
Superparamagnetic Iron Oxide Nanoparticles for Immunotherapy of Cancers through Macrophages and Magnetic Hyperthermia

 ,
,  ,
,
Abstract
:1. Introduction
2. Immunity, Cancer, and SPION Nanoparticles
2.1. Immune Targets in Cancer
2.1.1. Immune Cells and Tumor Microenvironment at a Glance
2.1.2. Innate and Adaptive Immunity in Cancer
2.2. Opportunities for Targeting Immune System with SPION Nanoparticles
2.2.1. Rational for Targeting Immune System with SPION Nanoparticles
2.2.2. Nanoparticles to Target Immune TME (iTME)
2.2.3. Nanoparticles to Target Circulating Immune Cells
2.2.4. Nanoparticles to Target Myeloid and Lymphoid Immune Cell-Enriched Tissues
3. SPIONs: An Overview
3.1. Biophysical Properties of Superparamagnetic Materials
3.1.1. Magnetic Behavior of Ferromagnetic and Ferrimagnetic Materials
3.1.2. From Ferromagnetic and Ferrimagnetic to Superparamagnetic Behavior
3.2. Overview of the Use of SPIONs as MRI Contrast Agents
3.3. Basic Aspects of Magnetic Hyperthermia (MHT)
3.3.1. Biological Aspects of Hyperthermia
3.3.2. Heat Production Mechanisms in MHT
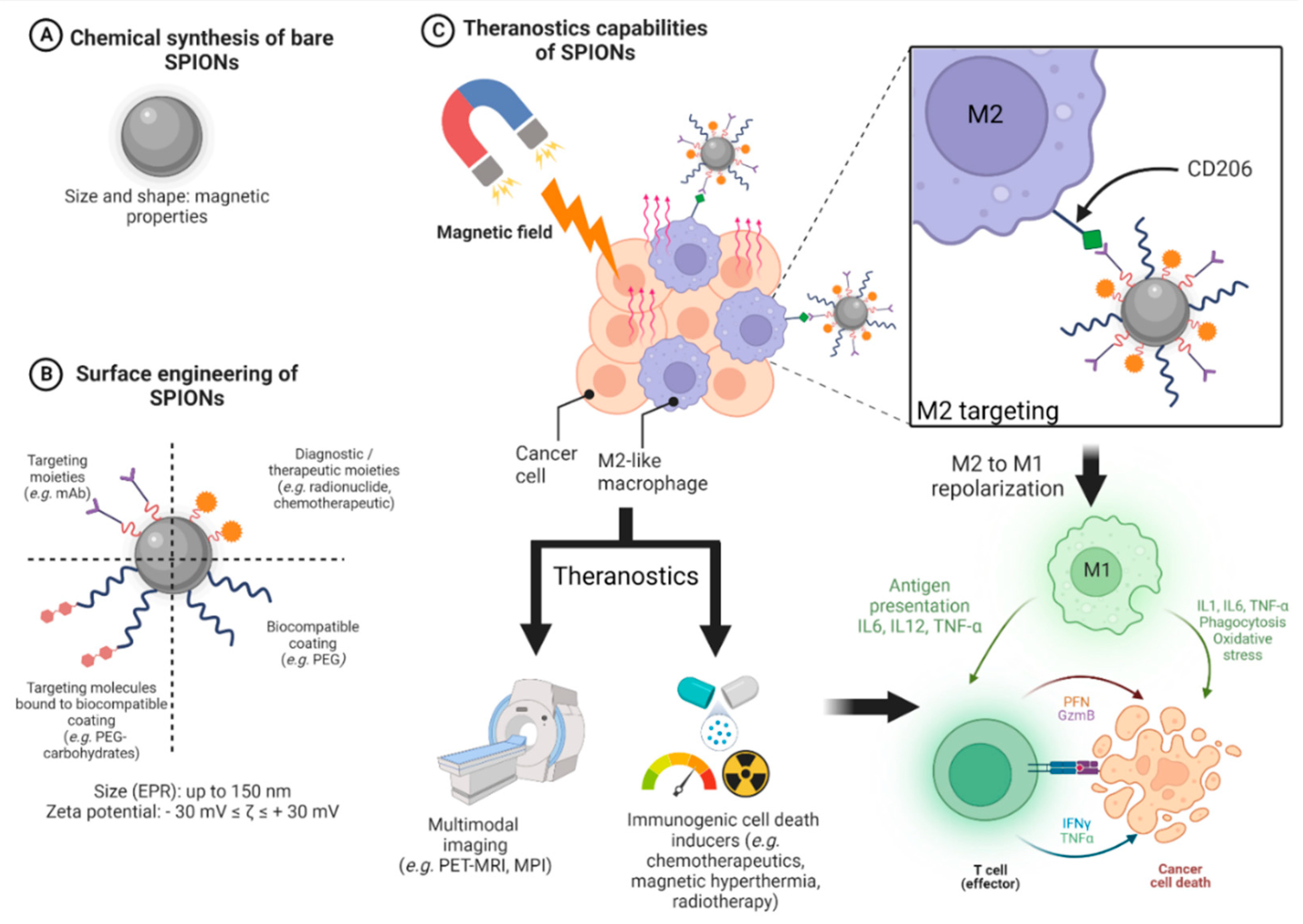
3.4. Design of SPIONs
3.4.1. General Design of Cancer Nanomedicines
3.4.2. Design of SPIONs Suitable for Hyperthermia and Immune System Targeting
4. Targeting the Immune System with SPIONs
4.1. Magnetic Hyperthermia Based on SPIONs as an Immune Trigger against Tumors
4.2. SPIONs and Immunomodulation of the Monocyte-Macrophage Axis
4.2.1. Solid Tumors and TME
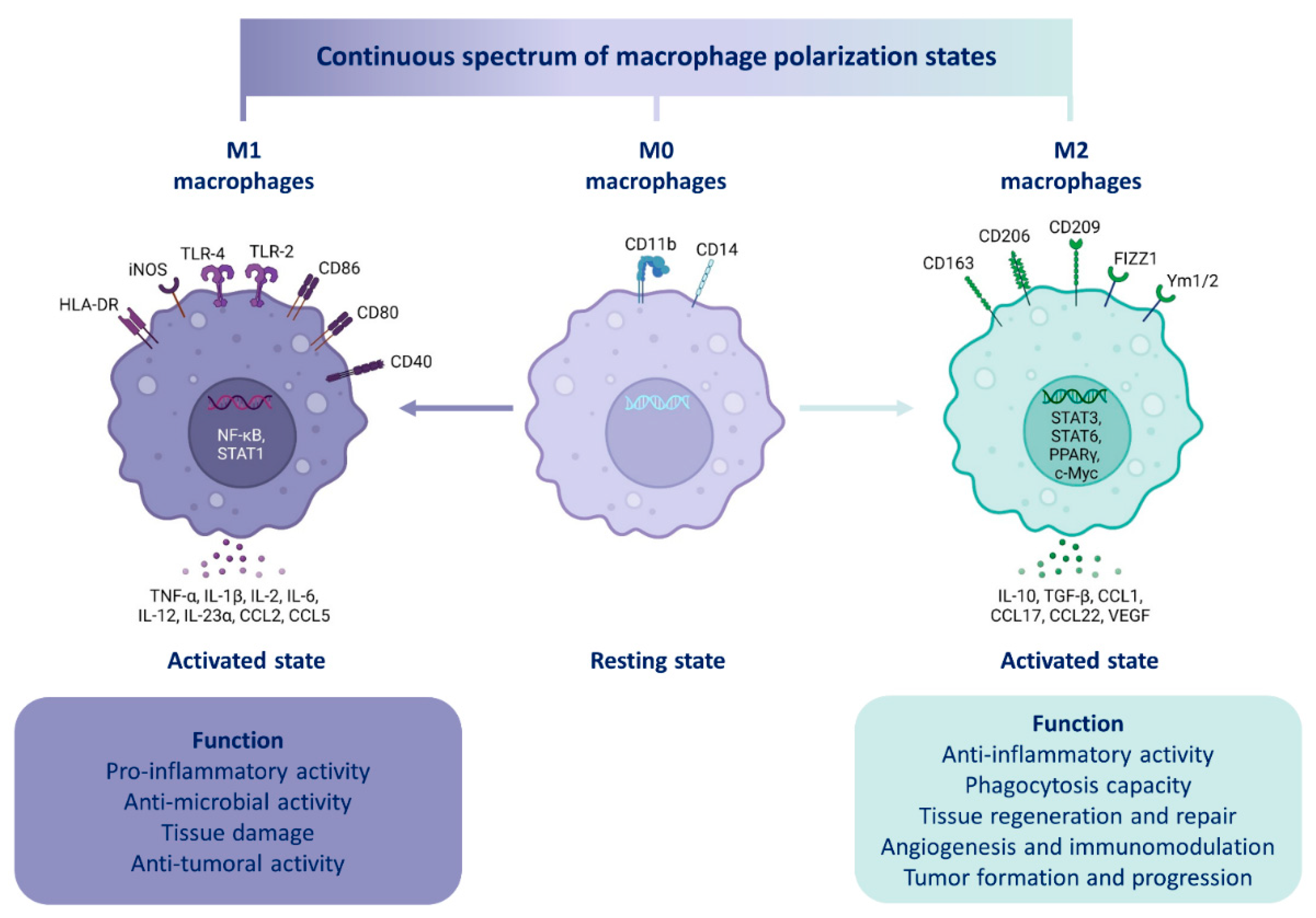
4.2.2. Macrophage Polarization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule Family | Polarization Marker | M0, M1 or M2 Marker | Species | References |
