
Development of a Peptide-Based Nano-Sized Cathepsin B Inhibitor for Anticancer Therapy
 and
and 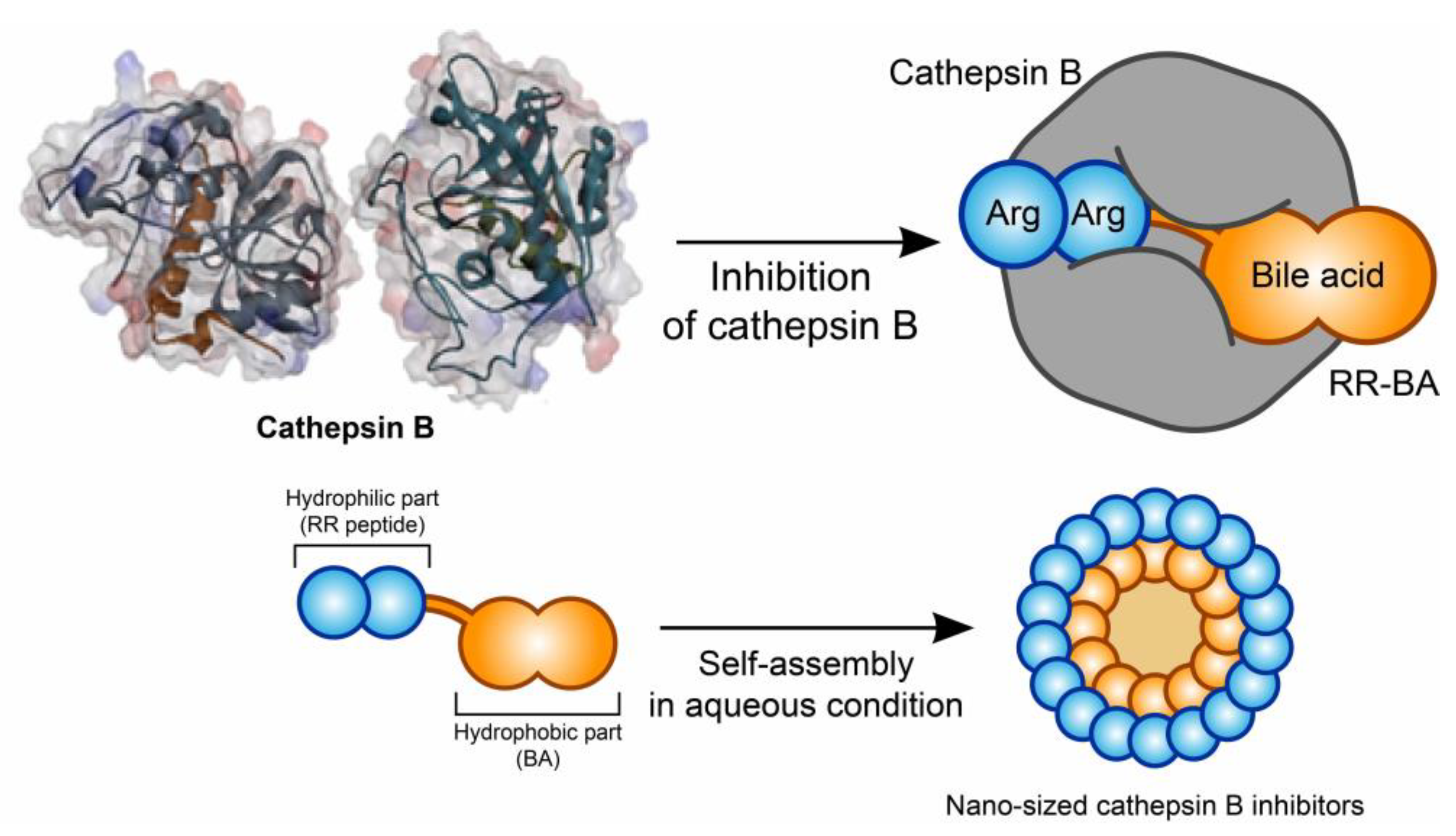{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis
2.3. Characterizations
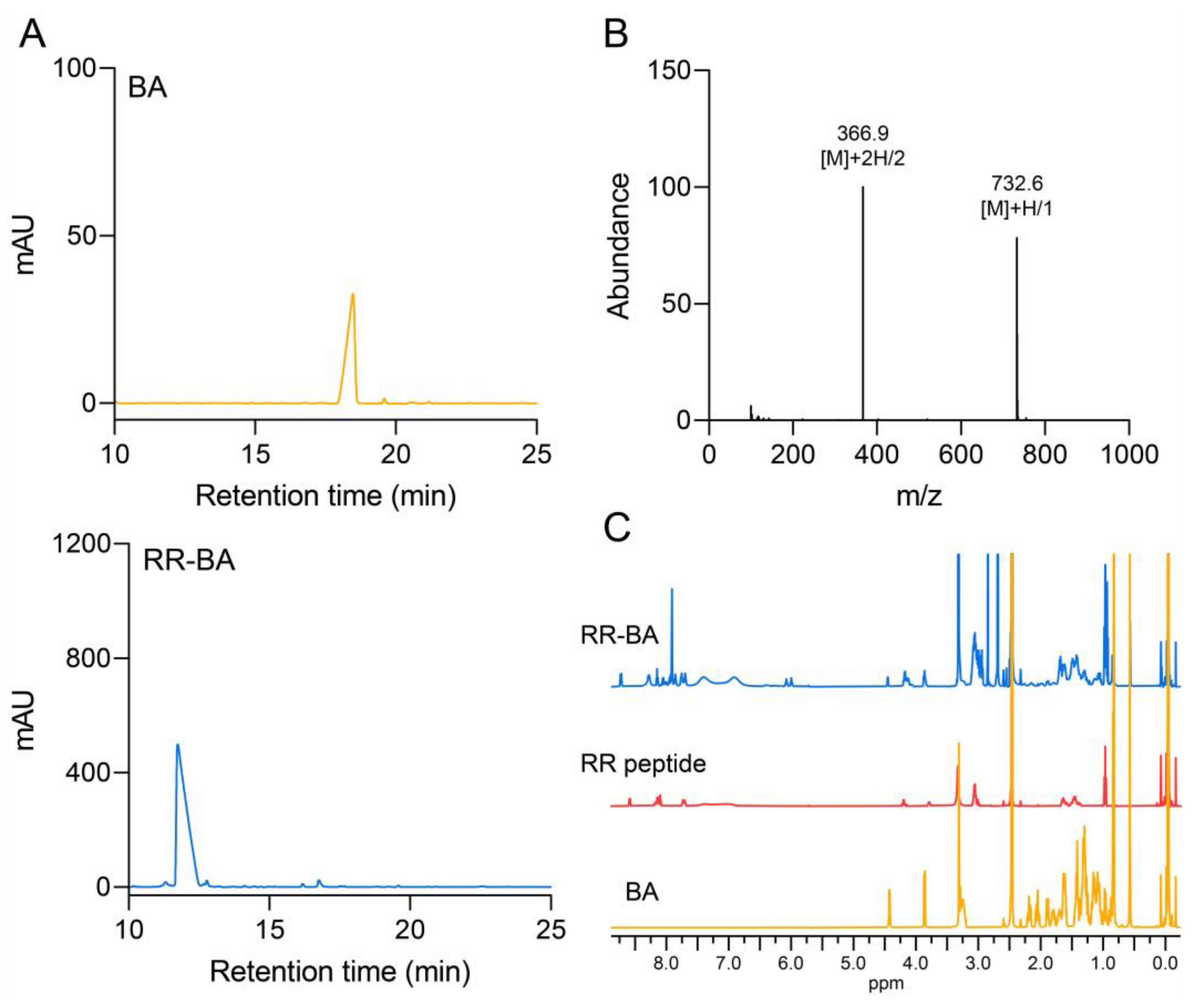
2.3.1. RP-HPLC Analysis
2.3.2. HPLC-MS and NMR Measurement
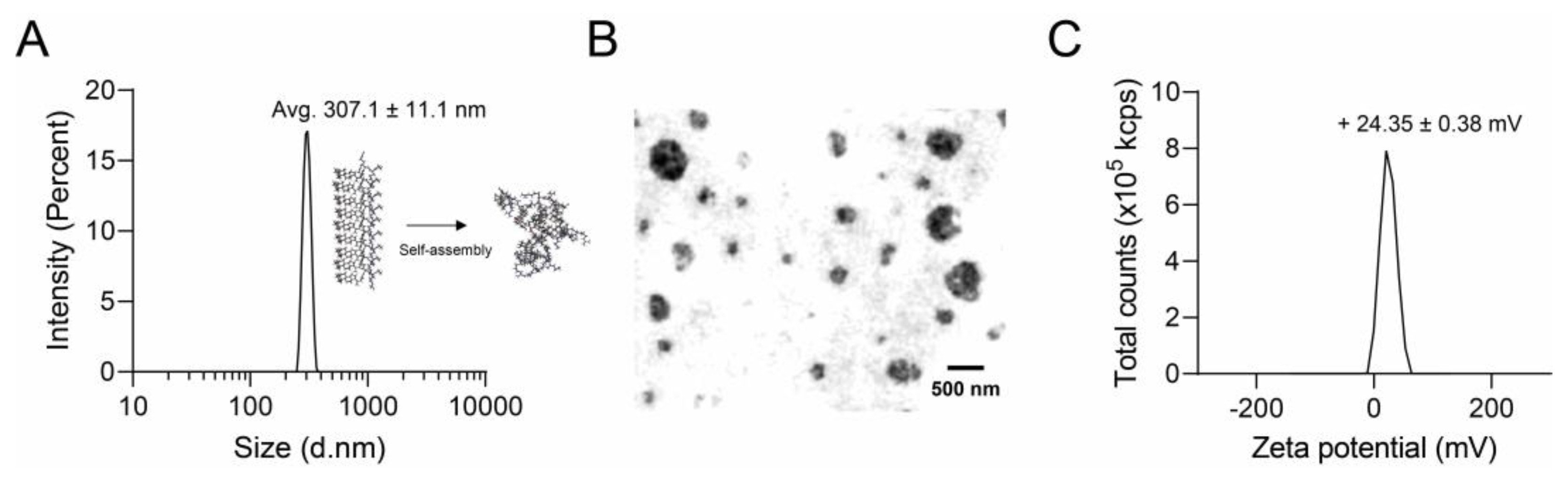
2.4. Nanoparticle Analysis
2.5. Cathepsin B Inhibition Assay
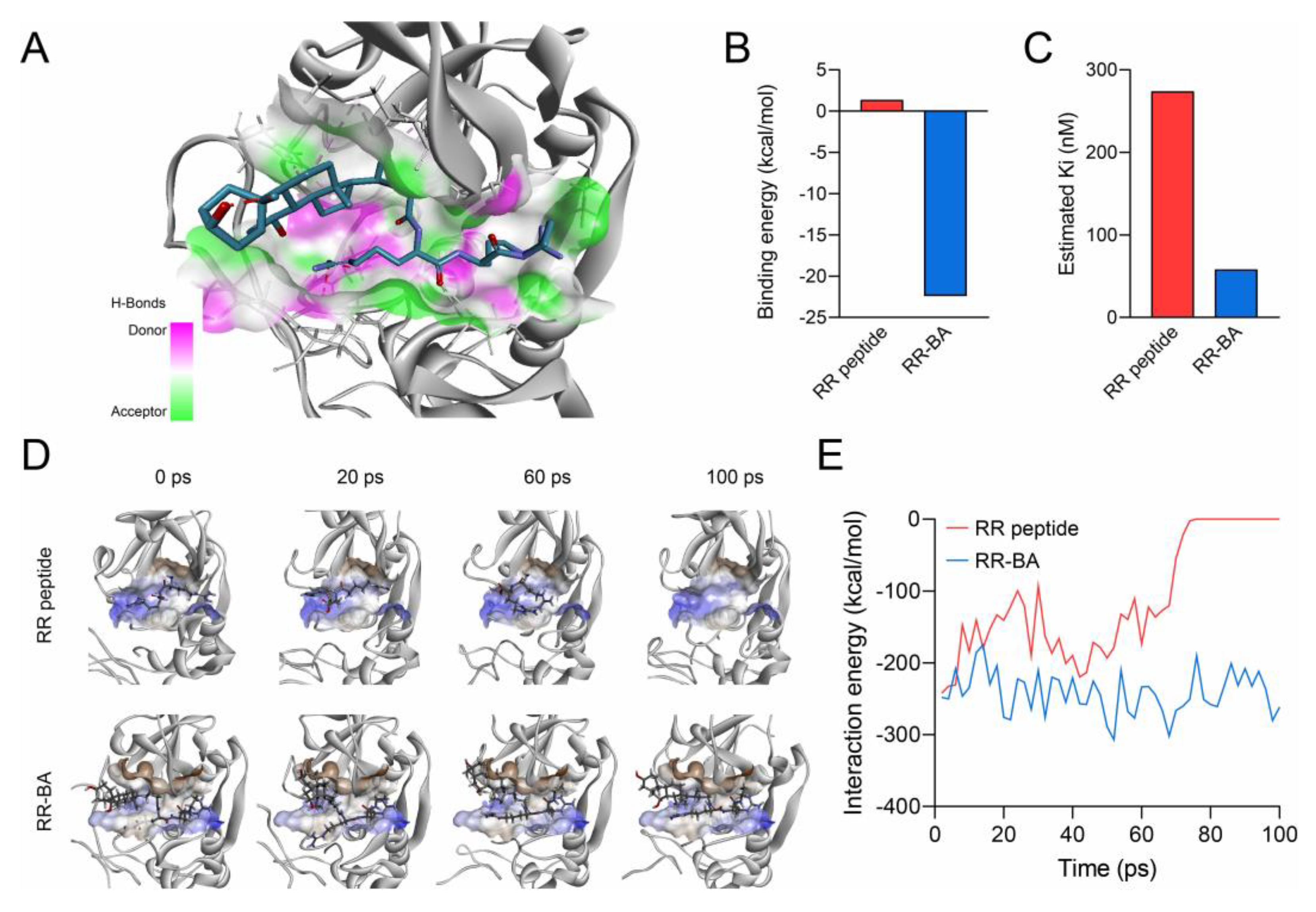
2.6. In Silico Computer Simulation for Molecular Binding
2.7. In Vitro Cytotoxicity Study
2.8. In Vivo Antitumor Effect of RR–BA Nanoparticles in Tumor-Bearing Mice
3. Results and Discussion
3.1. Synthesis and Characterization of RR–BA as a Cathepsin B Inhibitor
3.2. Nano Characterization of RR–BA
3.3. Computer Simulation of RR–BA with Cathepsin B
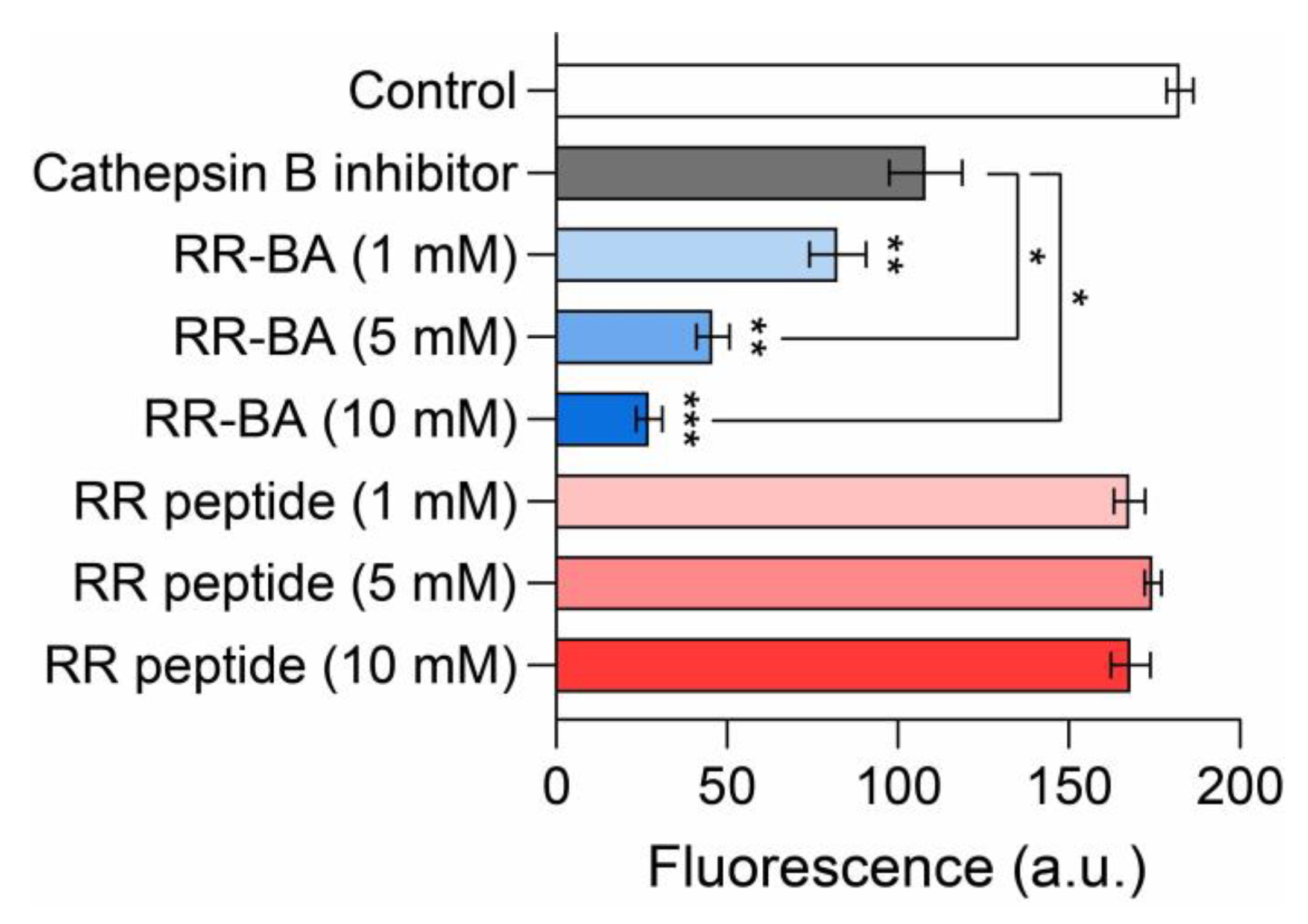
3.4. Inhibition Assay Using a Cathepsin B Activity Assay Kit
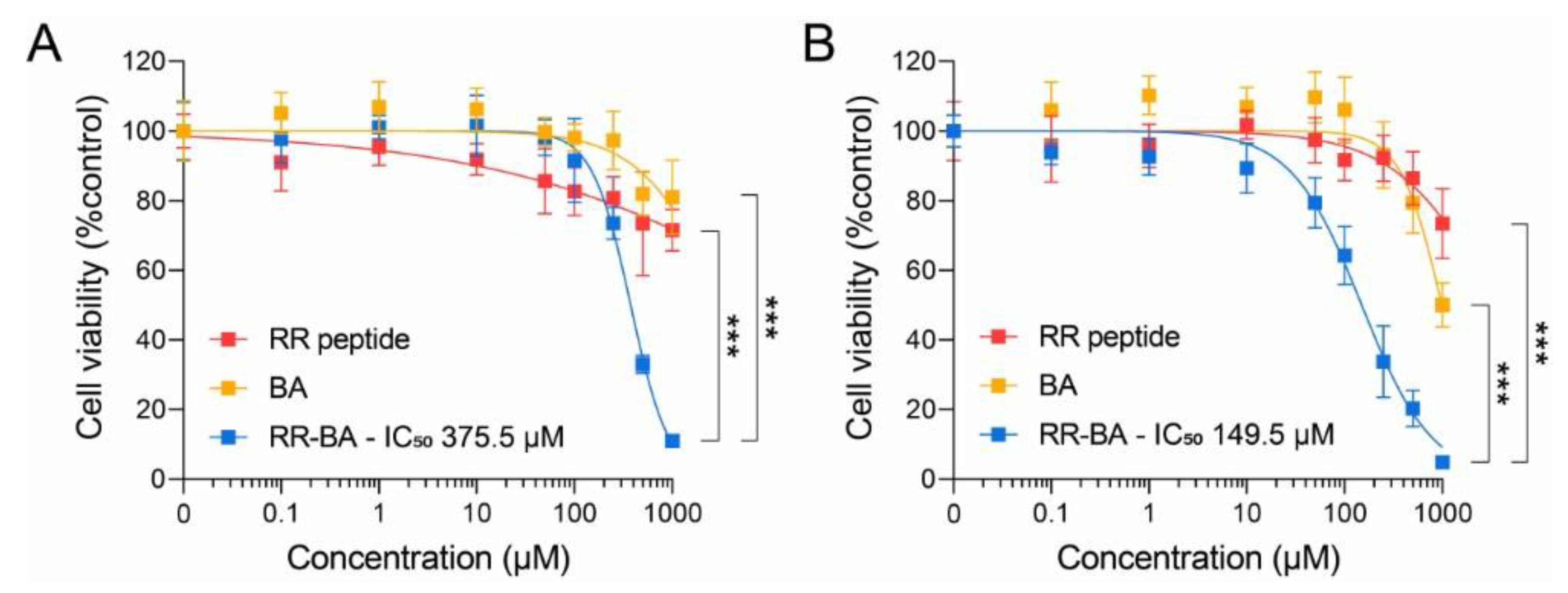
3.5. Cathepsin-B-Dependent Cytotoxic Effect
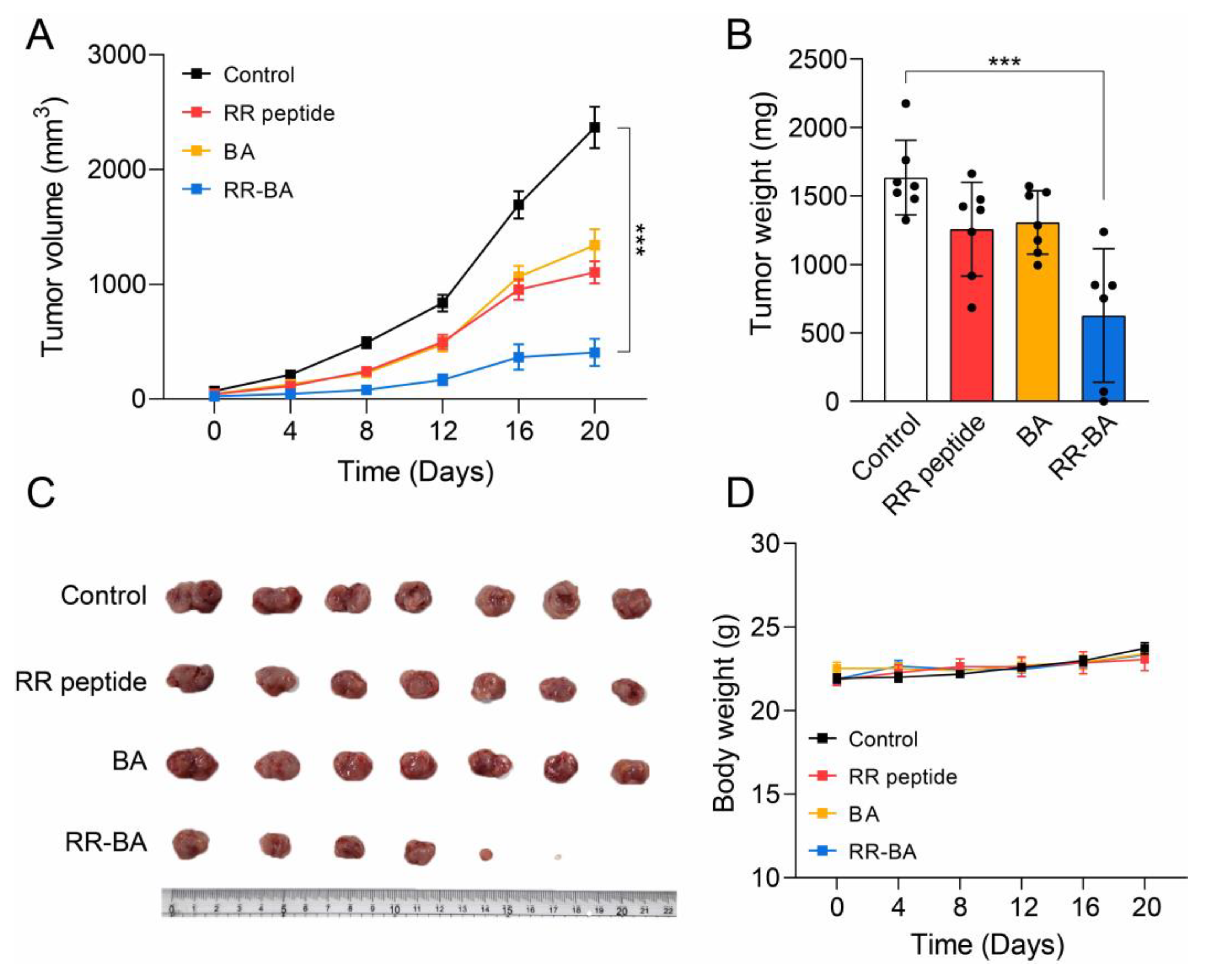
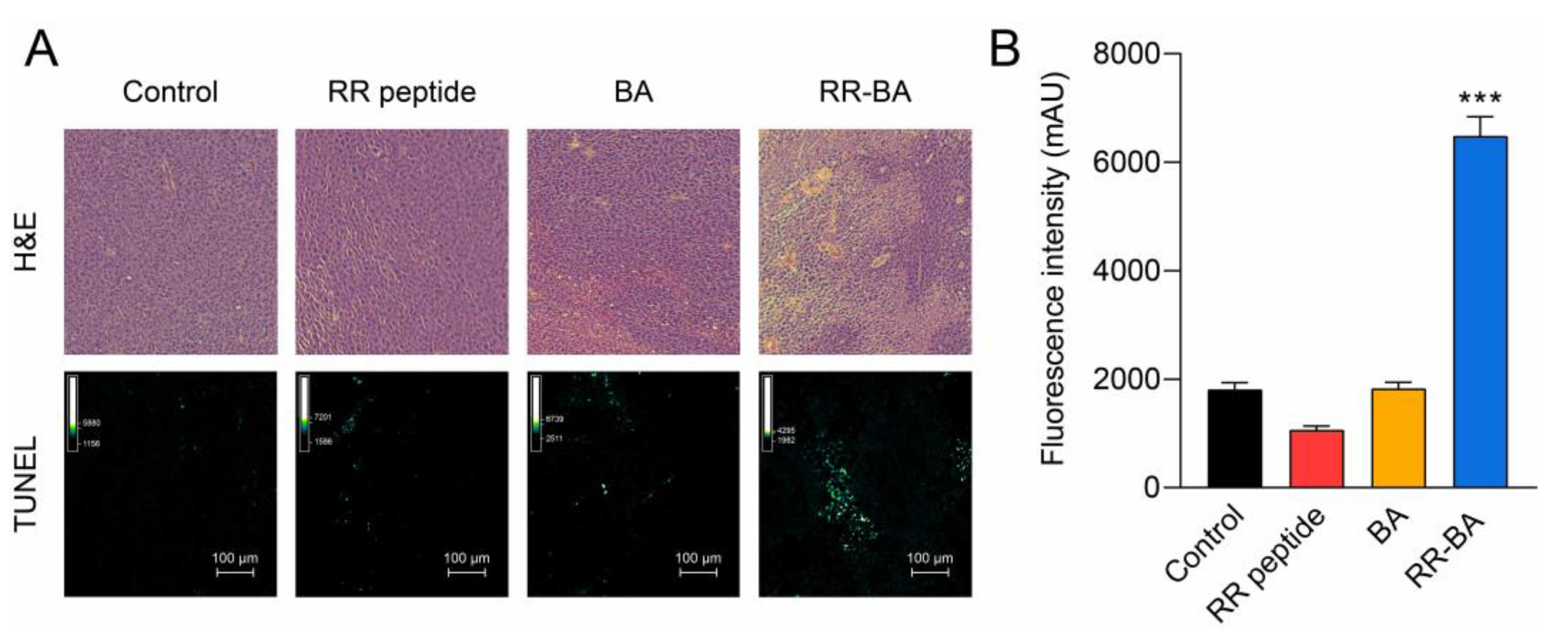
3.6. In Vivo Anticancer Effect of RR–BA in Tumor-Bearing Mice
