Scholar] [CrossRef] [Green Version]
- McNamara, J.O.; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell Type–Specific Delivery of SiRNAs with Aptamer-SiRNA Chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.K.; Willoughby, J.L.S.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N -Acetylgalactosamine-Conjugated SiRNA Localizes in Hepatocytes and Elicits Robust RNAi-Mediated Gene Silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, S.; Keiser, K.; Nair, J.K.; Charisse, K.; Manoharan, R.M.; Kretschmer, P.; Peng, C.G.; Kel’in, A.V.; Kandasamy, P.; Willoughby, J.L.S.; et al. SiRNA Conjugates Carrying Sequentially Assembled Trivalent N-Acetylgalactosamine Linked Through Nucleosides Elicit Robust Gene Silencing In Vivo in Hepatocytes. ACS Chem. Biol. 2015, 10, 1181–1187. [Google Scholar] [CrossRef]
- Brown, K.M.; Nair, J.K.; Janas, M.M.; Anglero-Rodriguez, Y.I.; Dang, L.T.H.; Peng, H.; Theile, C.S.; Castellanos-Rizaldos, E.; Brown, C.; Foster, D.; et al. Expanding RNAi Therapeutics to Extrahepatic Tissues with Lipophilic Conjugates. Nat. Biotechnol. 2022, 40, 1500–1508. [Google Scholar] [CrossRef]
- Cuellar, T.L.; Barnes, D.; Nelson, C.; Tanguay, J.; Yu, S.-F.; Wen, X.; Scales, S.J.; Gesch, J.; Davis, D.; van Brabant Smith, A.; et al. Systematic Evaluation of Antibody-Mediated SiRNA Delivery Using an Industrial Platform of THIOMAB–SiRNA Conjugates. Nucleic Acids Res. 2015, 43, 1189–1203. [Google Scholar] [CrossRef]
- Brown, C.R.; Gupta, S.; Qin, J.; Racie, T.; He, G.; Lentini, S.; Malone, R.; Yu, M.; Matsuda, S.; Shulga-Morskaya, S.; et al. Investigating the Pharmacodynamic Durability of GalNAc–SiRNA Conjugates. Nucleic Acids Res. 2020, 48, 11827–11844. [Google Scholar] [CrossRef] [PubMed]
- McClorey, G.; Banerjee, S. Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines 2018, 6, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehto, T.; Ezzat, K.; Wood, M.J.A.; El Andaloussi, S. Peptides for Nucleic Acid Delivery. Adv. Drug Deliv. Rev. 2016, 106, 172–182. [Google Scholar] [CrossRef]
- Averick, S.E.; Dey, S.K.; Grahacharya, D.; Matyjaszewski, K.; Das, S.R. Solid-Phase Incorporation of an ATRP Initiator for Polymer-DNA Biohybrids. Angew. Chem. Int. Ed. 2014, 53, 2739–2744. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.E.; Winssinger, N. Synthesis of Protein-Oligonucleotide Conjugates. Biomolecules 2022, 12, 1523. [Google Scholar] [CrossRef] [PubMed]
- Farzan, V.M.; Ulashchik, E.A.; Martynenko-Makaev, Y.V.; Kvach, M.V.; Aparin, I.O.; Brylev, V.A.; Prikazchikova, T.A.; Maklakova, S.Y.; Majouga, A.G.; Ustinov, A.V.; et al. Automated Solid-Phase Click Synthesis of Oligonucleotide Conjugates: From Small Molecules to Diverse N-Acetylgalactosamine Clusters. Bioconjug. Chem. 2017, 28, 2599–2607. [Google Scholar] [CrossRef]
- Godeau, G.; Staedel, C.; Barthélémy, P. Lipid-Conjugated Oligonucleotides via “Click Chemistry” Efficiently Inhibit Hepatitis C Virus Translation. J. Med. Chem. 2008, 51, 4374–4376. [Google Scholar] [CrossRef]
- Nishina, K.; Unno, T.; Uno, Y.; Kubodera, T.; Kanouchi, T.; Mizusawa, H.; Yokota, T. Efficient In Vivo Delivery of SiRNA to the Liver by Conjugation of α-Tocopherol. Mol. Ther. 2008, 16, 734–740. [Google Scholar] [CrossRef]
- Pooga, M.; Langel, Ü. Classes of Cell-Penetrating Peptides. Methods Mol. Biol. 2015, 1324, 3–28. [Google Scholar]
- Zorko, M.; Langel, Ü. Cell-Penetrating Peptides. Methods Mol. Biol. 2022, 2383, 3–32. [Google Scholar] [CrossRef]
- Derakhshankhah, H.; Jafari, S. Cell Penetrating Peptides: A Concise Review with Emphasis on Biomedical Applications. Biomed. Pharmacother. 2018, 108, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Hirose, H.; Takeuchi, T.; Osakada, H.; Pujals, S.; Katayama, S.; Nakase, I.; Kobayashi, S.; Haraguchi, T.; Futaki, S. Transient Focal Membrane Deformation Induced by Arginine-Rich Peptides Leads to Their Direct Penetration into Cells. Mol. Ther. 2012, 20, 984–993. [Google Scholar] [CrossRef] [Green Version]
- Palm-Apergi, C.; Lönn, P.; Dowdy, S.F. Do Cell-Penetrating Peptides Actually “Penetrate” Cellular Membranes? Mol. Ther. 2012, 20, 695–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of Antimicrobial Dermaseptin and Its Fluorescently Labeled Analogs with Phospholipid Membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef]
- Thennarasu, S.; Tan, A.; Penumatchu, R.; Shelburne, C.E.; Heyl, D.L.; Ramamoorthy, A. Antimicrobial and Membrane Disrupting Activities of a Peptide Derived from the Human Cathelicidin Antimicrobial Peptide LL37. Biophys. J. 2010, 98, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herce, H.D.; Garcia, A.E.; Cardoso, M.C. Fundamental Molecular Mechanism for the Cellular Uptake of Guanidinium-Rich Molecules. J. Am. Chem. Soc. 2014, 136, 17459–17467. [Google Scholar] [CrossRef]
- Geller, B.L.; Li, L.; Martinez, F.; Sully, E.; Sturge, C.R.; Daly, S.M.; Pybus, C.; Greenberg, D.E. Morpholino Oligomers Tested in Vitro, in Biofilm and in Vivo against Multidrug-Resistant Klebsiella Pneumoniae. J. Antimicrob. Chemother. 2018, 73, 1611–1619. [Google Scholar] [CrossRef] [Green Version]
- Betts, C.A.; McClorey, G.; Healicon, R.; Hammond, S.M.; Manzano, R.; Muses, S.; Ball, V.; Godfrey, C.; Merritt, T.M.; Westering, T.; et al. Cmah-Dystrophin Deficient Mdx Mice Display an Accelerated Cardiac Phenotype That Is Improved Following Peptide-PMO Exon Skipping Treatment. Hum. Mol. Genet. 2018, 28, 396–406. [Google Scholar] [CrossRef] [Green Version]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic Peptide-Mediated Oligonucleotide Therapy Improves Long-Term Survival in Spinal Muscular Atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 10962–10967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Moulton, H.M.; Iversen, P.L.; Jiang, J.; Li, J.; Li, J.; Spurney, C.F.; Sali, A.; Guerron, A.D.; Nagaraju, K.; et al. Effective Rescue of Dystrophin Improves Cardiac Function in Dystrophin-Deficient Mice by a Modified Morpholino Oligomer. Proc. Natl. Acad. Sci. USA 2008, 105, 14814–14819. [Google Scholar] [CrossRef] [Green Version]
- McClorey, G.; Moulton, H.M.; Iversen, P.L.; Fletcher, S.; Wilton, S.D. Antisense Oligonucleotide-Induced Exon Skipping Restores Dystrophin Expression in Vitro in a Canine Model of DMD. Gene Ther. 2006, 13, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Aslesh, T.; Erkut, E.; Ren, J.; Lim, K.R.Q.; Woo, S.; Hatlevig, S.; Moulton, H.M.; Gosgnach, S.; Greer, J.; Maruyama, R.; et al. DG9-Conjugated Morpholino Rescues Phenotype in SMA Mice by Reaching the CNS via a Subcutaneous Administration. JCI Insight 2023, 8, e160516. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Woo, S.; Melo, D.; Huang, Y.; Dzierlega, K.; Shah, M.N.A.; Aslesh, T.; Roshmi, R.R.; Echigoya, Y.; Maruyama, R.; et al. Development of DG9 Peptide-Conjugated Single- and Multi-Exon Skipping Therapies for the Treatment of Duchenne Muscular Dystrophy. Proc. Natl. Acad. Sci. USA 2022, 119, e2112546119. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iverson, P.; Wood, M.J.A. Cell-Penetrating Peptide-Conjugated Antisense Oligonucleotides Restore Systemic Muscle and Cardiac Dystrophin Expression and Function. Hum. Mol. Genet. 2008, 17, 3909–3918. [Google Scholar] [CrossRef] [Green Version]
- Gait, M.J.; Arzumanov, A.A.; McClorey, G.; Godfrey, C.; Betts, C.; Hammond, S.; Wood, M.J.A. Cell-Penetrating Peptide Conjugates of Steric Blocking Oligonucleotides as Therapeutics for Neuromuscular Diseases from a Historical Perspective to Current Prospects of Treatment. Nucleic Acid Ther. 2019, 29, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westering, T.L.E.; Lomonosova, Y.; Coenen-Stass, A.M.L.; Betts, C.A.; Bhomra, A.; Hulsker, M.; Clark, L.E.; McClorey, G.; Aartsma-Rus, A.; Putten, M.; et al. Uniform Sarcolemmal Dystrophin Expression Is Required to Prevent Extracellular MicroRNA Release and Improve Dystrophic Pathology. J. Cachexia Sarcopenia Muscle 2020, 11, 578–593. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Saleh, A.F.; Betts, C.; Camelliti, P.; Seow, Y.; Ashraf, S.; Arzumanov, A.; Hammond, S.; Merritt, T.; Gait, M.J.; et al. Pip5 Transduction Peptides Direct High Efficiency Oligonucleotide-Mediated Dystrophin Exon Skipping in Heart and Phenotypic Correction in Mdx Mice. Mol. Ther. 2011, 19, 1295–1303. [Google Scholar] [CrossRef]