|---|---|---|---|---|
| Enzyme | Arg1 | M2 | Murine | [154] |
| iNOS | M2 | Murine | [154] | |
| Membrane receptors | CD11b | M0 | Human/Murine | [155] |
| CD14 | M0 | Human | [155] | |
| CD40 | M1 | Human/Murine | [156,157] | |
| CD80 | M1 | Human/Murine | [158] | |
| CD86 | M1 | Human/Murine | [158] | |
| CD163 | M2 | Human/Murine | [155] | |
| CD206 | M2 | Human/Murine | [155] | |
| F4/80 | M0 | Murine | [155] | |
| Cytokines | IL-1β | M1 | Human/Murine | [26,159] |
| IL-2 | M1 | Human/Murine | [26] | |
| IL-6 | M1 | Human/Murine | [154] | |
| IL-10 | M2 | Human/Murine | [158] | |
| IL-12 | M1 | Human/Murine | [26] | |
| IL-23α | M1 | Human/Murine | [26] | |
| CCL2 | M1 | Human/Murine | [160] | |
| TNF-α | M1 | Human/Murine | [154] | |
| TGF-β | M2 | Human/Murine | [158] | |
| VEGF | M2 | Human/Murine | [161] |
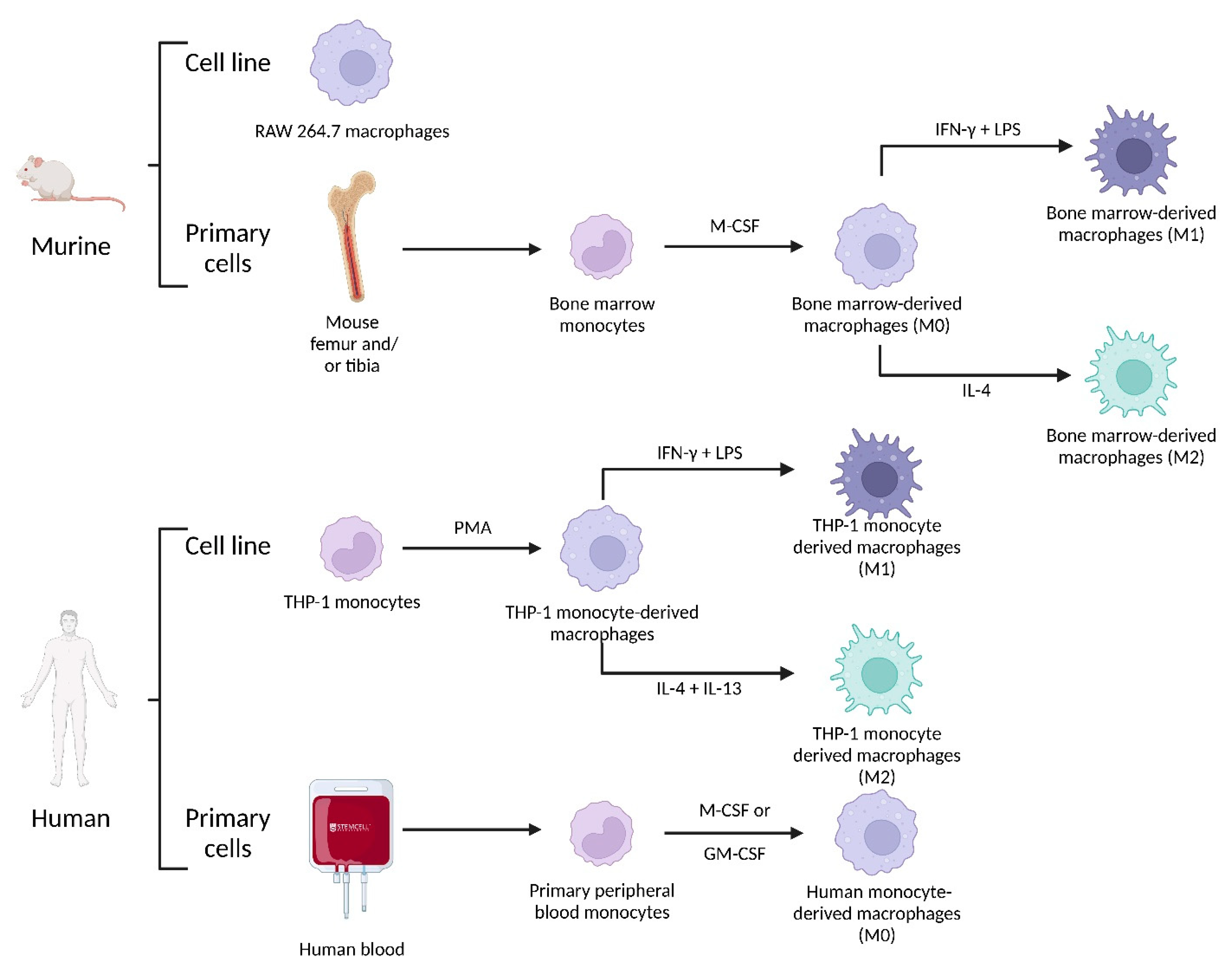
4.2.3. Macrophage Origin
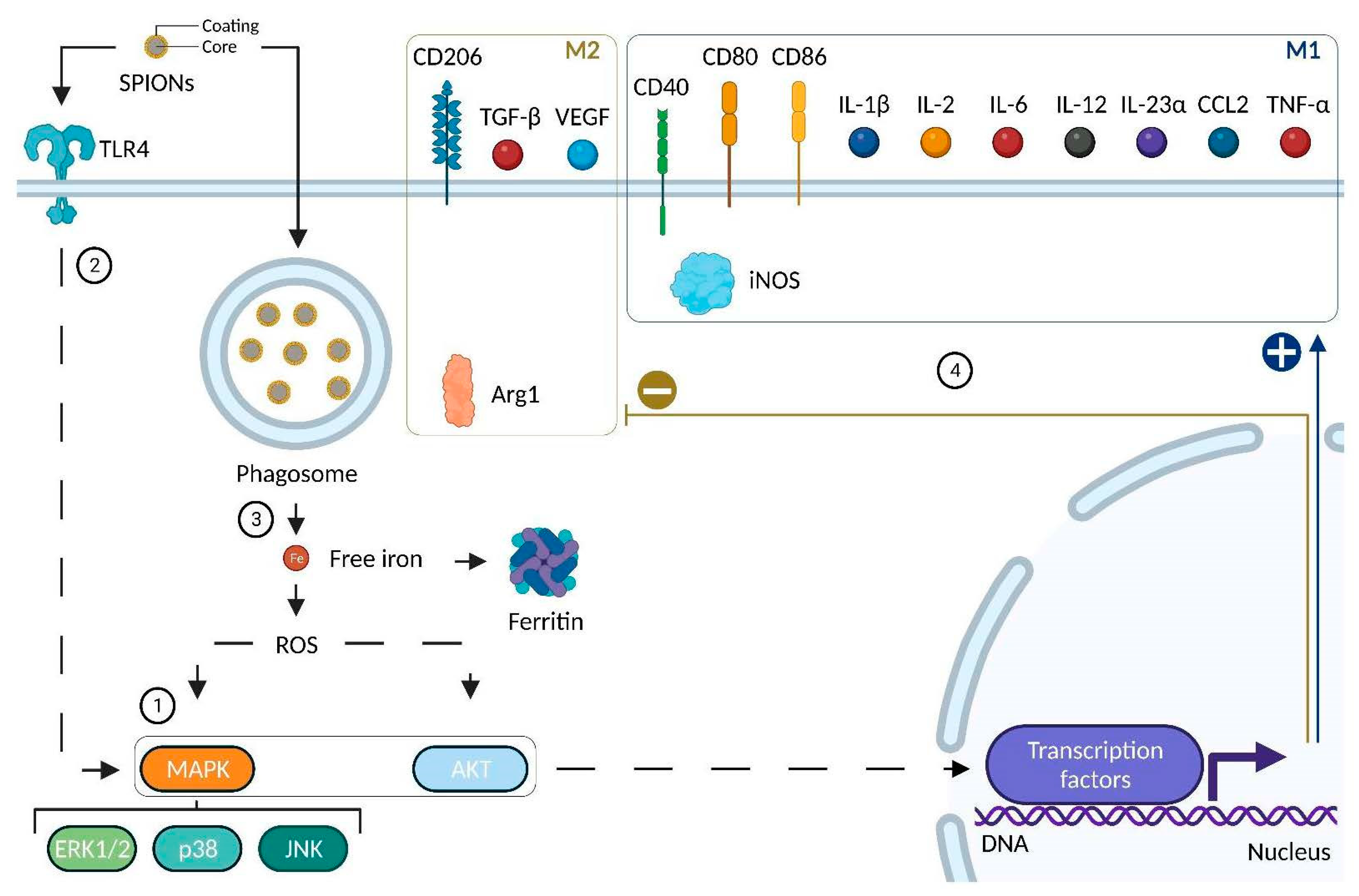
4.2.4. Impact of SPIONs on Monocytes and Macrophages
| Species | Cell Model | SPIONs | Biological Responses | Polarization Markers | References |
|---|---|---|---|---|---|
| Murine | RAW 264.7 | SPIONs (+): +44.72 mV Size: 19.4 nm | Important uptake Cytotoxicity | Increase in iNOS and TNF-α Decrease in IL-10 and VEGF Increase in CD80 and decrease in CD206 in vivo | [114] |
| Murine | RAW 264.7 | SPIONs (−): −27.31 mV Size: 21.3 nm | Important uptake Cytotoxicity | Increase in iNOS and TNF-α Decrease in IL-10 and VEGF Decrease in CD206 in vivo | [114] |