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ruiz-Blazquez, P.; Pistorio, V.; Fernandez-Fernandez, M.; Moles, A. The multifaceted role of cathepsins in liver disease. J. Hepatol. 2021, 75, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Lakka, S.S.; Gondi, C.S.; Yanamandra, N.; Olivero, W.C.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene 2004, 23, 4681–4689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanamandra, N.; Gumidyala, K.V.; Waldron, K.G.; Gujrati, M.; Olivero, W.C.; Dinh, D.; Rao, J.S.; Mohanam, S. Blockade of cathepsin B expression in human glioblastoma cells is associated with suppression of angiogenesis. Oncogene 2004, 23, 2224–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habibollahi, P.; Figueiredo, J.L.; Heidari, P.; Dulak, A.M.; Imamura, Y.; Bass, A.J.; Ogino, S.; Chan, A.T.; Mahmood, U. Optical Imaging with a Cathepsin B Activated Probe for the Enhanced Detection of Esophageal Adenocarcinoma by Dual Channel Fluorescent Upper GI Endoscopy. Theranostics 2012, 2, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Kryczka, J.; Papiewska-Pajak, I.; Kowalska, M.A.; Boncela, J. Cathepsin B Is Upregulated and Mediates ECM Degradation in Colon Adenocarcinoma HT29 Cells Overexpressing Snail. Cells 2019, 8, 203. [Google Scholar] [CrossRef] [Green Version]