- Li, X.; Feng, K.; Li, L.; Yang, L.; Pan, X.; Yazd, H.S.; Cui, C.; Li, J.; Moroz, L.; Sun, Y.; et al. Lipid–Oligonucleotide Conjugates for Bioapplications. Natl. Sci. Rev. 2020, 7, 1933–1953. [Google Scholar] [CrossRef]
- Wang, S.; Allen, N.; Prakash, T.P.; Liang, X.; Crooke, S.T. Lipid Conjugates Enhance Endosomal Release of Antisense Oligonucleotides Into Cells. Nucleic Acid Ther. 2019, 29, 245–255. [Google Scholar] [CrossRef]
- Osborn, M.F.; Coles, A.H.; Biscans, A.; Haraszti, R.A.; Roux, L.; Davis, S.; Ly, S.; Echeverria, D.; Hassler, M.R.; Godinho, B.M.D.C.; et al. Hydrophobicity Drives the Systemic Distribution of Lipid-Conjugated SiRNAs via Lipid Transport Pathways. Nucleic Acids Res. 2019, 47, 1070–1081. [Google Scholar] [CrossRef] [Green Version]
- Chernikov, I.V.; Gladkikh, D.V.; Meschaninova, M.I.; Ven’yaminova, A.G.; Zenkova, M.A.; Vlassov, V.V.; Chernolovskaya, E.L. Cholesterol-Containing Nuclease-Resistant SiRNA Accumulates in Tumors in a Carrier-Free Mode and Silences MDR1 Gene. Mol. Ther. Nucleic Acids 2017, 6, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic Silencing of an Endogenous Gene by Systemic Administration of Modified SiRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Weber, H.; DiMuzio, J.; Matter, A.; Dogdas, B.; Shah, T.; Thankappan, A.; Disa, J.; Jadhav, V.; Lubbers, L.; et al. Silencing Myostatin Using Cholesterol-Conjugated SiRNAs Induces Muscle Growth. Mol. Ther. Nucleic Acids 2016, 5, e342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, T.P.; Mullick, A.E.; Lee, R.G.; Yu, J.; Yeh, S.T.; Low, A.; Chappell, A.E.; Østergaard, M.E.; Murray, S.; Gaus, H.J.; et al. Fatty Acid Conjugation Enhances Potency of Antisense Oligonucleotides in Muscle. Nucleic Acids Res. 2019, 47, 6029–6044. [Google Scholar] [CrossRef]
- Wolfrum, C.; Shi, S.; Jayaprakash, K.N.; Jayaraman, M.; Wang, G.; Pandey, R.K.; Rajeev, K.G.; Nakayama, T.; Charrise, K.; Ndungo, E.M.; et al. Mechanisms and Optimization of in Vivo Delivery of Lipophilic SiRNAs. Nat. Biotechnol. 2007, 25, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, C.; Tang, X. Cholesterol-Modified Caged SiRNAs for Photoregulating Exogenous and Endogenous Gene Expression. Bioconjug. Chem. 2018, 29, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Debacker, A.J.; Voutila, J.; Catley, M.; Blakey, D.; Habib, N. Delivery of Oligonucleotides to the Liver with GalNAc: From Research to Registered Therapeutic Drug. Mol. Ther. 2020, 28, 1759–1771. [Google Scholar] [CrossRef]
- D’Souza, A.A.; Devarajan, P.V. Asialoglycoprotein Receptor Mediated Hepatocyte Targeting—Strategies and Applications. J. Control. Release 2015, 203, 126–139. [Google Scholar] [CrossRef]
- Park, E.I.; Mi, Y.; Unverzagt, C.; Gabius, H.-J.; Baenziger, J.U. The Asialoglycoprotein Receptor Clears Glycoconjugates Terminating with Sialic Acidα2,6GalNAc. Proc. Natl. Acad. Sci. USA 2005, 102, 17125–17129. [Google Scholar] [CrossRef] [Green Version]
- Steirer, L.M.; Park, E.I.; Townsend, R.R.; Baenziger, J.U. The Asialoglycoprotein Receptor Regulates Levels of Plasma Glycoproteins Terminating with Sialic Acid A2,6-Galactose. J. Biol. Chem. 2009, 284, 3777–3783. [Google Scholar] [CrossRef] [Green Version]
- Tanowitz, M.; Hettrick, L.; Revenko, A.; Kinberger, G.A.; Prakash, T.P.; Seth, P.P. Asialoglycoprotein Receptor 1 Mediates Productive Uptake of N-Acetylgalactosamine-Conjugated and Unconjugated Phosphorothioate Antisense Oligonucleotides into Liver Hepatocytes. Nucleic Acids Res. 2017, 45, 12388–12400. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Zhu, X.; Li, S.; Wang, P.; Fang, J. Liver-Targeted Delivery of Oligonucleotides with N-Acetylgalactosamine Conjugation. ACS Omega 2021, 6, 16259–16265. [Google Scholar] [CrossRef]
- Sharma, V.K.; Osborn, M.F.; Hassler, M.R.; Echeverria, D.; Ly, S.; Ulashchik, E.A.; Martynenko-Makaev, Y.V.; Shmanai, V.V.; Zatsepin, T.S.; Khvorova, A.; et al. Novel Cluster and Monomer-Based GalNAc Structures Induce Effective Uptake of SiRNAs In Vitro and In Vivo. Bioconjug. Chem. 2018, 29, 2478–2488. [Google Scholar] [CrossRef]
- Prakash, T.P.; Graham, M.J.; Yu, J.; Carty, R.; Low, A.; Chappell, A.; Schmidt, K.; Zhao, C.; Aghajan, M.; Murray, H.F.; et al. Targeted Delivery of Antisense Oligonucleotides to Hepatocytes Using Triantennary N-Acetyl Galactosamine Improves Potency 10-Fold in Mice. Nucleic Acids Res. 2014, 42, 8796–8807. [Google Scholar] [CrossRef] [Green Version]
- Kinberger, G.A.; Prakash, T.P.; Yu, J.; Vasquez, G.; Low, A.; Chappell, A.; Schmidt, K.; Murray, H.M.; Gaus, H.; Swayze, E.E.; et al. Conjugation of Mono and Di-GalNAc Sugars Enhances the Potency of Antisense Oligonucleotides via ASGR Mediated Delivery to Hepatocytes. Bioorg. Med. Chem. Lett. 2016, 26, 3690–3693. [Google Scholar] [CrossRef]
- Schmidt, K.; Prakash, T.P.; Donner, A.J.; Kinberger, G.A.; Gaus, H.J.; Low, A.; Østergaard, M.E.; Bell, M.; Swayze, E.E.; Seth, P.P. Characterizing the Effect of GalNAc and Phosphorothioate Backbone on Binding of Antisense Oligonucleotides to the Asialoglycoprotein Receptor. Nucleic Acids Res. 2017, 45, 2294–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.Z.; Gunawan, R.; Post, N.; Zanardi, T.; Hall, S.; Burkey, J.; Kim, T.-W.; Graham, M.J.; Prakash, T.P.; Seth, P.P.; et al. Disposition and Pharmacokinetics of a GalNAc3-Conjugated Antisense Oligonucleotide Targeting Human Lipoprotein (a) in Monkeys. Nucleic Acid Ther. 2016, 26, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.Z.; Graham, M.J.; Post, N.; Riney, S.; Zanardi, T.; Hall, S.; Burkey, J.; Shemesh, C.S.; Prakash, T.P.; Seth, P.P.; et al. Disposition and Pharmacology of a GalNAc3-Conjugated ASO Targeting Human Lipoprotein (a) in Mice. Mol. Ther. Nucleic Acids 2016, 5, e317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yu, R.Z.; Henry, S.; Geary, R.S. Pharmacokinetics and Clinical Pharmacology Considerations of GalNAc 3 -Conjugated Antisense Oligonucleotides. Expert Opin. Drug Metab. Toxicol. 2019, 15, 475–485. [Google Scholar] [CrossRef]
- Sugo, T.; Terada, M.; Oikawa, T.; Miyata, K.; Nishimura, S.; Kenjo, E.; Ogasawara-Shimizu, M.; Makita, Y.; Imaichi, S.; Murata, S.; et al. Development of Antibody-SiRNA Conjugate Targeted to Cardiac and Skeletal Muscles. J. Control. Release 2016, 237, 1–13. [Google Scholar] [CrossRef]
- Soldevilla, M.; Meraviglia-Crivelli de Caso, D.; Menon, A.; Pastor, F. Aptamer-IRNAs as Therapeutics for Cancer Treatment. Pharmaceuticals 2018, 11, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catuogno, S.; Esposito, C.; de Franciscis, V. Aptamer-Mediated Targeted Delivery of Therapeutics: An Update. Pharmaceuticals 2016, 9, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, E.L.; Senter, P.D. Antibody-Drug Conjugates in Cancer Therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Sun, T.; Huang, S.; Dou, S.; Lin, L.; Chen, J.; Ruan, J.; Mao, C.; Yu, F.; Zeng, M.; et al. Targeted Delivery of PLK1-SiRNA by ScFv Suppresses Her2 + Breast Cancer Growth and Metastasis. Sci. Transl. Med. 2012, 4, 130ra48. [Google Scholar] [CrossRef]
- Arnold, A.E.; Malek-Adamian, E.; Le, P.U.; Meng, A.; Martínez-Montero, S.; Petrecca, K.; Damha, M.J.; Shoichet, M.S. Antibody-Antisense Oligonucleotide Conjugate Downregulates a Key Gene in Glioblastoma Stem Cells. Mol. Ther. Nucleic Acids 2018, 11, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Astriab-Fisher, A.; Fisher, M.H.; Juliano, R.; Herdewijn, P. Increased Uptake of Antisense Oligonucleotides by Delivery as Double Stranded Complexes. Biochem. Pharmacol. 2004, 68, 403–407. [Google Scholar] [CrossRef]
- Hong, S.; Sun, N.; Liu, M.; Wang, J.; Pei, R. Building a Chimera of Aptamer–Antisense Oligonucleotide for Silencing Galectin-1 Gene. RSC Adv. 2016, 6, 112445–112450. [Google Scholar] [CrossRef]
- Nuzzo, S.; Roscigno, G.; Affinito, A.; Ingenito, F.; Quintavalle, C.; Condorelli, G. Potential and Challenges of Aptamers as Specific Carriers of Therapeutic Oligonucleotides for Precision Medicine in Cancer. Cancers 2019, 11, 1521. [Google Scholar] [CrossRef] [Green Version]
- Dyne Therapeutics Targeting Muscle to Stop or Reverse Disease Progression. Available online: https://www.dyne-tx.com/our-forcetm-platform/ (accessed on 18 January 2023).