| Murine | RAW 264.7 | SPIONs (N): −0.282 mV Size: 15.9 nm | Uptake No cytotoxicity | Increase in TNF-α Decrease in IL-10 and VEGF Slight decrease in CD206 in vivo | [114] |
| Murine | RAW 264.7 | PEI-SPIONs (+): from +52.2 to +67.1 mV Size: 139–144 nm | Activation of TLR4, MAPK (p44/p42; p38; JNK) ROS production Modulation of podosome formation | Increase in CD40, CD80, CD86 and IL-12 Increase in IL10 | [174] |
| Murine | Bone Marrow-Derived Macrophages | DEAE-Dextran 1:4 (+): +16.8 mV Size: 68 nm | Low cell viability reduction Low cytotoxicity High iron uptake No impact on phagocytosis | Increase in CD86, IL-1β, IL-12β and TNF-α Decrease in CD206 and Arg1 | [173] |
| Murine | Bone Marrow-Derived Macrophages | CM-Dextran (−): −11.6 mV Size: 34.3 nm | Reduction in cell viability Low cytotoxicity Low iron uptake | NR | [173] |
| Murine | Bone Marrow-Derived Macrophages | Dextran (N): −3.3 mV Size: 36 nm | Reduction in cell viability Low cytotoxicity Low iron uptake | NR | [173] |
| Murine | Bone Marrow-Derived Macrophages | Resovist®: Ferucarbotran Carboxydextrane-coated SPIONs Size: 45–60 nm; core size: 5.8 nm | Activation of TLR4 | Increase in IL-1β, IL-2, IL-12, CCL2 and TNF-α | [175] |
| Murine | Bone Marrow-Derived Macrophages | DMSA SPIONs (−): −29.3 mV Size: 65 nm; core size: 10 nm | Fast uptake No reduction in cell viability Activation of MAPK (ERK) and AKT Decrease in transferrin receptor ROS production | Increase in IL-23α and CCL2 No variation in IL-12 Increase in IL-10 | [176] |
| Murine | Bone Marrow-Derived Macrophages | APS SPIONs (+): +33.3 Mv Size: 54 nm; core size: 8.3 nm | Fast uptake No reduction in cell viability Activation of MAPK (ERK) and AKT Decrease in transferrin receptor Important ROS production | Increase in IL-23α and CCL2 No variation in IL-12 Increase in IL-10 | [176] |