- Ruan, J.; Zheng, H.; Rong, X.; Rong, X.; Zhang, J.; Fang, W.; Zhao, P.; Luo, R. Over-expression of cathepsin B in hepatocellular carcinomas predicts poor prognosis of HCC patients. Mol. Cancer 2016, 15, 17. [Google Scholar] [CrossRef] [Green Version]
- Mijanovic, O.; Brankovic, A.; Panin, A.N.; Savchuk, S.; Timashev, P.; Ulasov, I.; Lesniak, M.S. Cathepsin B: A sellsword of cancer progression. Cancer Lett. 2019, 449, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Gornowicz, A.; Szymanowska, A.; Mojzych, M.; Czarnomysy, R.; Bielawski, K.; Bielawska, A. The Anticancer Action of a Novel 1,2,4-Triazine Sulfonamide Derivative in Colon Cancer Cells. Molecules 2021, 26, 2045. [Google Scholar] [CrossRef]
- Shen, Y.K.; Li, X. Cathepsin B as a target in cancer therapy and imaging. New J. Chem. 2022, 46, 19593–19611. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, S.; Ma, P.; Jiang, Y.; Cheng, K.; Yu, Y.; Jiang, N.; Miao, H.; Tang, Q.; Liu, F.; et al. Drug conjugate-based anticancer therapy—Current status and perspectives. Cancer Lett. 2023, 552, 215969. [Google Scholar] [CrossRef]
- Ruan, H.; Hao, S.; Young, P.; Zhang, H. Targeting Cathepsin B for Cancer Therapies. Horiz. Cancer Res. 2015, 56, 23–40. [Google Scholar] [PubMed]