- Avidity Biosciences Platform|Overview Delivering on the RNA Revolution. Available online: https://www.aviditybiosciences.com/platform/overview/ (accessed on 18 January 2023).
- Mullard, A. Antibody–Oligonucleotide Conjugates Enter the Clinic. Nat. Rev. Drug Discov. 2022, 21, 6–8. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, K. PEGylation of Therapeutic Oligonucletides: From Linear to Highly Branched PEG Architectures. Nano Res. 2018, 11, 5519–5534. [Google Scholar] [CrossRef]
- Ng, E.W.M.; Shima, D.T.; Calias, P.; Cunningham, E.T.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a Targeted Anti-VEGF Aptamer for Ocular Vascular Disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Nagasaki, Y. Impacts of PEGylation on the Gene and Oligonucleotide Delivery System. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Hoang Thi, T.T.; Pilkington, E.H.; Nguyen, D.H.; Lee, J.S.; Park, K.D.; Truong, N.P. The Importance of Poly(Ethylene Glycol) Alternatives for Overcoming PEG Immunogenicity in Drug Delivery and Bioconjugation. Polymers 2020, 12, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishburn, C.S. The Pharmacology of PEGylation: Balancing PD with PK to Generate Novel Therapeutics. J. Pharm. Sci. 2008, 97, 4167–4183. [Google Scholar] [CrossRef]
- Haruta, K.; Otaki, N.; Nagamine, M.; Kayo, T.; Sasaki, A.; Hiramoto, S.; Takahashi, M.; Hota, K.; Sato, H.; Yamazaki, H. A Novel PEGylation Method for Improving the Pharmacokinetic Properties of Anti-Interleukin-17A RNA Aptamers. Nucleic Acid Ther. 2017, 27, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Hao, X.-Y.; Li, Y.-J.; Wu, J.; Xiang, D.-X.; Luo, S. Nonviral Delivery Systems for Antisense Oligonucleotide Therapeutics. Biomater. Res. 2022, 26, 49. [Google Scholar] [CrossRef]
- van der Koog, L.; Gandek, T.B.; Nagelkerke, A. Liposomes and Extracellular Vesicles as Drug Delivery Systems: A Comparison of Composition, Pharmacokinetics, and Functionalization. Adv. Healthc. Mater. 2022, 11, 2100639. [Google Scholar] [CrossRef]
- Cha, W.; Fan, R.; Miao, Y.; Zhou, Y.; Qin, C.; Shan, X.; Wan, X.; Li, J. Mesoporous Silica Nanoparticles as Carriers for Intracellular Delivery of Nucleic Acids and Subsequent Therapeutic Applications. Molecules 2017, 22, 782. [Google Scholar] [CrossRef] [Green Version]
- Steinbacher, J.L.; Landry, C.C. Adsorption and Release of SiRNA from Porous Silica. Langmuir 2014, 30, 4396–4405. [Google Scholar] [CrossRef]
- Morgan, E.; Wupperfeld, D.; Morales, D.; Reich, N. Shape Matters: Gold Nanoparticle Shape Impacts the Biological Activity of SiRNA Delivery. Bioconjug. Chem. 2019, 30, 853–860. [Google Scholar] [CrossRef]
- Ding, Y.; Jiang, Z.; Saha, K.; Kim, C.S.; Kim, S.T.; Landis, R.F.; Rotello, V.M. Gold Nanoparticles for Nucleic Acid Delivery. Mol. Ther. 2014, 22, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Akash, M.S.H.; Rehman, K.; Chen, S. Polymeric-Based Particulate Systems for Delivery of Therapeutic Proteins. Pharm. Dev. Technol. 2016, 21, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Tenchov, R.; Bird, R.; Curtze, A.E.; Zhou, Q. Lipid Nanoparticles—From Liposomes to MRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano 2021, 15, 16982–17015. [Google Scholar] [CrossRef] [PubMed]
- Tam, Y.; Chen, S.; Cullis, P. Advances in Lipid Nanoparticles for SiRNA Delivery. Pharmaceutics 2013, 5, 498–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.-E.; Hee Kim, M.; Davidson, B.L.; Kyung Lee, S.; Shankar, P.; Manjunath, N. Transvascular Delivery of Small Interfering RNA to the Central Nervous System. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef] [PubMed]
- K Balamurugan; P Chintamani Lipid Nano Particulate Drug Delivery: An Overview of the Emerging Trend. Pharm. Innov. 2018, 7, 779–789.