| Murine | Bone Marrow-Derived Macrophages | AD SPIONs (+): +40.3 nm Size: 150 nm; core size: 6.8 nm | Fast uptake No reduction in cell viability Activation of MAPK (ERK) and AKT Decrease in transferrin receptor Important ROS production | Increase in IL-23α and CCL2 No variation in IL-12 No variation in IL-10 | [176] |
| Human | THP-1 monocytes | Dextran-coated SPIONs Size: 83.5 and 86 nm; core size: 6.48 nm | Fast uptake | No increase in CD14, CD11b or CD86 Increase in IL-1β secretion Slight decrease in IL-10 secretion | [170] |
| Human | THP-1 Monocyte-derived macrophages | Dextran-coated SPIONs Size: 83.5 and 86 nm; core size: 6.48 nm | Fast uptake | No variation in CD14, CD11b or CD86 No variation in IL-1β No variation in IL-10 secretion | [170] |
| Human | THP-1 Monocyte-derived macrophages | Resovist®: Ferucarbotran Carboxydextrane-coated SPIONs Size: 45–60 nm; core size: 5.8 nm | Increase in Ferritin | Increase in CD86 and TNF-α on M2 macrophages | [177] |
| Human | THP-1 Monocyte-derived macrophages | DMSA SPIONs (−): −29.3 mV Size: 65 nm; core size: 10 nm | Fast uptake No reduction in cell viability Activation of MAPK (ERK) and AKT No activation of p38 nor JNK Decrease in transferrin receptor ROS production | Increase in CD86, TGF-β No variation in IL-12, IL-23α nor CCL2Increase in IL-10 | [176] |
