
mRNA-Loaded Lipid Nanoparticles Targeting Dendritic Cells for Cancer Immunotherapy


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Mice and Cell Cultures
2.3. In Vitro Transcription of mRNA
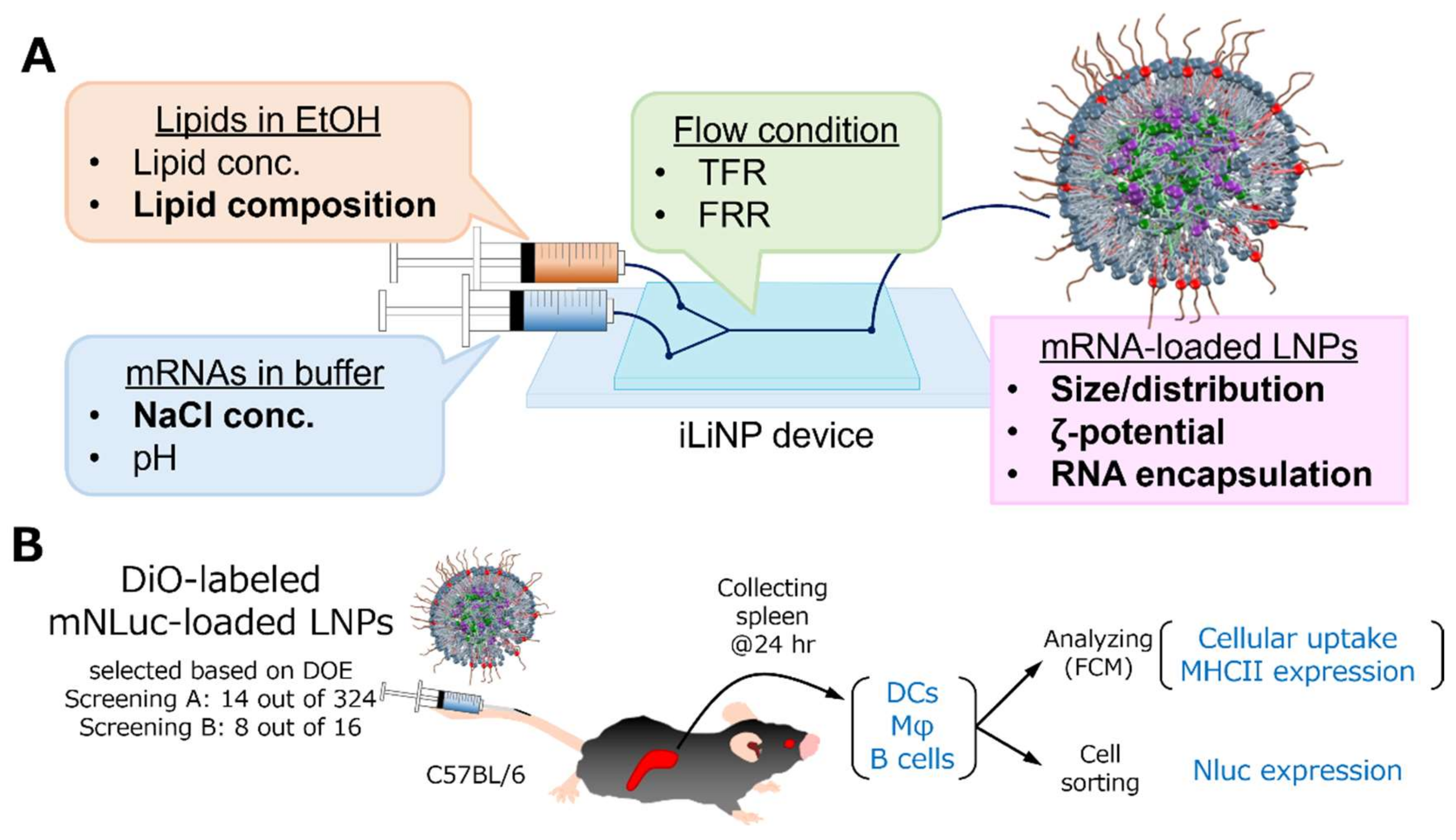
2.4. Preparation of RNA-Loaded LNPs
2.5. Preparation of RNA-Lipoplexes (RNA-LPX)
2.6. Characterization of RNA-Loaded LNPs
2.7. In Vivo Screening of RNA-Loaded LNPs
2.8. Comparison of Transgene Expression Activity
2.9. Measurement of Activation Markers in Splenocytes
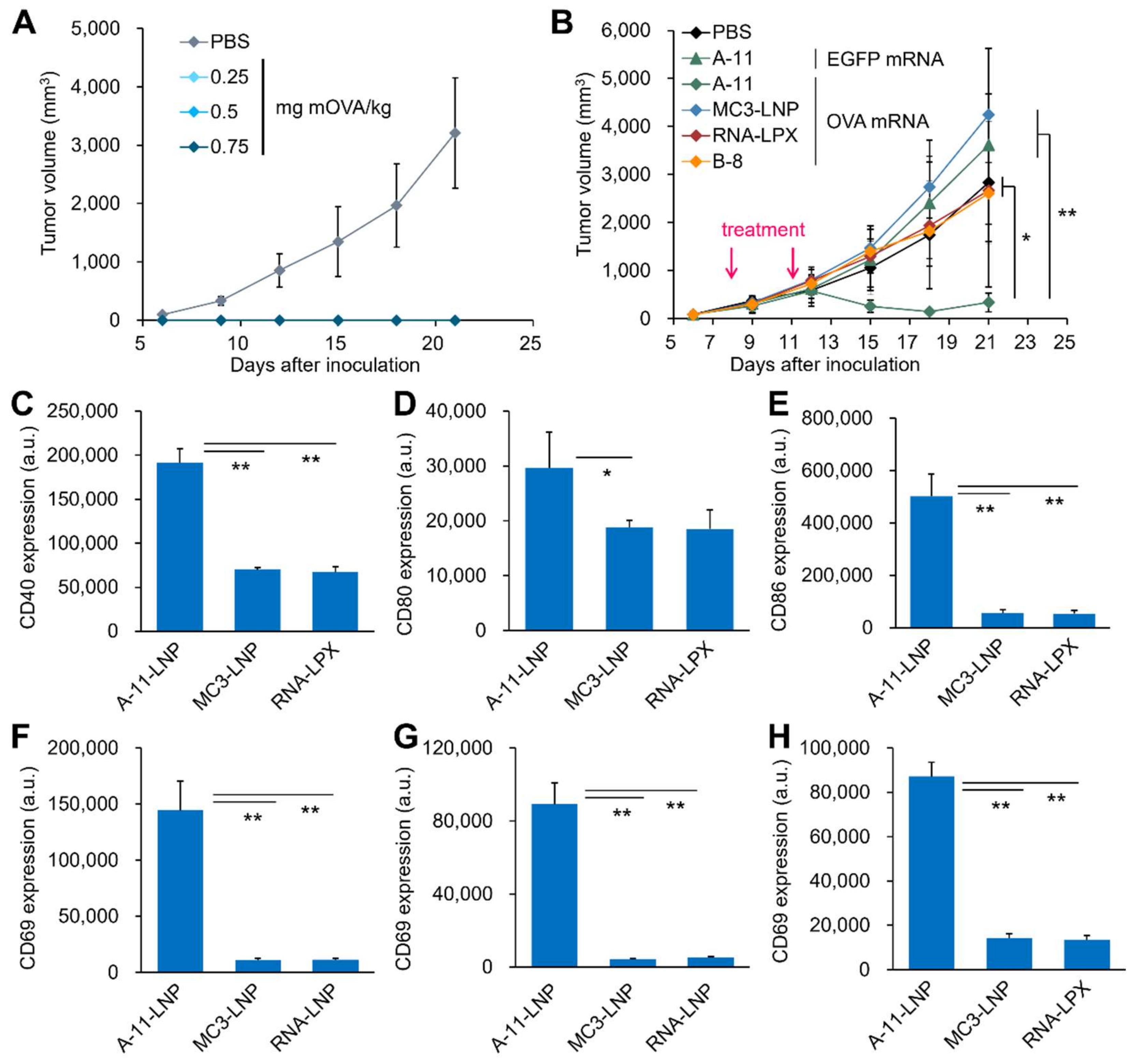
2.10. Prophylactic and Therapeutic Antitumor Effect of mRNA-Loaded LNPs on Mice
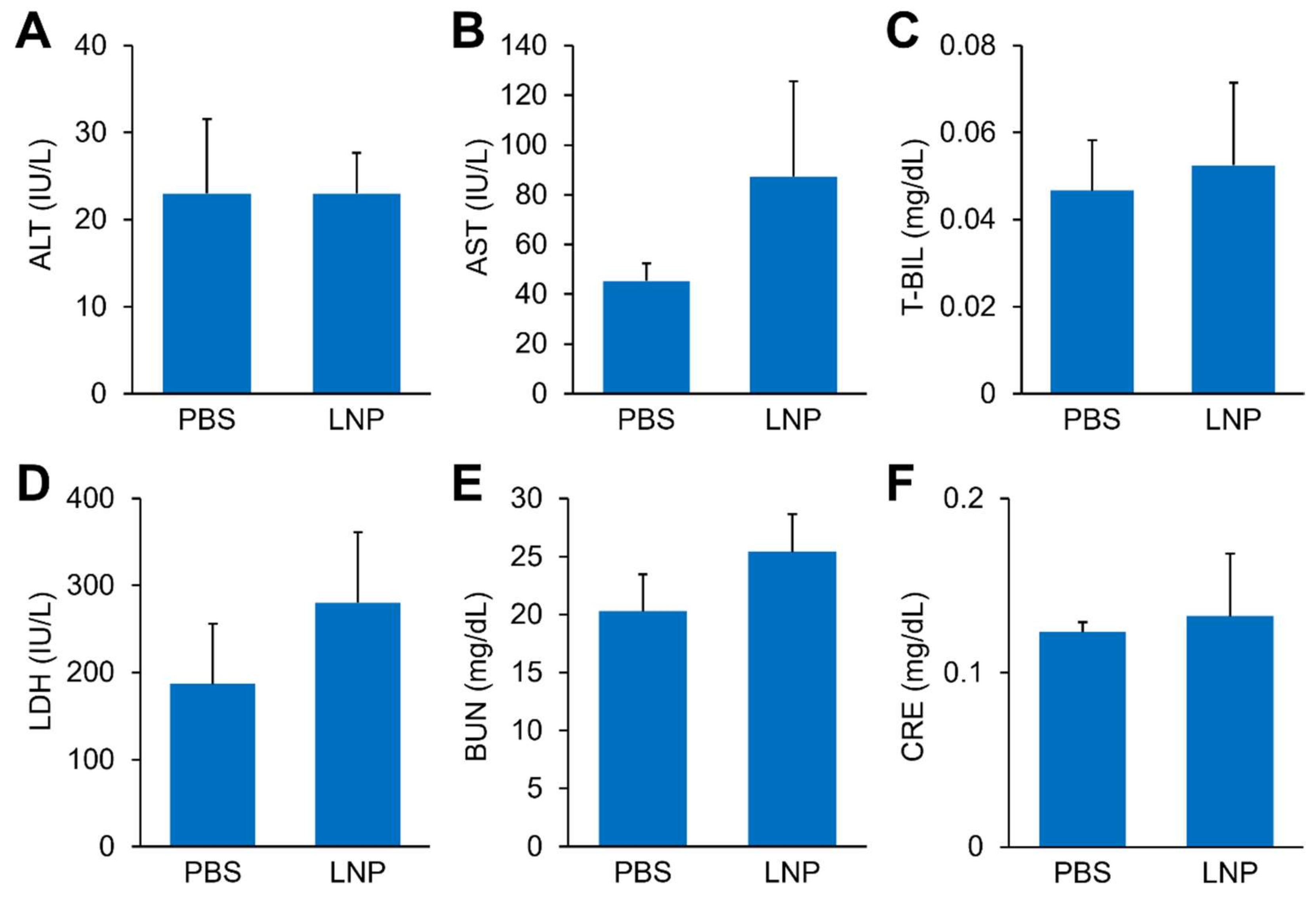
2.11. Toxicity Test
2.12. Statistical Analyses
3. Results
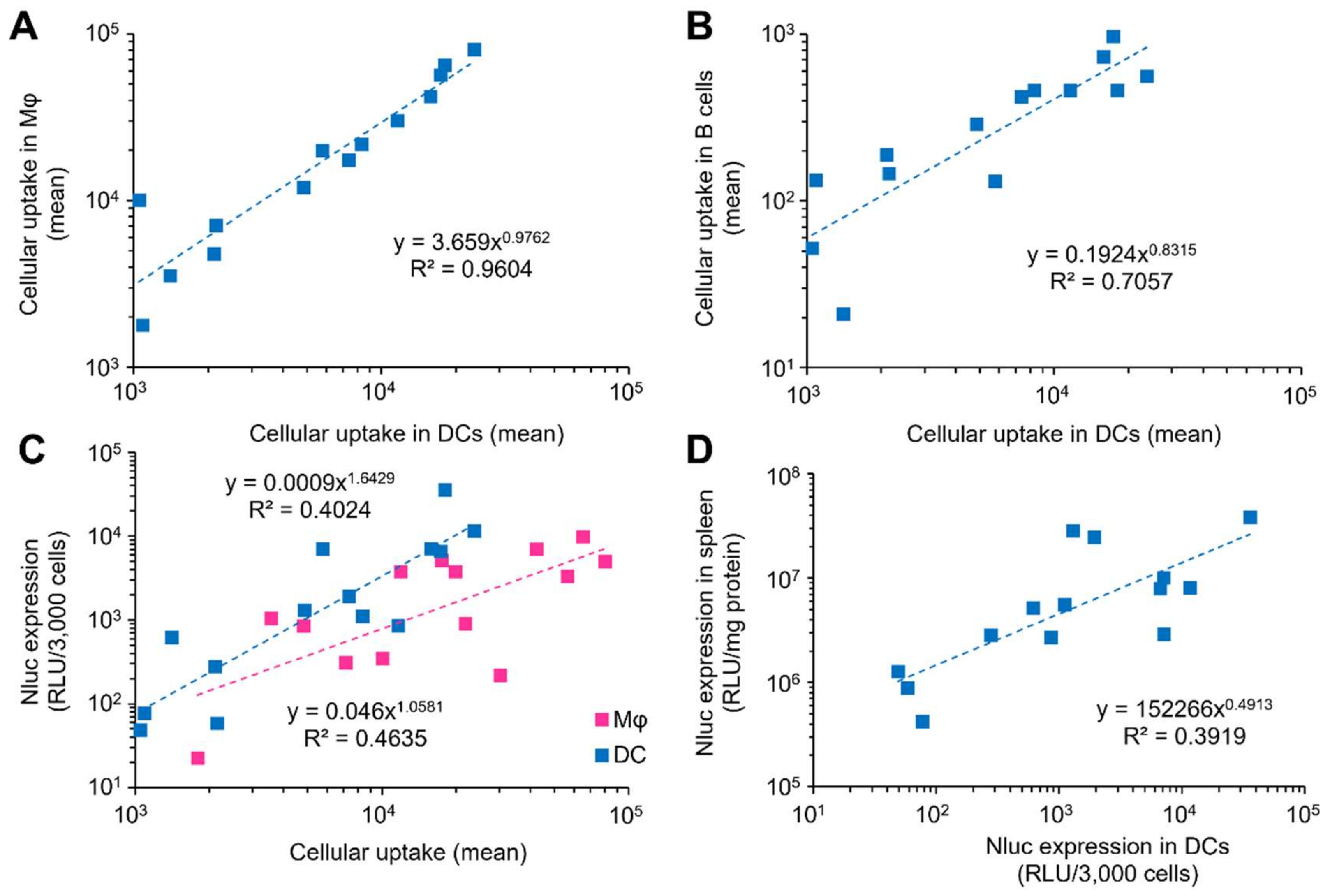
3.1. Optimization Strategy for Splenic DCs
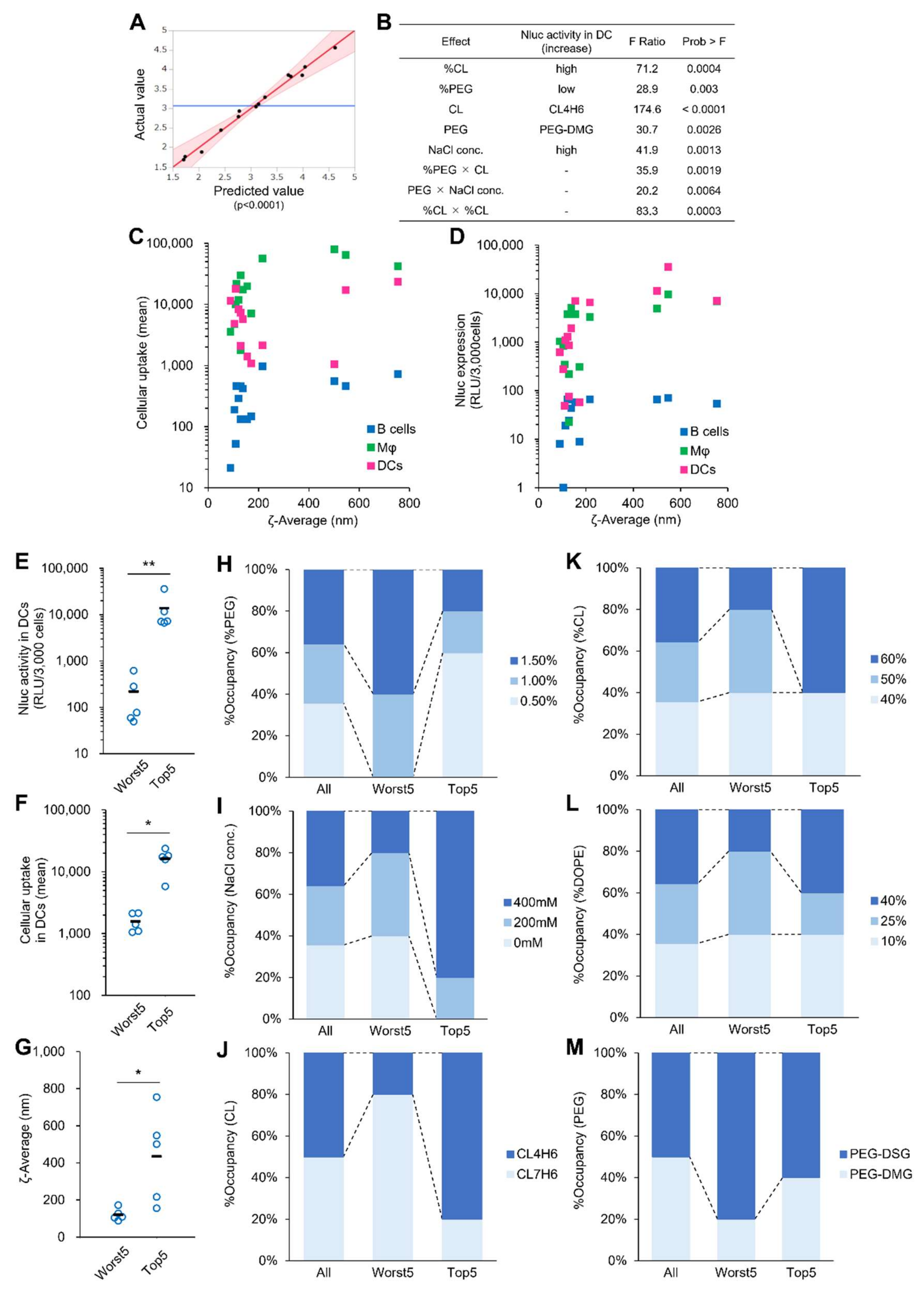
3.2. DOE-Based Optimization of LNPs
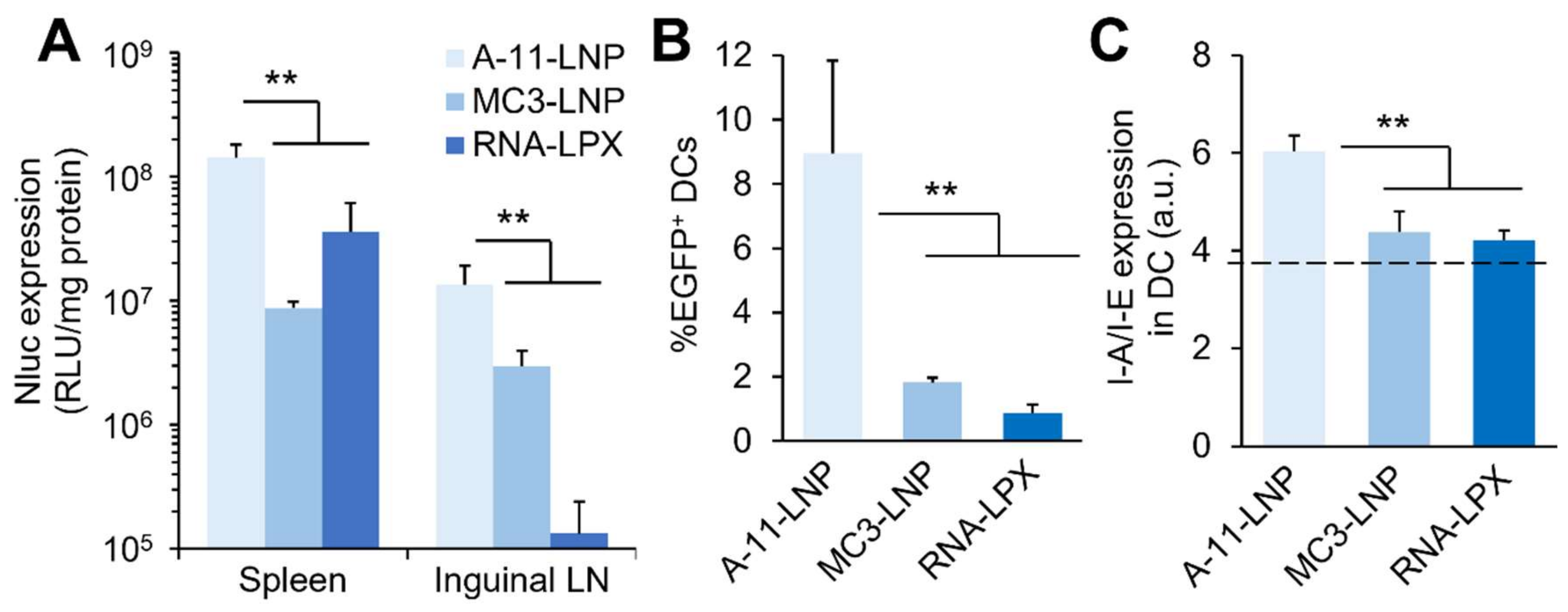
3.3. Comparison with Clinically Relevant Formulations
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moingeon, P.; Lombardi, V.; Saint-Lu, N.; Tourdot, S.; Bodo, V.; Mascarell, L. Adjuvants and Vector Systems for Allergy Vaccines. Immunol. Allergy Clin. N. Am. 2011, 31, 407–419. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Bogers, W.M.; Oostermeijer, H.; Mooij, P.; Koopman, G.; Verschoor, E.J.; Davis, D.; Ulmer, J.B.; Brito, L.A.; Cu, Y.; Banerjee, K.; et al. Potent Immune Responses in Rhesus Macaques Induced by Nonviral Delivery of a Self-amplifying RNA Vaccine Expressing HIV Type 1 Envelope with a Cationic Nanoemulsion. J. Infect. Dis. 2014, 211, 947–955. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Richner, J.M.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.M.; Julander, J.G.; Tang, W.W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell 2017, 168, 1114–1125.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, Y.N. BNT162b2 mRNA COVID-19 Vaccine: First Approval. Drugs 2021, 81, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Topol, E.J. Messenger RNA vaccines against SARS-CoV-2. Cell 2021, 184, 1401. [Google Scholar] [CrossRef]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J.A. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.; Kim, M.; Seo, Y.; Moon, Y.S.; Lee, H.J.; Lee, K.; Lee, H. Emergence of synthetic mRNA: In vitro synthesis of mRNA and its applications in regenerative medicine. Biomaterials 2018, 156, 172–193. [Google Scholar] [CrossRef] [PubMed]
- Pollard, C.; Rejman, J.; De Haes, W.; Verrier, B.; Van Gulck, E.; Naessens, T.; De Smedt, S.; Bogaert, P.; Grooten, J.; Vanham, G.; et al. Type I IFN Counteracts the Induction of Antigen-Specific Immune Responses by Lipid-Based Delivery of mRNA Vaccines. Mol. Ther. 2013, 21, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Qu, S.; Chen, X.; Wu, Q.; Shi, M. Promising Targets for Cancer Immunotherapy: TLRs, RLRs, and STING-Mediated Innate Immune Pathways. Int. J. Mol. Sci. 2017, 18, 404. [Google Scholar] [CrossRef] [PubMed]
- Rettig, L.; Haen, S.P.; Bittermann, A.G.; von Boehmer, L.; Curioni, A.; Krämer, S.D.; Knuth, A.; Pascolo, S. Particle size and activation threshold: A new dimension of danger signaling. Blood 2010, 115, 4533–4541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, T.; Luft, D.; Abraham, M.K.; Reinhardt, S.; Salinas Medina, M.L.; Kurz, J.; Schaller, M.; Avci-Adali, M.; Schlensak, C.; Peter, K.; et al. Cationic Nanoliposomes Meet mRNA: Efficient Delivery of Modified mRNA Using Hemocompatible and Stable Vectors for Therapeutic Applications. Mol. Ther. Nucleic Acids 2017, 8, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habrant, D.; Peuziat, P.; Colombani, T.; Dallet, L.; Gehin, J.; Goudeau, E.; Evrard, B.; Lambert, O.; Haudebourg, T.; Pitard, B. Design of Ionizable Lipids To Overcome the Limiting Step of Endosomal Escape: Application in the Intracellular Delivery of mRNA, DNA, and siRNA. J. Med. Chem. 2016, 59, 3046–3062. [Google Scholar] [CrossRef]
- Hajj, K.A.; Melamed, J.R.; Chaudhary, N.; Lamson, N.G.; Ball, R.L.; Yerneni, S.S.; Whitehead, K.A. A Potent Branched-Tail Lipid Nanoparticle Enables Multiplexed mRNA Delivery and Gene Editing In Vivo. Nano Lett. 2020, 20, 5167–5175. [Google Scholar] [CrossRef]