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Yang, J. Patisiran for the Treatment of Hereditary Transthyretin-Mediated Amyloidosis. Expert Rev. Clin. Pharmacol. 2019, 12, 95–99. [Google Scholar] [CrossRef]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A Highly Efficient, Lipid-Mediated DNA-Transfection Procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, J.A.; Darjuan, M.M.; Mercer, J.E.; Chen, S.; van der Meel, R.; Thewalt, J.L.; Tam, Y.Y.C.; Cullis, P.R. On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and SiRNA. ACS Nano 2018, 12, 4787–4795. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.; Kim, H.; Leal, C. Self-Organization of Nucleic Acids in Lipid Constructs. Curr. Opin. Colloid Interface Sci. 2016, 26, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the Potency of SiRNA Lipid Nanoparticles for Hepatic Gene Silencing In Vivo. Angew. Chem. Int. Ed. 2012, 51, 8529–8533. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Tam, Y.Y.C.; Lin, P.J.C.; Sung, M.M.H.; Tam, Y.K.; Cullis, P.R. Influence of Particle Size on the in Vivo Potency of Lipid Nanoparticle Formulations of SiRNA. J. Control. Release 2016, 235, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Keough, E.; Matter, A.; Leander, K.; Young, S.; Carlini, E.; Sachs, A.B.; Tao, W.; Abrams, M.; Howell, B.; et al. Biodistribution of Small Interfering RNA at the Organ and Cellular Levels after Lipid Nanoparticle-Mediated Delivery. J. Histochem. Cytochem. 2011, 59, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted Delivery of RNAi Therapeutics with Endogenous and Exogenous Ligand-Based Mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Habrant, D.; Peuziat, P.; Colombani, T.; Dallet, L.; Gehin, J.; Goudeau, E.; Evrard, B.; Lambert, O.; Haudebourg, T.; Pitard, B. Design of Ionizable Lipids To Overcome the Limiting Step of Endosomal Escape: Application in the Intracellular Delivery of MRNA, DNA, and SiRNA. J. Med. Chem. 2016, 59, 3046–3062. [Google Scholar] [CrossRef] [PubMed]
- Boisguérin, P.; Deshayes, S.; Gait, M.J.; O’Donovan, L.; Godfrey, C.; Betts, C.A.; Wood, M.J.A.; Lebleu, B. Delivery of Therapeutic Oligonucleotides with Cell Penetrating Peptides. Adv. Drug Deliv. Rev. 2015, 87, 52–67. [Google Scholar] [CrossRef]
- Sharma, R.; Shivpuri, S.; Anand, A.; Kulshreshtha, A.; Ganguli, M. Insight into the Role of Physicochemical Parameters in a Novel Series of Amphipathic Peptides for Efficient DNA Delivery. Mol. Pharm. 2013, 10, 2588–2600. [Google Scholar] [CrossRef]
- Kurrikoff, K.; Langel, Ü. Recent CPP-Based Applications in Medicine. Expert Opin. Drug Deliv. 2019, 16, 1183–1191. [Google Scholar] [CrossRef]
- El Andaloussi, S.; Lehto, T.; Mäger, I.; Rosenthal-Aizman, K.; Oprea, I.I.; Simonson, O.E.; Sork, H.; Ezzat, K.; Copolovici, D.M.; Kurrikoff, K.; et al. Design of a Peptide-Based Vector, PepFect6, for Efficient Delivery of SiRNA in Cell Culture and Systemically in Vivo. Nucleic Acids Res. 2011, 39, 3972–3987. [Google Scholar] [CrossRef] [Green Version]
- Wyman, T.B.; Nicol, F.; Zelphati, O.; Scaria, P.V.; Plank, C.; Szoka, F.C. Design, Synthesis, and Characterization of a Cationic Peptide That Binds to Nucleic Acids and Permeabilizes Bilayers. Biochemistry 1997, 36, 3008–3017. [Google Scholar] [CrossRef]
- McCarthy, H.O.; McCaffrey, J.; McCrudden, C.M.; Zholobenko, A.; Ali, A.A.; McBride, J.W.; Massey, A.S.; Pentlavalli, S.; Chen, K.-H.; Cole, G.; et al. Development and Characterization of Self-Assembling Nanoparticles Using a Bio-Inspired Amphipathic Peptide for Gene Delivery. J. Control. Release 2014, 189, 141–149. [Google Scholar] [CrossRef]
- Mäe, M.; El Andaloussi, S.; Lundin, P.; Oskolkov, N.; Johansson, H.J.; Guterstam, P.; Langel, Ü. A Stearylated CPP for Delivery of Splice Correcting Oligonucleotides Using a Non-Covalent Co-Incubation Strategy. J. Control. Release 2009, 134, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Freitag, F.; Wagner, E. Optimizing Synthetic Nucleic Acid and Protein Nanocarriers: The Chemical Evolution Approach. Adv. Drug Deliv. Rev. 2021, 168, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Zhu, X.; Qiu, L. Electroneutral Composite Polymersomes Self-Assembled by Amphiphilic Polyphosphazenes for Effective MiR-200c in Vivo Delivery to Inhibit Drug Resistant Lung Cancer. Biomaterials 2016, 106, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.; Csaba, N.; Alexander, C.; Garcia-Fuentes, M. Polyphosphazenes for the Delivery of Biopharmaceuticals. J. Appl. Polym. Sci. 2020, 137, 48688. [Google Scholar] [CrossRef]
- Teasdale, I. Stimuli-Responsive Phosphorus-Based Polymers. Eur. J. Inorg. Chem. 2019, 2019, 1445–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezvantalab, S.; Drude, N.I.; Moraveji, M.K.; Güvener, N.; Koons, E.K.; Shi, Y.; Lammers, T.; Kiessling, F. PLGA-Based Nanoparticles in Cancer Treatment. Front. Pharmacol. 2018, 9, 1260. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Xu, Y.; Solis, L.M.; Tao, W.; Wang, L.; Behrens, C.; Xu, X.; Zhao, L.; Liu, D.; Wu, J.; et al. Long-Circulating SiRNA Nanoparticles for Validating Prohibitin1-Targeted Non-Small Cell Lung Cancer Treatment. Proc. Natl. Acad. Sci. USA 2015, 112, 7779–7784. [Google Scholar] [CrossRef] [Green Version]
- Oyaghire, S.N.; Quijano, E.; Piotrowski-Daspit, A.S.; Saltzman, W.M.; Glazer, P.M. Poly(Lactic-Co-Glycolic Acid) Nanoparticle Delivery of Peptide Nucleic Acids In Vivo. Methods Mol. Biol. 2020, 2105, 261–281. [Google Scholar]
- Babu, A.; Wang, Q.; Muralidharan, R.; Shanker, M.; Munshi, A.; Ramesh, R. Chitosan Coated Polylactic Acid Nanoparticle-Mediated Combinatorial Delivery of Cisplatin and SiRNA/Plasmid DNA Chemosensitizes Cisplatin-Resistant Human Ovarian Cancer Cells. Mol. Pharm. 2014, 11, 2720–2733. [Google Scholar] [CrossRef] [PubMed]
- Kolonko, A.K.; Bangel-Ruland, N.; Goycoolea, F.M.; Weber, W.-M. Chitosan Nanocomplexes for the Delivery of ENaC Antisense Oligonucleotides to Airway Epithelial Cells. Biomolecules 2020, 10, 553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and Biomedical Applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taetz, S.; Nafee, N.; Beisner, J.; Piotrowska, K.; Baldes, C.; Mürdter, T.E.; Huwer, H.; Schneider, M.; Schaefer, U.F.; Klotz, U. The Influence of Chitosan Content in Cationic Chitosan/PLGA Nanoparticles on the Delivery Efficiency of Antisense 2′-O-Methyl-RNA Directed against Telomerase in Lung Cancer Cells. Eur. J. Pharm. Biopharm. 2009, 72, 358–369. [Google Scholar] [CrossRef]
- Thanki, K.; Zeng, X.; Justesen, S.; Tejlmann, S.; Falkenberg, E.; van Driessche, E.; Mørck Nielsen, H.; Franzyk, H.; Foged, C. Engineering of Small Interfering RNA-Loaded Lipidoid-Poly(DL-Lactic-Co-Glycolic Acid) Hybrid Nanoparticles for Highly Efficient and Safe Gene Silencing: A Quality by Design-Based Approach. Eur. J. Pharm. Biopharm. 2017, 120, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Penichet, M.L.; Kang, Y.-S.; Pardridge, W.M.; Morrison, S.L.; Shin, S.-U. An Antibody-Avidin Fusion Protein Specific for the Transferrin Receptor Serves as a Delivery Vehicle for Effective Brain Targeting: Initial Applications in Anti-HIV Antisense Drug Delivery to the Brain. J. Immunol. 1999, 163, 4421–4426. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Boado, R.J. Enhanced Cellular Uptake of Biotinylated Antisense Oligonucleotide or Peptide Mediated by Avidin, a Cationic Protein. FEBS Lett. 1991, 288, 30–32. [Google Scholar] [CrossRef] [Green Version]
- Mirochnik, Y.; Rubenstein, M.; Guinan, P. Targeting of Biotinylated Oligonucleotides to Prostate Tumors with Antibody-Based Delivery Vehicles. J. Drug Target. 2007, 15, 342–350. [Google Scholar] [CrossRef]