- Zhong, Y.J.; Shao, L.H.; Li, Y. Cathepsin B-cleavable doxorubicin prodrugs for targeted cancer therapy (Review). Int. J. Oncol. 2013, 42, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Song, S.J.; Lee, J.; Ha, T.H.; Choi, J.S. Cathepsin B-Responsive Liposomes for Controlled Anticancer Drug Delivery in Hep G2 Cells. Pharmaceutics 2020, 12, 876. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Choi, J.S. Enzyme-Responsive Amphiphilic Peptide Nanoparticles for Biocompatible and Efficient Drug Delivery. Pharmaceutics 2022, 14, 143. [Google Scholar] [CrossRef] [PubMed]
- Kratz, F.; Muller, I.A.; Ryppa, C.; Warnecke, A. Prodrug strategies in anticancer chemotherapy. ChemMedChem 2008, 3, 20–53. [Google Scholar] [CrossRef]
- Thanou, M.; Duncan, R. Polymer-protein and polymer-drug conjugates in cancer therapy. Curr. Opin. Investig. Drugs 2003, 4, 701–709. [Google Scholar]
- Cho, H.; Shim, M.K.; Moon, Y.; Song, S.; Kim, J.; Choi, J.; Kim, J.; Lee, Y.; Park, J.Y.; Kim, Y.; et al. Tumor-Specific Monomethyl Auristatin E (MMAE) Prodrug Nanoparticles for Safe and Effective Chemotherapy. Pharmaceutics 2022, 14, 2131. [Google Scholar] [CrossRef]
- Jain, M.; Bouilloux, J.; Borrego, I.; Cook, S.; van den Bergh, H.; Lange, N.; Wagnieres, G.; Giraud, M.N. Cathepsin B-Cleavable Polymeric Photosensitizer Prodrug for Selective Photodynamic Therapy: In Vitro Studies. Pharmaceuticals 2022, 15, 564. [Google Scholar] [CrossRef]