- Miao, L.; Li, L.; Huang, Y.; Delcassian, D.; Chahal, J.; Han, J.; Shi, Y.; Sadtler, K.; Gao, W.; Lin, J.; et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol. 2019, 37, 1174–1185. [Google Scholar] [CrossRef]
- Cornebise, M.; Narayanan, E.; Xia, Y.; Acosta, E.; Ci, L.; Koch, H.; Milton, J.; Sabnis, S.; Salerno, T.; Benenato, K.E. Discovery of a Novel Amino Lipid That Improves Lipid Nanoparticle Performance through Specific Interactions with mRNA. Adv. Funct. Mater. 2022, 32, 2106727. [Google Scholar] [CrossRef]
- Semple, S.C.; Chonn, A.; Cullis, P.R. Influence of cholesterol on the association of plasma proteins with liposomes. Biochemistry 1996, 35, 2521–2525. [Google Scholar] [CrossRef] [PubMed]
- Senior, J.; Gregoriadis, G. Stability of small unilamellar liposomes in serum and clearance from the circulation: The effect of the phospholipid and cholesterol components. Life Sci. 1982, 30, 2123–2136. [Google Scholar] [CrossRef]
- Schroeder, A.; Levins, C.G.; Cortez, C.; Langer, R.; Anderson, D.G. Lipid-based nanotherapeutics for siRNA delivery. J. Intern. Med. 2010, 267, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Mui, B.L.; Tam, Y.K.; Jayaraman, M.; Ansell, S.M.; Du, X.; Tam, Y.Y.; Lin, P.J.; Chen, S.; Narayanannair, J.K.; Rajeev, K.G.; et al. Influence of Polyethylene Glycol Lipid Desorption Rates on Pharmacokinetics and Pharmacodynamics of siRNA Lipid Nanoparticles. Mol. Ther. Nucleic Acids 2013, 2, e139. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Note, Y.; Maeki, M.; Kaji, N.; Baba, Y.; Tokeshi, M.; Harashima, H. Elucidation of the physicochemical properties and potency of siRNA-loaded small-sized lipid nanoparticles for siRNA delivery. J. Control. Release 2016, 229, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y. Development of Lipid Nanoparticles for the Delivery of Macromolecules Based on the Molecular Design of pH-Sensitive Cationic Lipids. Chem. Pharm. Bull. 2021, 69, 1141–1159. [Google Scholar] [CrossRef] [PubMed]