- Song, E.; Zhu, P.; Lee, S.-K.; Chowdhury, D.; Kussman, S.; Dykxhoorn, D.M.; Feng, Y.; Palliser, D.; Weiner, D.B.; Shankar, P.; et al. Antibody Mediated in Vivo Delivery of Small Interfering RNAs via Cell-Surface Receptors. Nat. Biotechnol. 2005, 23, 709–717. [Google Scholar] [CrossRef]
- Dugal-Tessier, J.; Thirumalairajan, S.; Jain, N. Antibody-Oligonucleotide Conjugates: A Twist to Antibody-Drug Conjugates. J. Clin. Med. 2021, 10, 838. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological Properties of Extracellular Vesicles and Their Physiological Functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooijmans, S.A.A.; de Jong, O.G.; Schiffelers, R.M. Exploring Interactions between Extracellular Vesicles and Cells for Innovative Drug Delivery System Design. Adv. Drug Deliv. Rev. 2021, 173, 252–278. [Google Scholar] [CrossRef] [PubMed]
- Popowski, K.; Lutz, H.; Hu, S.; George, A.; Dinh, P.; Cheng, K. Exosome Therapeutics for Lung Regenerative Medicine. J. Extracell. Vesicles 2020, 9, 1785161. [Google Scholar] [CrossRef]
- Rodríguez, D.A.; Vader, P. Extracellular Vesicle-Based Hybrid Systems for Advanced Drug Delivery. Pharmaceutics 2022, 14, 267. [Google Scholar] [CrossRef]
- Giebel, B.; Kordelas, L.; Börger, V. Clinical Potential of Mesenchymal Stem/Stromal Cell-Derived Extracellular Vesicles. Stem Cell Investig. 2017, 4, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.K.; Choo, A.; Chen, T.S.; Salto-Tellez, M.; Timmers, L.; Lee, C.N.; el Oakley, R.M.; et al. Exosome Secreted by MSC Reduces Myocardial Ischemia/Reperfusion Injury. Stem Cell Res. 2010, 4, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordelas, L.; Rebmann, V.; Ludwig, A.-K.; Radtke, S.; Ruesing, J.; Doeppner, T.R.; Epple, M.; Horn, P.A.; Beelen, D.W.; Giebel, B. MSC-Derived Exosomes: A Novel Tool to Treat Therapy-Refractory Graft-versus-Host Disease. Leukemia 2014, 28, 970–973. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Brennan, M.Á.; Lötvall, J.; Breakefield, X.O.; el Andaloussi, S. Advances in Therapeutic Applications of Extracellular Vesicles. Sci. Transl. Med. 2019, 11, eaav8521. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma Microvesicles Transport RNA and Proteins That Promote Tumour Growth and Provide Diagnostic Biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-Mediated Transfer of MRNAs and MicroRNAs Is a Novel Mechanism of Genetic Exchange between Cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes Facilitate Therapeutic Targeting of Oncogenic KRAS in Pancreatic Cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of SiRNA to the Mouse Brain by Systemic Injection of Targeted Exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Didiot, M.-C.; Hall, L.M.; Coles, A.H.; Haraszti, R.A.; Godinho, B.M.; Chase, K.; Sapp, E.; Ly, S.; Alterman, J.F.; Hassler, M.R.; et al. Exosome-Mediated Delivery of Hydrophobically Modified SiRNA for Huntingtin MRNA Silencing. Mol. Ther. 2016, 24, 1836–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraszti, R.A.; Miller, R.; Didiot, M.-C.; Biscans, A.; Alterman, J.F.; Hassler, M.R.; Roux, L.; Echeverria, D.; Sapp, E.; DiFiglia, M.; et al. Optimized Cholesterol-SiRNA Chemistry Improves Productive Loading onto Extracellular Vesicles. Mol. Ther. 2018, 26, 1973–1982. [Google Scholar] [CrossRef] [Green Version]
- Lamichhane, T.N.; Jeyaram, A.; Patel, D.B.; Parajuli, B.; Livingston, N.K.; Arumugasaamy, N.; Schardt, J.S.; Jay, S.M. Oncogene Knockdown via Active Loading of Small RNAs into Extracellular Vesicles by Sonication. Cell Mol. Bioeng. 2016, 9, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.-C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and Testing of Clinical-Grade Exosomes for Pancreatic Cancer. JCI Insight 2018, 3, e99263. [Google Scholar] [CrossRef]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from Marrow Stromal Cells Expressing MiR-146b Inhibit Glioma Growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor MicroRNA to Breast Cancer Cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, X.; Chen, X.; Wang, L.; Yang, G. Exosome Mediated Delivery of MiR-124 Promotes Neurogenesis after Ischemia. Mol. Ther. Nucleic Acids 2017, 7, 278–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.M.; Wiklander, P.B.O.; Nordin, J.Z.; Al-Shawi, R.; Wood, M.J.; Vithlani, M.; Schapira, A.H.V.; Simons, J.P.; El-Andaloussi, S.; Alvarez-Erviti, L. Systemic Exosomal SiRNA Delivery Reduced Alpha-Synuclein Aggregates in Brains of Transgenic Mice. Mov. Disord. 2014, 29, 1476–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciferri, M.C.; Quarto, R.; Tasso, R. Extracellular Vesicles as Biomarkers and Therapeutic Tools: From Pre-Clinical to Clinical Applications. Biology 2021, 10, 359. [Google Scholar] [CrossRef] [PubMed]
- Cutler, J.I.; Auyeung, E.; Mirkin, C.A. Spherical Nucleic Acids. J. Am. Chem. Soc. 2012, 134, 1376–1391. [Google Scholar] [CrossRef]
- Kapadia, C.H.; Melamed, J.R.; Day, E.S. Spherical Nucleic Acid Nanoparticles: Therapeutic Potential. BioDrugs 2018, 32, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Hill, H.D.; Millstone, J.E.; Banholzer, M.J.; Mirkin, C.A. The Role Radius of Curvature Plays in Thiolated Oligonucleotide Loading on Gold Nanoparticles. ACS Nano 2009, 3, 418–424. [Google Scholar] [CrossRef] [Green Version]
- Park, S.S.; Urbach, Z.J.; Brisbois, C.A.; Parker, K.A.; Partridge, B.E.; Oh, T.; Dravid, V.P.; de la Olvera Cruz, M.; Mirkin, C.A. DNA- and Field-Mediated Assembly of Magnetic Nanoparticles into High-Aspect Ratio Crystals. Adv. Mater. 2020, 32, 1906626. [Google Scholar] [CrossRef]
- Cutler, J.I.; Zhang, K.; Zheng, D.; Auyeung, E.; Prigodich, A.E.; Mirkin, C.A. Polyvalent Nucleic Acid Nanostructures. J. Am. Chem. Soc. 2011, 133, 9254–9257. [Google Scholar] [CrossRef] [Green Version]
- Rosi, N.L.; Giljohann, D.A.; Thaxton, C.S.; Lytton-Jean, A.K.R.; Han, M.S.; Mirkin, C.A. Oligonucleotide-Modified Gold Nanoparticles for Intracellular Gene Regulation. Science 2006, 312, 1027–1030. [Google Scholar] [CrossRef]
- Seferos, D.S.; Prigodich, A.E.; Giljohann, D.A.; Patel, P.C.; Mirkin, C.A. Polyvalent DNA Nanoparticle Conjugates Stabilize Nucleic Acids. Nano Lett. 2009, 9, 308–311. [Google Scholar] [CrossRef]
- Giljohann, D.A.; Seferos, D.S.; Prigodich, A.E.; Patel, P.C.; Mirkin, C.A. Gene Regulation with Polyvalent SiRNA−Nanoparticle Conjugates. J. Am. Chem. Soc. 2009, 131, 2072–2073. [Google Scholar] [CrossRef] [Green Version]
- Jensen, S.A.; Day, E.S.; Ko, C.H.; Hurley, L.A.; Luciano, J.P.; Kouri, F.M.; Merkel, T.J.; Luthi, A.J.; Patel, P.C.; Cutler, J.I.; et al. Spherical Nucleic Acid Nanoparticle Conjugates as an RNAi-Based Therapy for Glioblastoma. Sci. Transl. Med. 2013, 5, 209ra152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemati, H.; Ghahramani, M.-H.; Faridi-Majidi, R.; Izadi, B.; Bahrami, G.; Madani, S.-H.; Tavoosidana, G. Using SiRNA-Based Spherical Nucleic Acid Nanoparticle Conjugates for Gene Regulation in Psoriasis. J. Control. Release 2017, 268, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Randeria, P.S.; Seeger, M.A.; Wang, X.-Q.; Wilson, H.; Shipp, D.; Mirkin, C.A.; Paller, A.S. SiRNA-Based Spherical Nucleic Acids Reverse Impaired Wound Healing in Diabetic Mice by Ganglioside GM3 Synthase Knockdown. Proc. Natl. Acad. Sci. USA 2015, 112, 5573–5578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinen, A.B.; Guan, C.M.; Ko, C.H.; Mirkin, C.A. The Impact of Protein Corona Formation on the Macrophage Cellular Uptake and Biodistribution of Spherical Nucleic Acids. Small 2017, 13, 1603847. [Google Scholar] [CrossRef] [Green Version]
- Exicure Inc Science and Technology|Exicure’s Approach. Available online: https://www.exicuretx.com/science-technology/index.php (accessed on 18 January 2023).