- Raghuram, S.; Mackeyev, Y.; Symons, J.; Zahra, Y.; Gonzalez, V.; Mahadevan, K.K.; Requejo, K.I.; Liopo, A.; Derry, P.; Zubarev, E.; et al. Uncloaking cell-impermeant gold nanorods via tumor microenvironmental cathepsin B facilitates cancer cell penetration and potent radiosensitization. Biomaterials 2022, 291, 121887. [Google Scholar] [CrossRef]
- Moskowitz, C.H.; Nademanee, A.; Masszi, T. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 385, 1853–1862, Erratum in Lancet 2015, 386, 532. [Google Scholar] [CrossRef]
- Yang, S.B.; Banik, N.; Han, B.; Lee, D.N.; Park, J. Peptide-Based Bioconjugates and Therapeutics for Targeted Anticancer Therapy. Pharmaceutics 2022, 14, 1378. [Google Scholar] [CrossRef]
- Cho, H.; Shim, M.K.; Yang, S.; Song, S.; Moon, Y.; Kim, J.; Byun, Y.; Ahn, C.H.; Kim, K. Cathepsin B-Overexpressed Tumor Cell Activatable Albumin-Binding Doxorubicin Prodrug for Cancer-Targeted Therapy. Pharmaceutics 2021, 14, 83. [Google Scholar] [CrossRef]
- Shim, M.K.; Park, J.; Yoon, H.Y.; Lee, S.; Um, W.; Kim, J.H.; Kang, S.W.; Seo, J.W.; Hyun, S.W.; Park, J.H.; et al. Carrier-free nanoparticles of cathepsin B-cleavable peptide-conjugated doxorubicin prodrug for cancer targeting therapy. J. Control. Release 2019, 294, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Shim, N.; Jeon, S.I.; Yang, S.; Park, J.Y.; Jo, M.; Kim, J.; Choi, J.; Yun, W.S.; Kim, J.; Lee, Y.; et al. Comparative study of cathepsin B-cleavable linkers for the optimal design of cathepsin B-specific doxorubicin prodrug nanoparticles for targeted cancer therapy. Biomaterials 2022, 289, 121806. [Google Scholar] [CrossRef]
- Jeon, S.I.; Yang, S.; Shim, M.K.; Kim, K. Cathepsin B-responsive prodrugs for cancer-targeted therapy: Recent advances and progress for clinical translation. Nano Res. 2022, 15, 7247–7266. [Google Scholar] [CrossRef]
- Liow, K.Y.; Chow, S.C. The cathepsin B inhibitor z-FA-CMK induces cell death in leukemic T cells via oxidative stress. Naunyn Schmiedebergs Arch. Pharmacol. 2018, 391, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Gondi, C.S.; Rao, J.S. Cathepsin B as a cancer target. Expert. Opin. Ther. Tar. 2013, 17, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Azziz, I.A.; Amin, N.H.; El-Saadi, M.T.; Abdel-Rahman, H.M. Design, synthesis and mechanistic studies of benzophenones hydrazone derivatives as cathepsin inhibitors. J. Mol. Struct. 2023, 1274, 134583. [Google Scholar] [CrossRef]