| Human | THP-1 Monocyte-derived macrophages | APS SPIONs (+): +33.3 Mv Size: 54 nm; core size: 8.3 nm | Fast uptake No reduction in cell viability Activation of MAPK (ERK) and AKT No activation of p38 nor JNK Decrease in transferrin receptor and FPN-1 ROS production | Increase in CD86, TGF-β No variation in IL-12, IL-23α nor CCL2 Increase in IL-10 | [176] |
| Human | THP-1 Monocyte-derived macrophages | AD SPIONs (+): +40.3 nm Size: 150 nm; core size: 6.8 nm | Fast uptake No reduction in cell viability Activation of MAPK (ERK) and AKT No activation of p38 nor JNK Decrease in transferrin receptor and FPN-1 ROS production | No variation in CD86, IL-12, IL-23α nor CCL2 | [176] |
| Human | Primary peripheral blood monocytes | Dextran-coated SPIONs Size: 83.5 and 86 nm; core size: 6.48 nm | Fast uptake | NR | [170] |
| Human | Primary peripheral blood monocytes | Starch-coated SPIONs (−) Size: 200 nm | Low uptake No toxic effects Disruption of actin skeleton | Decrease in IL-6 No variation in IL-1β No variation in IL-10 | [172] |
| Human | Primary peripheral blood monocytes | Dextran SPIONs (−): −11 mV Size: 62,8 nm | Uptake in phagosomes or cytoplasm No decrease in cell viability nor cytotoxicity Activation of MAPK (ERK; p38; JNK) | Increase in IL-1β and TNF-α | [171] |
| Human | Human Monocyte-derived macrophages | DEAE-Dextran 1:4 (+): +16.8 mV Size: 68 nm | Important cell viability reduction Cytotoxicity Iron uptake | NR | [173] |
| Human | Human Monocyte-derived macrophages | Resovist®: Ferucarbotran Carboxydextrane-coated SPIONs Size: 45 and 60 nm; core size: 5.8 nm | Increase in Ferritin | NR | [177] |
5. Conclusions-Perspectives-Outlook
- -
- Good magnetic properties for imaging (MRI and MPI) and hyperthermia (magnetic core > 10 nm).