- Semple, S.C.; Klimuk, S.K.; Harasym, T.O.; Dos Santos, N.; Ansell, S.M.; Wong, K.F.; Maurer, N.; Stark, H.; Cullis, P.R.; Hope, M.J.; et al. Efficient encapsulation of antisense oligonucleotides in lipid vesicles using ionizable aminolipids: Formation of novel small multilamellar vesicle structures. Biochim. Biophys. Acta 2001, 1510, 152–166. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Hatakeyama, H.; Hyodo, M.; Harashima, H. Relationship Between the Physicochemical Properties of Lipid Nanoparticles and the Quality of siRNA Delivery to Liver Cells. Mol. Ther. 2016, 24, 788–795. [Google Scholar] [CrossRef] [Green Version]
- Hafez, I.M.; Maurer, N.; Cullis, P.R. On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Ther. 2001, 8, 1188–1196. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Hatakeyama, H.; Sakurai, Y.; Hyodo, M.; Akita, H.; Harashima, H. A pH-sensitive cationic lipid facilitates the delivery of liposomal siRNA and gene silencing activity in vitro and in vivo. J. Control. Release 2012, 163, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashiba, A.; Toyooka, M.; Sato, Y.; Maeki, M.; Tokeshi, M.; Harashima, H. The use of design of experiments with multiple responses to determine optimal formulations for in vivo hepatic mRNA delivery. J. Control. Release 2020, 327, 467–476. [Google Scholar] [CrossRef]
- Suzuki, Y.; Onuma, H.; Sato, R.; Sato, Y.; Hashiba, A.; Maeki, M.; Tokeshi, M.; Kayesh, M.E.H.; Kohara, M.; Tsukiyama-Kohara, K.; et al. Lipid nanoparticles loaded with ribonucleoprotein-oligonucleotide complexes synthesized using a microfluidic device exhibit robust genome editing and hepatitis B virus inhibition. J. Control. Release 2021, 330, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Shobaki, N.; Sato, Y.; Harashima, H. Mixing lipids to manipulate the ionization status of lipid nanoparticles for specific tissue targeting. Int. J. Nanomed. 2018, 13, 8395–8410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef]