- Seeman, N.C. DNA Nanotechnology at 40. Nano Lett. 2020, 20, 1477–1478. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lu, X.; Wu, X.; Li, Y.; Tang, W.; Yang, C.; Liu, J.; Ding, B. Chemically Modified DNA Nanostructures for Drug Delivery. Innovation 2022, 3, 100217. [Google Scholar] [CrossRef]
- Xu, F.; Xia, Q.; Wang, P. Rationally Designed DNA Nanostructures for Drug Delivery. Front. Chem. 2020, 8, 751. [Google Scholar] [CrossRef]
- Kim, K.-R.; Kim, H.Y.; Lee, Y.-D.; Ha, J.S.; Kang, J.H.; Jeong, H.; Bang, D.; Ko, Y.T.; Kim, S.; Lee, H.; et al. Self-Assembled Mirror DNA Nanostructures for Tumor-Specific Delivery of Anticancer Drugs. J. Control. Release 2016, 243, 121–131. [Google Scholar] [CrossRef]
- Mohri, K.; Nishikawa, M.; Takahashi, N.; Shiomi, T.; Matsuoka, N.; Ogawa, K.; Endo, M.; Hidaka, K.; Sugiyama, H.; Takahashi, Y.; et al. Design and Development of Nanosized DNA Assemblies in Polypod-Like Structures as Efficient Vehicles for Immunostimulatory CpG Motifs to Immune Cells. ACS Nano 2012, 6, 5931–5940. [Google Scholar] [CrossRef]
- Lee, H.; Lytton-Jean, A.K.R.; Chen, Y.; Love, K.T.; Park, A.I.; Karagiannis, E.D.; Sehgal, A.; Querbes, W.; Zurenko, C.S.; Jayaraman, M.; et al. Molecularly Self-Assembled Nucleic Acid Nanoparticles for Targeted in Vivo SiRNA Delivery. Nat. Nanotechnol. 2012, 7, 389–393. [Google Scholar] [CrossRef]
- Torchilin, V.P. Multifunctional, Stimuli-Sensitive Nanoparticulate Systems for Drug Delivery. Nat. Rev. Drug Discov. 2014, 13, 813–827. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.; He, Z.; Xiong, J.; Chao, J. Smart Drug Delivery Systems Based on DNA Nanotechnology. Chempluschem 2022, 87, e202100548. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; He, J.; Yi, H.; Xiang, G.; Chen, K.; Fu, B.; Yang, Y.; Chen, G. SiRNA Suppression of HTERT Using Activatable Cell-Penetrating Peptides in Hepatoma Cells. Biosci. Rep. 2015, 35, e00181. [Google Scholar] [CrossRef] [PubMed]
- Rozema, D.B.; Lewis, D.L.; Wakefield, D.H.; Wong, S.C.; Klein, J.J.; Roesch, P.L.; Bertin, S.L.; Reppen, T.W.; Chu, Q.; Blokhin, A.V.; et al. Dynamic PolyConjugates for Targeted In Vivo Delivery of SiRNA to Hepatocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 12982–12987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, G.D.; Kamaly, N.; Kalber, T.L.; Brody, L.P.; Sahuri, M.; Shamsaei, E.; Miller, A.D.; Bell, J.D. Novel Multifunctional Nanoparticle Mediates SiRNA Tumour Delivery, Visualisation and Therapeutic Tumour Reduction In Vivo. J. Control. Release 2011, 149, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.M.; Bachelet, I.; Church, G.M. A Logic-Gated Nanorobot for Targeted Transport of Molecular Payloads. Science 2012, 335, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yu, C.; Xu, T.; Yao, D.; Zhu, L.; Shen, Z.; Huang, X. Self-Assembly of DNA Nanostructure Containing Cell-Specific Aptamer as a Precise Drug Delivery System for Cancer Therapy in Non-Small Cell Lung Cancer. J. Nanobiotechnol. 2022, 20, 486. [Google Scholar] [CrossRef]