- -
- Suitable size for passive tumor targeting through EPR effect (typically below 100 nm).
- -
- Surface chemistry: coating to avoid aggregation, conjugations: with targeting moieties if pharmacological selectivity is desired, bifunctional chelating agents for radiolabeling purposes (nuclear imaging, targeted radionuclide therapy), fluorophores, photosensitizers, etc.
- -
- The shape has to be considered since it is recognized as a parameter affecting the immunological response.
- -
- A requirement for standard and optimized zeta potential values: typically, the higher, the better (good stability with absolute zeta values > |30| mV).
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: http://who.int/data/gho/data/themes/mortality-and-global-health-estimates/ghe-leading-causes-of-death (accessed on 23 May 2022).
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- Atallah-Yunes, S.A.; Robertson, M.J. Cytokine Based Immunotherapy for Cancer and Lymphoma: Biology, Challenges and Future Perspectives. Front. Immunol. 2022, 13, 872010. [Google Scholar] [CrossRef]
- Mayes, P.A.; Hance, K.W.; Hoos, A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 2018, 17, 509–527. [Google Scholar] [CrossRef]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef]
- Nel, A.R.E.; Meng, H. New Insights into “Permeability” as in the Enhanced Permeability and Retention Effect of Cancer Nanotherapeutics. ACS Nano 2017, 11, 9567–9569. [Google Scholar] [CrossRef]
- Tuma, P.; Hubbard, A.L. Transcytosis: Crossing cellular barriers. Physiol. Rev. 2003, 83, 871–932. [Google Scholar] [CrossRef]
- Heshmati Aghda, N.; Dabbaghianamiri, M.; Tunnell, J.W.; Betancourt, T. Design of smart nanomedicines for effective cancer treatment. Int. J. Pharm. 2022, 621, 121791. [Google Scholar] [CrossRef]
- Niculescu, A.G.; Grumezescu, A.M. Novel Tumor-Targeting Nanoparticles for Cancer Treatment-A Review. Int. J. Mol. Sci. 2022, 23, 5253. [Google Scholar] [CrossRef]
- Halwani, A.A. Development of Pharmaceutical Nanomedicines: From the Bench to the Market. Pharmaceutics 2022, 14, 106. [Google Scholar] [CrossRef]
- Sun, D.; Zhou, S.; Gao, W. What Went Wrong with Anticancer Nanomedicine Design and How to Make It Right. ACS Nano 2020, 14, 12281–12290. [Google Scholar] [CrossRef]
- Martin, J.D.; Cabral, H.; Stylianopoulos, T.; Jain, R.K. Improving cancer immunotherapy using nanomedicines: Progress, opportunities and challenges. Nat. Rev. Clin. Oncol. 2020, 17, 251–266. [Google Scholar] [CrossRef]
- Soetaert, F.; Korangath, P.; Serantes, D.; Fiering, S.; Ivkov, R. Cancer therapy with iron oxide nanoparticles: Agents of thermal and immune therapies. Adv. Drug Deliv. Rev. 2020, 163–164, 65–83. [Google Scholar] [CrossRef]
- Canese, R.; Vurro, F.; Marzola, P. Iron Oxide Nanoparticles as Theranostic Agents in Cancer Immunotherapy. Nanomaterials 2021, 11, 1950. [Google Scholar] [CrossRef]
- Dulinska-Litewka, J.; Lazarczyk, A.; Halubiec, P.; Szafranski, O.; Karnas, K.; Karewicz, A. Superparamagnetic Iron Oxide Nanoparticles-Current and Prospective Medical Applications. Materials 2019, 12, 617. [Google Scholar] [CrossRef]
- Montiel Schneider, M.G.; Martin, M.J.; Otarola, J.; Vakarelska, E.; Simeonov, V.; Lassalle, V.; Nedyalkova, M. Biomedical Applications of Iron Oxide Nanoparticles: Current Insights Progress and Perspectives. Pharmaceutics 2022, 14, 204. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Wilczynski, J.R.; Nowak, M. Cancer Immunoediting: Elimination, Equilibrium, and Immune Escape in Solid Tumors. Exp. Suppl. 2022, 113, 1–57. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.; Son, S.; Park, K.S.; Zou, W.; Shea, L.D.; Moon, J.J. Cancer nanomedicine for combination cancer immunotherapy. Nat. Rev. Mater. 2019, 4, 398–414. [Google Scholar] [CrossRef]