- Warashina, S.; Nakamura, T.; Sato, Y.; Fujiwara, Y.; Hyodo, M.; Hatakeyama, H.; Harashima, H. A lipid nanoparticle for the efficient delivery of siRNA to dendritic cells. J. Control. Release 2016, 225, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Z.; Zhao, X.; Song, X.R. Ex vivo pulsed dendritic cell vaccination against cancer. Acta Pharm. Sin. 2020, 41, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Tacken, P.J.; de Vries, I.J.M.; Torensma, R.; Figdor, C.G. Dendritic-cell immunotherapy: From ex vivo loading to in vivo targeting. Nat. Rev. Immunol. 2007, 7, 790–802. [Google Scholar] [CrossRef]
- Phua, K.K. Towards Targeted Delivery Systems: Ligand Conjugation Strategies for mRNA Nanoparticle Tumor Vaccines. J. Immunol. Res. 2015, 2015, 680620. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Hashiba, K.; Sasaki, K.; Maeki, M.; Tokeshi, M.; Harashima, H. Understanding structure-activity relationships of pH-sensitive cationic lipids facilitates the rational identification of promising lipid nanoparticles for delivering siRNAs in vivo. J. Control. Release 2019, 295, 140–152. [Google Scholar] [CrossRef]
- Okuda, K.; Sato, Y.; Iwakawa, K.; Sasaki, K.; Okabe, N.; Maeki, M.; Tokeshi, M.; Harashima, H. On the size-regulation of RNA-loaded lipid nanoparticles synthesized by microfluidic device. J. Control. Release 2022, 348, 648–659. [Google Scholar] [CrossRef]
- Kimura, N.; Maeki, M.; Sato, Y.; Note, Y.; Ishida, A.; Tani, H.; Harashima, H.; Tokeshi, M. Development of the iLiNP Device: Fine Tuning the Lipid Nanoparticle Size within 10 nm for Drug Delivery. ACS Omega 2018, 3, 5044–5051. [Google Scholar] [CrossRef]
- Sato, Y.; Kinami, Y.; Hashiba, K.; Harashima, H. Different kinetics for the hepatic uptake of lipid nanoparticles between the apolipoprotein E/low density lipoprotein receptor and the N-acetyl-d-galactosamine/asialoglycoprotein receptor pathway. J. Control. Release 2020, 322, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Maeki, M.; Kimura, N.; Sato, Y.; Harashima, H.; Tokeshi, M. Advances in microfluidics for lipid nanoparticles and extracellular vesicles and applications in drug delivery systems. Adv. Drug Deliv. Rev. 2018, 128, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Belliveau, N.M.; Huft, J.; Lin, P.J.; Chen, S.; Leung, A.K.; Leaver, T.J.; Wild, A.W.; Lee, J.B.; Taylor, R.J.; Tam, Y.K.; et al. Microfluidic Synthesis of Highly Potent Limit-size Lipid Nanoparticles for In Vivo Delivery of siRNA. Mol. Ther. Nucleic Acids 2012, 1, e37. [Google Scholar] [CrossRef] [PubMed]