- Pan, Q.; Nie, C.; Hu, Y.; Yi, J.; Liu, C.; Zhang, J.; He, M.; He, M.; Chen, T.; Chu, X. Aptamer-Functionalized DNA Origami for Targeted Codelivery of Antisense Oligonucleotides and Doxorubicin to Enhance Therapy in Drug-Resistant Cancer Cells. ACS Appl. Mater. Interfaces 2020, 12, 400–409. [Google Scholar] [CrossRef]
- Wu, P.; Grainger, D.W. Drug/Device Combinations for Local Drug Therapies and Infection Prophylaxis. Biomaterials 2006, 27, 2450–2467. [Google Scholar] [CrossRef]
- Batasheva, S.; Fakhrullin, R. Sequence Does Not Matter: The Biomedical Applications of DNA-Based Coatings and Cores. Int. J. Mol. Sci. 2021, 22, 12884. [Google Scholar] [CrossRef]
- Jewell, C.; Lynn, D. Multilayered Polyelectrolyte Assemblies as Platforms for the Delivery of DNA and Other Nucleic Acid-Based Therapeutics☆. Adv. Drug Deliv. Rev. 2008, 60, 979–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Kim, B.-S.; Kim, S.R.; Hammond, P.T.; Irvine, D.J. Layer-by-Layer-Assembled Multilayer Films for Transcutaneous Drug and Vaccine Delivery. ACS Nano 2009, 3, 3719–3729. [Google Scholar] [CrossRef] [PubMed]
- Hammond, P.T. Form and Function in Multilayer Assembly: New Applications at the Nanoscale. Adv. Mater. 2004, 16, 1271–1293. [Google Scholar] [CrossRef]
- Sukhorukov, G.B.; Rogach, A.L.; Garstka, M.; Springer, S.; Parak, W.J.; Muñoz-Javier, A.; Kreft, O.; Skirtach, A.G.; Susha, A.S.; Ramaye, Y.; et al. Multifunctionalized Polymer Microcapsules: Novel Tools for Biological and Pharmacological Applications. Small 2007, 3, 944–955. [Google Scholar] [CrossRef]
- Prasad, V. Nusinersen for Spinal Muscular Atrophy. JAMA Pediatr. 2018, 172, 123. [Google Scholar] [CrossRef]
- Agrawal, P.; Reifenberger, J.G.; Dorfman, K.D. 3D Printing-Enabled DNA Extraction for Long-Read Genomics. ACS Omega 2020, 5, 20817–20824. [Google Scholar] [CrossRef] [PubMed]





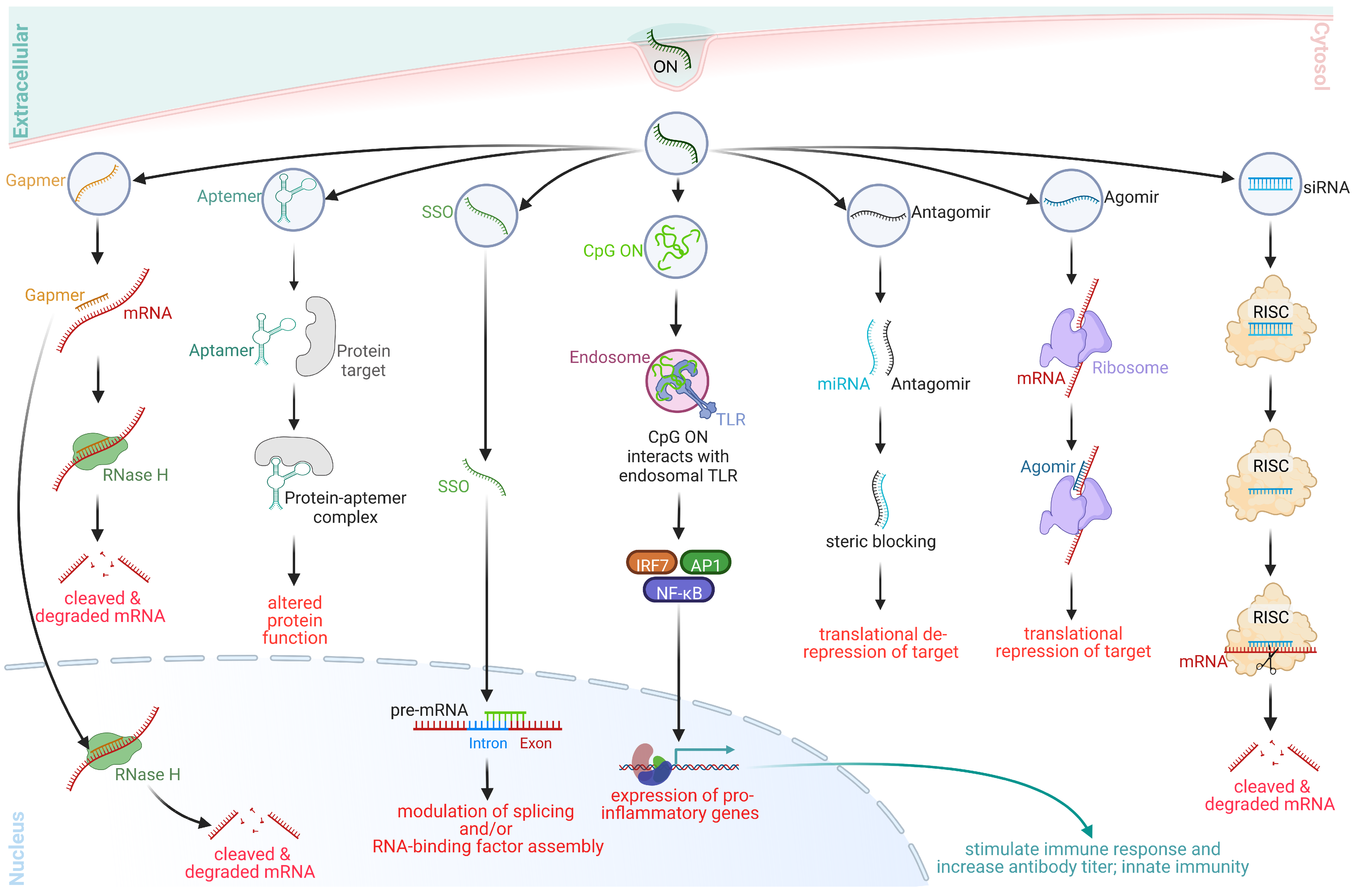{kind=link}
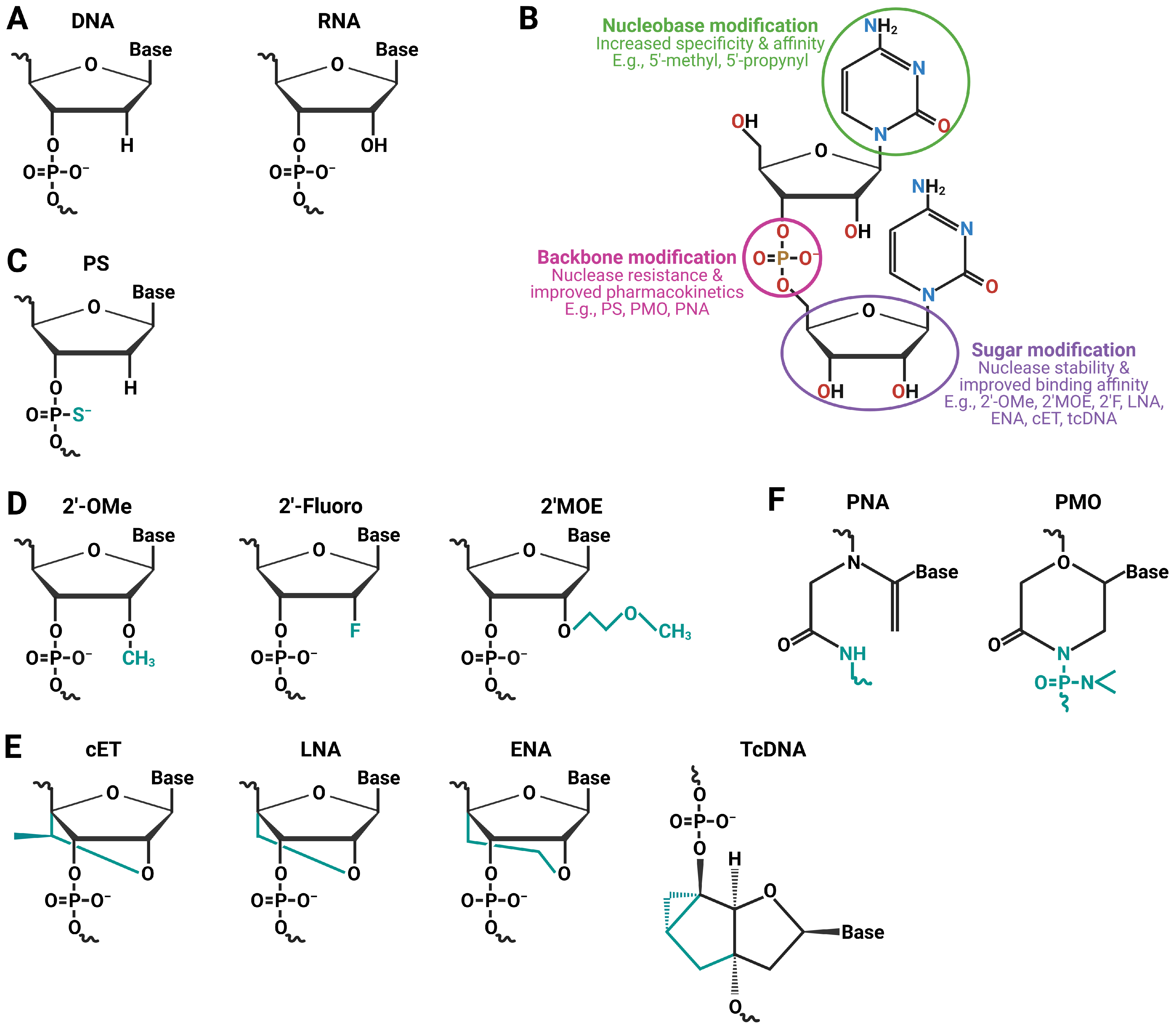{kind=link}
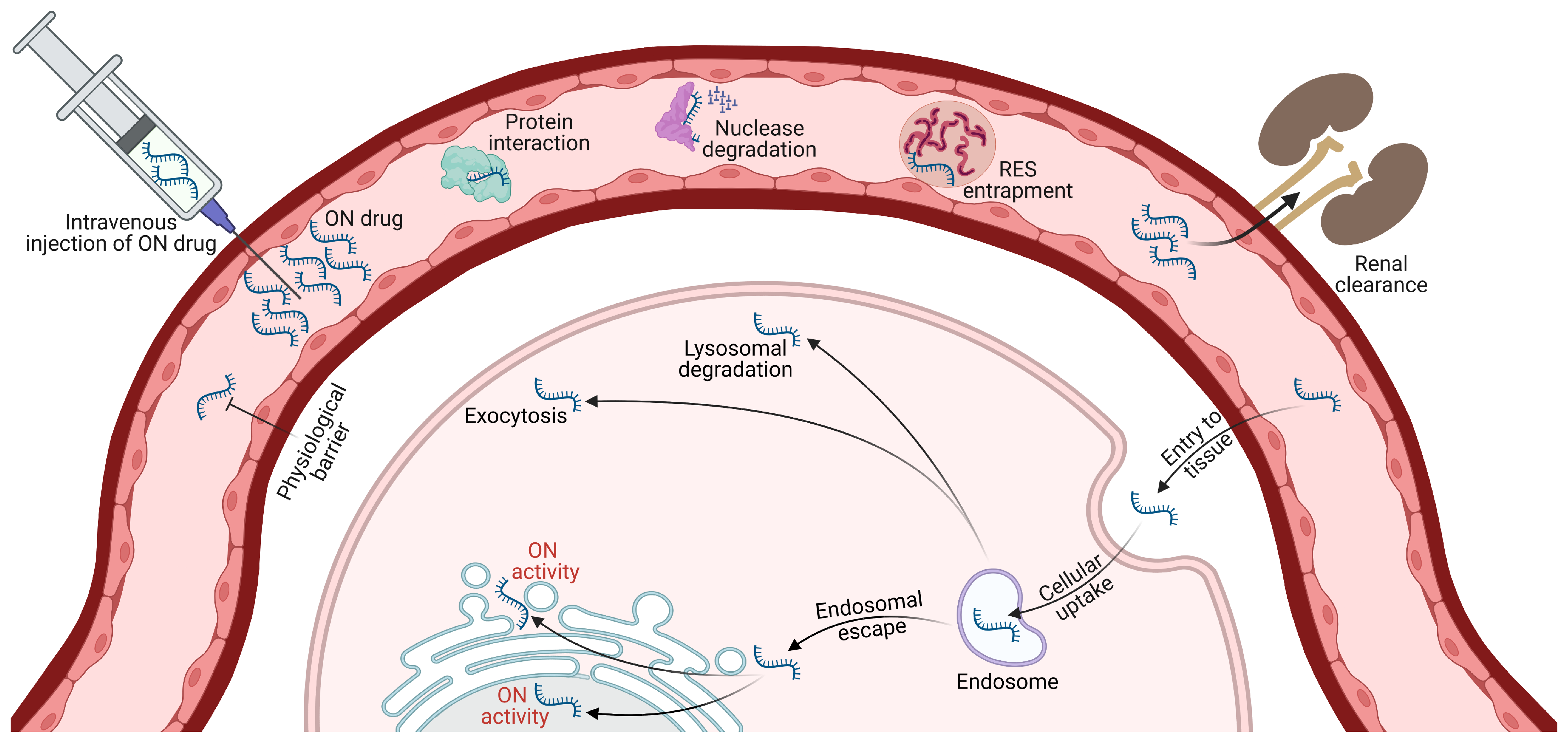{kind=link}
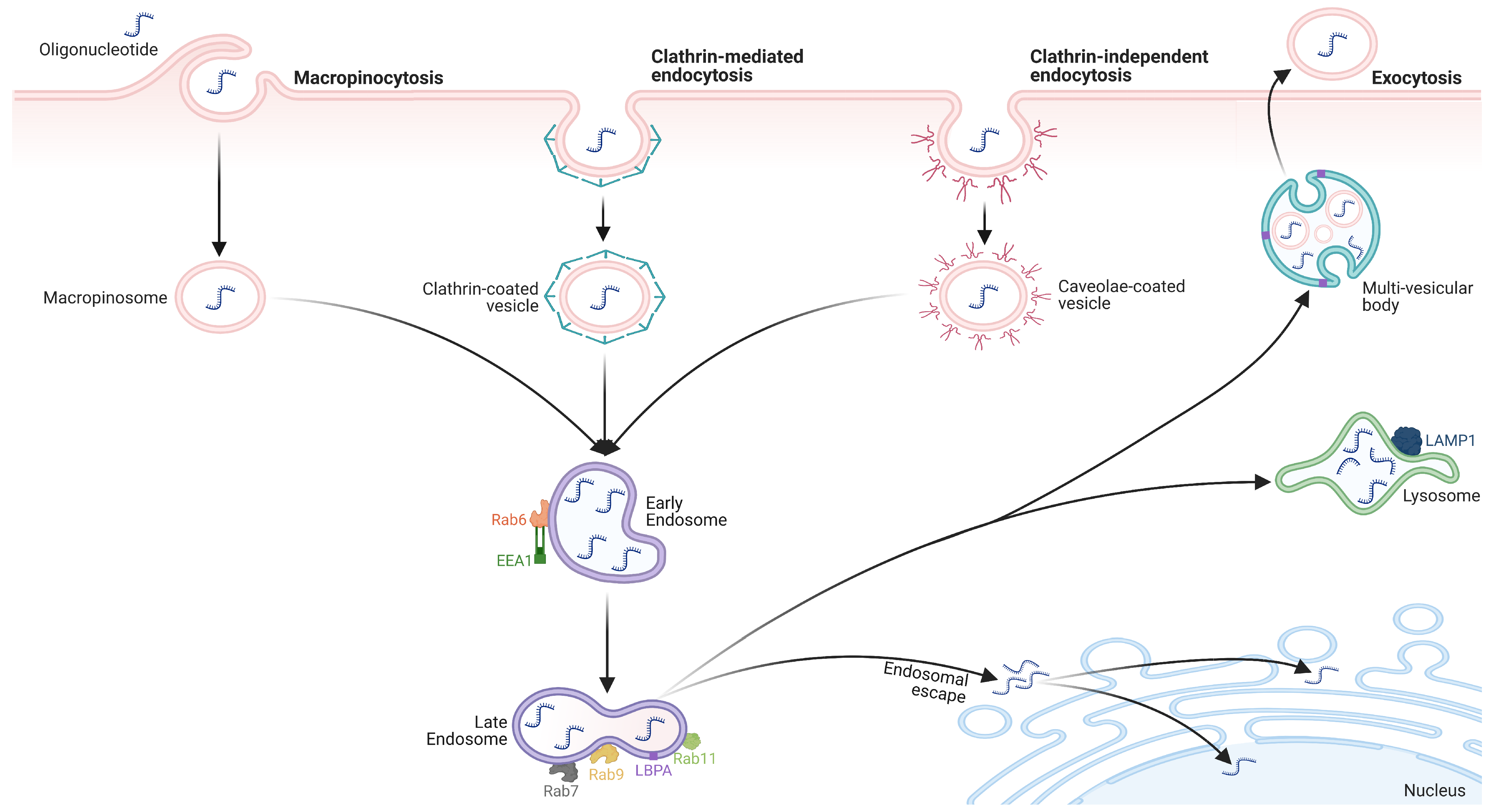{kind=link}
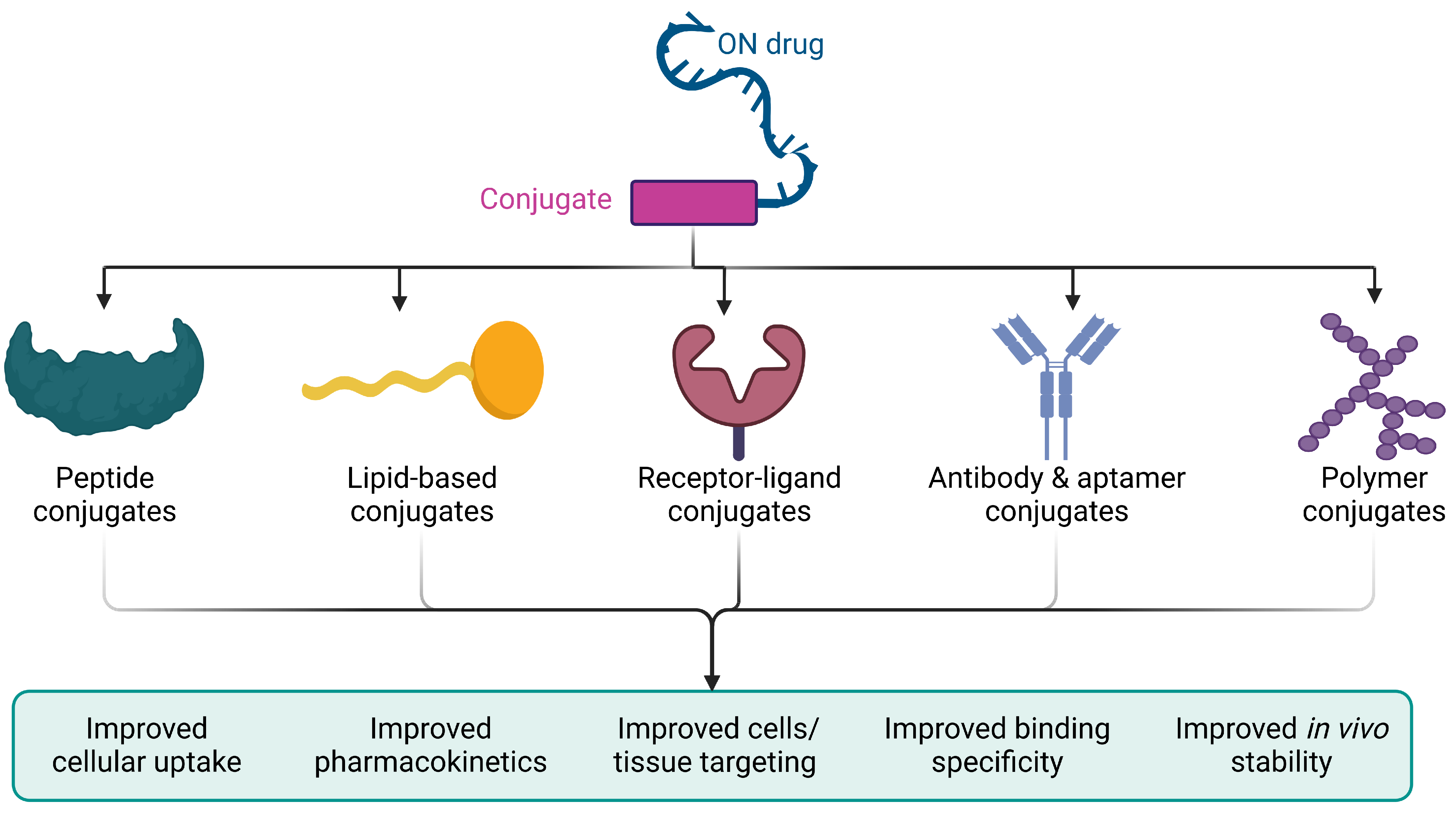{kind=link}
| Platform | Mechanism | Most Used ON Chemistry | Length (nt) | Primary Cellular/Tissue Target | Administration Route | Uptake Level | Off-Target Effects | Major Undesirable Effects |
|---|---|---|---|---|---|---|---|---|
| Gapmers | RNase H-mediated cleavage | PS, LNA, 2′OMe, 2′MOE | 12–30 | Cytoplasm, nucleus | SC, IV, IP | Moderate to high; high in liver, variable in other tissues depending on delivery method | Low with appropriate design and modifications | Variable hepatotoxicity depending on chemistry and dose, potential for immunogenicity |
| Aptamers | Binding and inhibiting target proteins via shape complementarity | RNA, DNA | 20–100 | Extracellular space, cell surface proteins | SC, IV, IP, oral | Low; high in plasma, low in tissues due to size and charge barriers | Low due to high specificity and affinity of binding | Rare hepatotoxicity, variable potential for immunogenicity, bleeding risk, rapid renal clearance |
| Steric blocking ONs | Binding and blocking target mRNA/pre-mRNA | PS, 2′Ome, 2′MOE, LNA, PMO, PNA | 8–50 | Cytoplasm, nucleus | SC, IV, IP, topical, oral, rectal | Variable depending on target accessibility and delivery vehicle | Low with appropriate design and modifications | Variable hepatotoxicity depending on chemistry and dose, low potential for immunogenicity, injection site reactions, RNase-activation |