- Doshi, A.S.; Asrani, K.H. Chapter Two-Innate and adaptive immunity in cancer. In Cancer Immunology and Immunotherapy; Amiji, M.M., Milane, L.S., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 19–61. [Google Scholar]
- Sofias, A.M.; Combes, F.; Koschmieder, S.; Storm, G.; Lammers, T. A paradigm shift in cancer nanomedicine: From traditional tumor targeting to leveraging the immune system. Drug Discov. Today 2021, 26, 1482–1489. [Google Scholar] [CrossRef]
- Ginefra, P.; Lorusso, G.; Vannini, N. Innate Immune Cells and Their Contribution to T-Cell-Based Immunotherapy. Int. J. Mol. Sci. 2020, 21, 4441. [Google Scholar] [CrossRef]
- Anfray, C.; Ummarino, A.; Andon, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Zuo, S.; Song, J.; Zhang, J.; He, Z.; Sun, B.; Sun, J. Nano-immunotherapy for each stage of cancer cellular immunity: Which, why, and what? Theranostics 2021, 11, 7471–7487. [Google Scholar] [CrossRef]
- Alexander, C.A.; Yang, Y.Y. Harnessing the combined potential of cancer immunotherapy and nanomedicine: A new paradigm in cancer treatment. Nanomedicine 2022, 40, 102492. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Viana, I.M.O.; Roussel, S.; Defrene, J.; Lima, E.M.; Barabe, F.; Bertrand, N. Innate and adaptive immune responses toward nanomedicines. Acta Pharm. Sin. B 2021, 11, 852–870. [Google Scholar] [CrossRef]
- Yu, S.; Wang, Y.; He, P.; Shao, B.; Liu, F.; Xiang, Z.; Yang, T.; Zeng, Y.; He, T.; Ma, J.; et al. Effective Combinations of Immunotherapy and Radiotherapy for Cancer Treatment. Front. Oncol. 2022, 12, 809304. [Google Scholar] [CrossRef]
- Hurwitz, M.D. Hyperthermia and immunotherapy: Clinical opportunities. Int. J. Hyperth. 2019, 36, 4–9. [Google Scholar] [CrossRef]
- Hartl, C.A.; Bertschi, A.; Puerto, R.B.; Andresen, C.; Cheney, E.M.; Mittendorf, E.A.; Guerriero, J.L.; Goldberg, M.S. Combination therapy targeting both innate and adaptive immunity improves survival in a pre-clinical model of ovarian cancer. J. Immunother. Cancer 2019, 7, 199. [Google Scholar] [CrossRef]
- Zhu, E.F.; Gai, S.A.; Opel, C.F.; Kwan, B.H.; Surana, R.; Mihm, M.C.; Kauke, M.J.; Moynihan, K.D.; Angelini, A.; Williams, R.T.; et al. Synergistic innate and adaptive immune response to combination immunotherapy with anti-tumor antigen antibodies and extended serum half-life IL-2. Cancer Cell 2015, 27, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Gowd, V.; Ahmad, A.; Tarique, M.; Suhail, M.; Zughaibi, T.A.; Tabrez, S.; Khan, R. Advancement of cancer immunotherapy using nanoparticles-based nanomedicine. Semin. Cancer Biol. 2022, 86, 624–644. [Google Scholar] [CrossRef]
- Hu, M.; Huang, L. Strategies targeting tumor immune and stromal microenvironment and their clinical relevance. Adv Drug Deliv. Rev. 2022, 183, 114137. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, Y.; Zhu, X.; Li, C.; Yan, M.; Pan, J.; Ma, G. Tumor microenvironment-responsive prodrug nanoplatform via co-self-assembly of photothermal agent and IDO inhibitor for enhanced tumor penetration and cancer immunotherapy. Biomaterials 2020, 242, 119933. [Google Scholar] [CrossRef]
- Phuengkham, H.; Song, C.; Lim, Y.T. A Designer Scaffold with Immune Nanoconverters for Reverting Immunosuppression and Enhancing Immune Checkpoint Blockade Therapy. Adv. Mater. 2019, 31, e1903242. [Google Scholar] [CrossRef]