- Roces, C.B.; Lou, G.; Jain, N.; Abraham, S.; Thomas, A.; Halbert, G.W.; Perrie, Y. Manufacturing Considerations for the Development of Lipid Nanoparticles Using Microfluidics. Pharmaceutics 2020, 12, 1095. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Kulkarni, J.A.; Huynh, A.; Chen, S.; van der Meel, R.; Tam, Y.Y.C.; Cullis, P.R. Characterization of Lipid Nanoparticles Containing Ionizable Cationic Lipids Using Design-of-Experiments Approach. Langmuir 2021, 37, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Hassett, K.J.; Higgins, J.; Woods, A.; Levy, B.; Xia, Y.; Hsiao, C.J.; Acosta, E.; Almarsson, Ö.; Moore, M.J.; Brito, L.A. Impact of lipid nanoparticle size on mRNA vaccine immunogenicity. J. Control. Release 2021, 335, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Diken, M.; Kreiter, S.; Selmi, A.; Britten, C.M.; Huber, C.; Türeci, Ö.; Sahin, U. Selective uptake of naked vaccine RNA by dendritic cells is driven by macropinocytosis and abrogated upon DC maturation. Gene Ther. 2011, 18, 702–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.P.; Mintern, J.D.; Gleeson, P.A. Macropinocytosis in Different Cell Types: Similarities and Differences. Membranes 2020, 10, 177. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barral, P.M.; Sarkar, D.; Su, Z.Z.; Barber, G.N.; DeSalle, R.; Racaniello, V.R.; Fisher, P.B. Functions of the cytoplasmic RNA sensors RIG-I and MDA-5: Key regulators of innate immunity. Pharmacol. Ther. 2009, 124, 219–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, J.; Sorensen, E.W.; Mintri, S.; Rabideau, A.E.; Zheng, W.; Besin, G.; Khatwani, N.; Su, S.V.; Miracco, E.J.; Issa, W.J.; et al. Impact of mRNA chemistry and manufacturing process on innate immune activation. Sci. Adv. 2020, 6, eaaz6893. [Google Scholar] [CrossRef] [PubMed]
- Baiersdörfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; Karikó, K. A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA. Mol. Ther. Nucleic Acids 2019, 15, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, D.P.; Wearsch, P.A.; Canaday, D.H.; Meyerson, H.J.; Liu, Y.C.; Wang, Y.; Boom, W.H.; Harding, C.V. Type I IFN drives a distinctive dendritic cell maturation phenotype that allows continued class II MHC synthesis and antigen processing. J. Immunol. 2012, 188, 3116–3126. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald-Bocarsly, P.; Feng, D. The role of type I interferon production by dendritic cells in host defense. Biochimie 2007, 89, 843–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalski, P.S.; Capasso Palmiero, U.; Huang, Y.; Rudra, A.; Langer, R.; Anderson, D.G. Ionizable Amino-Polyesters Synthesized via Ring Opening Polymerization of Tertiary Amino-Alcohols for Tissue Selective mRNA Delivery. Adv. Mater. 2018, 30, e1801151. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cheng, Q.; Wei, T.; Yu, X.; Johnson, L.T.; Farbiak, L.; Siegwart, D.J. Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR-Cas gene editing. Nat. Mater. 2021, 20, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 2012, 51, 8529–8533. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.; Hovdal, D.; Edmunds, N.; Nordberg, P.; Dahlén, A.; Dabkowska, A.; Arteta, M.Y.; Radulescu, A.; Kjellman, T.; Höijer, A.; et al. Functionalized lipid nanoparticles for subcutaneous administration of mRNA to achieve systemic exposures of a therapeutic protein. Mol. Ther. Nucleic Acids 2021, 24, 369–384. [Google Scholar] [CrossRef]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther. Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.H.; Park, H.B.; Choi, K. Enhanced Anti-tumor Reactivity of Cytotoxic T Lymphocytes Expressing PD-1 Decoy. Immune Netw. 2016, 16, 134–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataoka, K.; Shiraishi, Y.; Takeda, Y.; Sakata, S.; Matsumoto, M.; Nagano, S.; Maeda, T.; Nagata, Y.; Kitanaka, A.; Mizuno, S.; et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature 2016, 534, 402–406. [Google Scholar] [CrossRef]