- Schmitz, J.; Li, T.; Bartz, U.; Gutschow, M. Cathepsin B Inhibitors: Combining Dipeptide Nitriles with an Occluding Loop Recognition Element by Click Chemistry. ACS Med. Chem. Lett. 2016, 7, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Frlan, R.; Gobec, S. Inhibitors of cathepsin B. Curr. Med. Chem. 2006, 13, 2309–2327. [Google Scholar]
- Yamamoto, A.; Tomoo, K.; Hara, T.; Murata, M.; Kitamura, K.; Ishida, T. Substrate specificity of bovine cathepsin B and its inhibition by CA074, based on crystal structure refinement of the complex. J. Biochem. 2000, 127, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, U.; Weigert, M.; Broaddus, C.; Myers, G. Cell Detection with Star-Convex Polygons. Lect. Notes Comput. Sci. 2018, 11071, 265–273. [Google Scholar]
- Kos, J.; Mitrovic, A.; Mirkovic, B. The current stage of cathepsin B inhibitors as potential anticancer agents. Future Med. Chem. 2014, 6, 1355–1371. [Google Scholar] [CrossRef]
- Torchilin, V.P. Drug targeting. Eur. J. Pharm. Sci. 2000, 11 (Suppl. 2), S81–S91. [Google Scholar] [CrossRef]
- Park, J.; Choi, Y.; Chang, H.; Um, W.; Ryu, J.H.; Kwon, I.C. Alliance with EPR Effect: Combined Strategies to Improve the EPR Effect in the Tumor Microenvironment. Theranostics 2019, 9, 8073–8090. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Goossens, J.F.; Bailly, C. Ursodeoxycholic acid and cancer: From chemoprevention to chemotherapy. Pharmacol. Ther. 2019, 203, 107396. [Google Scholar] [CrossRef]