| Immunostimulant ONs | Activating innate immune cells via TLR recognition of unmethylated CpG motif sequences | CpG DNA, modified RNA | 16–28 | Toll-like receptor, dendritic cells, B cells | SC, IV, intratumoral | Low to high; high in immune cells expressing TLR9, low in other tissues | Low due to specific TLR recognition of CpG motifs | Rare hepatotoxicity, high potential for immunogenicity, cytokine induction, injection site reactions |
| Antagomirs | Binding and inhibiting endogenous miRNAs | 2′OMe, 2′MOE, 2′-F, LNA, PNA, PMO | 15–31 | Cytoplasm, endoplasmic reticulum, nucleus, and extracellular space | SC, IV | High; high in liver, variable in other tissues depending on delivery method | Low due to high specificity and affinity of binding | Variable hepatotoxicity depending on chemistry and dose, low potential for immunogenicity, bleeding risk, rapid renal clearance |
| Agomirs | Mimicking and enhancing endogenous miRNA activity | 2′OMe, 2′MOE, 2′-F, LNA, PNA, PMO | 19–23 | Cytoplasm, endoplasmic reticulum, nucleus, and extracellular space | SC, IV | High; variable depending on target accessibility and delivery methods | Low due to high specificity and affinity of binding | Variable hepatotoxicity depending on chemistry and dose, low potential for immunogenicity |
| siRNA | RNAi-mediated gene silencing | RNA, PS, 2′OMe, 2′MOE, 2′-F, LNA | 18–24 | Cytoplasm | SC, IV, topical | High; high in liver, variable in other tissues depending on delivery method | Low with appropriate design and modifications; sequence-specific RNAi | Rare hepatotoxicity, variable potential for immunogenicity, off-target gene deactivation |
| ON Delivery System | Advantages | Limitations |
|---|---|---|
| Lipoplex, liposomes, and lipid nanoparticles |
|
|
| Peptide-based delivery systems |
|
|
| Polymer-based delivery systems |
|
|
| Antibody complexation delivery systems |
|
|
| Extracellular vesicle-mediated delivery systems |
|
|
| Spherical nucleic acids |
|
|
| DNA nanostructures |
|
|
| Stimuli-responsive delivery technologies |
|
|
| Multilayered biopolymer nanofilms |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anwar, S.; Mir, F.; Yokota, T. Enhancing the Effectiveness of Oligonucleotide Therapeutics Using Cell-Penetrating Peptide Conjugation, Chemical Modification, and Carrier-Based Delivery Strategies. Pharmaceutics 2023, 15, 1130. https://doi.org/10.3390/pharmaceutics15041130
Anwar S, Mir F, Yokota T. Enhancing the Effectiveness of Oligonucleotide Therapeutics Using Cell-Penetrating Peptide Conjugation, Chemical Modification, and Carrier-Based Delivery Strategies. Pharmaceutics. 2023; 15(4):1130. https://doi.org/10.3390/pharmaceutics15041130
Chicago/Turabian StyleAnwar, Saeed, Farin Mir, and Toshifumi Yokota. 2023. "Enhancing the Effectiveness of Oligonucleotide Therapeutics Using Cell-Penetrating Peptide Conjugation, Chemical Modification, and Carrier-Based Delivery Strategies" Pharmaceutics 15, no. 4: 1130. https://doi.org/10.3390/pharmaceutics15041130