- Nakamura, T.; Haloho, S.E.E.; Harashima, H. Intravenous liposomal vaccine enhances CTL generation, but not until antigen presentation. J. Control. Release 2022, 343, 1–12. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | %CL | %DOPE | %PEG | NaCl Conc. (mM) | CL | PEG | ζ-Average (nm) | PdI | ζ-Potential (mV) | %RNA Encapsulation |
|---|---|---|---|---|---|---|---|---|---|---|
| A-1 | 40 | 40 | 0.5 | 0 | CL7H6 | PEG-DMG | 112 | 0.20 | −3.2 | 90.3 |
| A-2 | 50 | 10 | 0.5 | 0 | CL4H6 | PEG-DMG | 121 | 0.19 | −2.3 | 99.2 |
| A-3 | 60 | 10 | 1 | 0 | CL7H6 | PEG-DSG | 104 | 0.15 | −2.3 | 91.0 |
| A-4 | 60 | 40 | 1.5 | 0 | CL7H6 | PEG-DMG | 128 | 0.41 | −0.6 | 89.8 |
| A-5 | 40 | 25 | 1.5 | 0 | CL4H6 | PEG-DSG | 88 | 0.30 | −1.5 | 99.2 |
| A-6 | 60 | 40 | 0.5 | 200 | CL4H6 | PEG-DSG | 155 | 0.13 | 1.9 | 98.8 |
| A-7 | 50 | 25 | 1 | 200 | CL4H6 | PEG-DMG | 138 | 0.12 | −1.1 | 99.0 |
| A-8 | 40 | 10 | 1.5 | 200 | CL7H6 | PEG-DMG | 128 | 0.20 | −2.6 | 85.0 |
| A-9 | 50 | 25 | 1 | 200 | CL7H6 | PEG-DSG | 171 | 0.15 | −2.7 | 88.0 |
| A-10 | 40 | 40 | 1 | 400 | CL4H6 | PEG-DMG | 216 | 0.08 | 2.5 | 99.0 |
| A-11 | 60 | 10 | 1.5 | 400 | CL4H6 | PEG-DSG | 547 | 0.19 | −1.4 | 89.2 |
| A-12 | 50 | 40 | 1.5 | 400 | CL7H6 | PEG-DSG | 108 | 0.07 | −1.7 | 88.3 |
| A-13 | 60 | 25 | 0.5 | 400 | CL7H6 | PEG-DMG | 501 | 0.12 | −1.2 | 91.2 |
| A-14 | 40 | 10 | 0.5 | 400 | CL4H6 | PEG-DSG | 754 | 0.17 | −10.3 | 72.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sasaki, K.; Sato, Y.; Okuda, K.; Iwakawa, K.; Harashima, H. mRNA-Loaded Lipid Nanoparticles Targeting Dendritic Cells for Cancer Immunotherapy. Pharmaceutics 2022, 14, 1572. https://doi.org/10.3390/pharmaceutics14081572
Sasaki K, Sato Y, Okuda K, Iwakawa K, Harashima H. mRNA-Loaded Lipid Nanoparticles Targeting Dendritic Cells for Cancer Immunotherapy. Pharmaceutics. 2022; 14(8):1572. https://doi.org/10.3390/pharmaceutics14081572
Chicago/Turabian StyleSasaki, Kosuke, Yusuke Sato, Kento Okuda, Kazuki Iwakawa, and Hideyoshi Harashima. 2022. "mRNA-Loaded Lipid Nanoparticles Targeting Dendritic Cells for Cancer Immunotherapy" Pharmaceutics 14, no. 8: 1572. https://doi.org/10.3390/pharmaceutics14081572