- Shen, Y.; Lu, C.; Song, Z.; Qiao, C.; Wang, J.; Chen, J.; Zhang, C.; Zeng, X.; Ma, Z.; Chen, T.; et al. Ursodeoxycholic acid reduces antitumor immunosuppression by inducing CHIP-mediated TGF-beta degradation. Nat. Commun. 2022, 13, 3419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, H.J.; Zhang, C.L.; Tang, Q.L.; Bi, F. Ursodeoxycholic acid suppresses the malignant progression of colorectal cancer through TGR5-YAP axis. Cell. Death Discov. 2021, 7, 1–12. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.-H.; Lee, J.-H.; Yang, S.-B.; Lee, D.-N.; Kang, T.-B.; Park, J. Development of a Peptide-Based Nano-Sized Cathepsin B Inhibitor for Anticancer Therapy. Pharmaceutics 2023, 15, 1131. https://doi.org/10.3390/pharmaceutics15041131
Park S-H, Lee J-H, Yang S-B, Lee D-N, Kang T-B, Park J. Development of a Peptide-Based Nano-Sized Cathepsin B Inhibitor for Anticancer Therapy. Pharmaceutics. 2023; 15(4):1131. https://doi.org/10.3390/pharmaceutics15041131
Chicago/Turabian StylePark, So-Hyeon, Jun-Hyuck Lee, Seong-Bin Yang, Dong-Nyeong Lee, Tae-Bong Kang, and Jooho Park. 2023. "Development of a Peptide-Based Nano-Sized Cathepsin B Inhibitor for Anticancer Therapy" Pharmaceutics 15, no. 4: 1131. https://doi.org/10.3390/pharmaceutics15041131