- Wang-Bishop, L.; Wehbe, M.; Shae, D.; James, J.; Hacker, B.C.; Garland, K.; Chistov, P.P.; Rafat, M.; Balko, J.M.; Wilson, J.T. Potent STING activation stimulates immunogenic cell death to enhance antitumor immunity in neuroblastoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Viswanath, D.I.; Liu, H.C.; Huston, D.P.; Chua, C.Y.X.; Grattoni, A. Emerging biomaterial-based strategies for personalized therapeutic in situ cancer vaccines. Biomaterials 2022, 280, 121297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Tai, Z.; Cui, Z.; Chai, R.; Zhu, Q.; Chen, Z. Nano-engineered immune cells as “guided missiles” for cancer therapy. J. Control Release 2022, 341, 60–79. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Shen, M.Y.; Chen, H.H.; Lin, S.C.; Chiang, W.H.; Wu, P.H.; Chang, C.W.; Chiang, C.S.; Chiu, H.C. Monocytic delivery of therapeutic oxygen bubbles for dual-modality treatment of tumor hypoxia. J. Control Release 2015, 220, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Milligan, J.J.; Saha, S. A Nanoparticle’s Journey to the Tumor: Strategies to Overcome First-Pass Metabolism and Their Limitations. Cancers 2022, 14, 1741. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, G.; Cousins, A.; Pham, N.; Milanova, V.; Nelson, M.; Krishnan, S.; Shetty, A.; van den Berg, N.; Rosenthal, E.; Krishnan, S.; et al. Preclinical evaluation of a mannose-labeled magnetic tracer for enhanced sentinel lymph node retention in the head and neck. Nanomedicine 2022, 42, 102546. [Google Scholar] [CrossRef]
- Dennis, C.L.; Ivkov, R. Physics of heat generation using magnetic nanoparticles for hyperthermia. Int. J. Hyperth. 2013, 29, 715–729. [Google Scholar] [CrossRef]
- Sarychev, V.T. Electromagnetic field of a rotating magnetic dipole and electric-charge motion in this field. Radiophys. Quantum Electron. 2009, 52, 8. [Google Scholar] [CrossRef]
- Stöhr, J.; Siegmann, H.C. Magnetism: From Fundamentals to Nanoscale Dynamics; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2006; p. 820. [Google Scholar]
- Spaldin, N.A. Magnetic materials: Fundamentals and Applications, 2nd ed.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2011; p. 274. [Google Scholar]
- Nisticò, R. A synthetic guide toward the tailored production of magnetic iron oxide nanoparticles. Boletín Soc. Española Cerámica Y Vidr. 2021, 60, 29–40. [Google Scholar] [CrossRef]
- Rajan, A.; Sahu, N.K. Review on magnetic nanoparticle-mediated hyperthermia for cancer therapy. J. Nanopart Res. 2020, 22, 319. [Google Scholar] [CrossRef]
- Shaterabadi, Z.; Nabiyouni, G.; Soleymani, M. Physics responsible for heating efficiency and self-controlled temperature rise of magnetic nanoparticles in magnetic hyperthermia therapy. Prog. Biophys. Mol. Biol. 2018, 133, 9–19. [Google Scholar] [CrossRef]
- Cullity, B.D.; Graham, C.D. Introduction to Magnetic Materials, 2nd ed.; IEEE/Wiley: Hoboken, NJ, USA, 2009; p. 544. [Google Scholar]
- Williams, H.J.; Bozorth, R.M.; Shockley, W. Magnetic Domain Patterns on Single Crystals of Silicon Iron. Phys. Rev. 1949, 75, 155–178. [Google Scholar] [CrossRef]
- Laurent, S.; Dutz, S.; Häfeli, U.O.; Mahmoudi, M. Magnetic fluid hyperthermia: Focus on superparamagnetic iron oxide nanoparticles. Adv. Colloid Interface Sci. 2011, 166, 8–23. [Google Scholar] [CrossRef]
- Saslow, W.M. The Magnetism of Magnets. In Electricity, Magnetism, and Light; Elsevier: Amsterdam, The Netherlands, 2002; pp. 384–418. [Google Scholar]
- Aharoni, A. Introduction to the Theory of Ferromagnetism, 2nd ed.; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Gupta, A.K.; Wells, S. Surface-Modified Superparamagnetic Nanoparticles for Drug Delivery: Preparation, Characterization, and Cytotoxicity Studies. IEEE Trans. Nanobioscience 2004, 3, 66–73. [Google Scholar] [CrossRef]
- Kandasamy, G.; Maity, D. Recent advances in superparamagnetic iron oxide nanoparticles (SPIONs) for in vitro and in vivo cancer nanotheranostics. Int. J. Pharm. 2015, 496, 191–218. [Google Scholar] [CrossRef]
- Khot, V.M.; Salunkhe, A.B.; Ruso, J.M.; Pawar, S.H. Improved magnetic induction heating of nanoferrites for hyperthermia applications: Correlation with colloidal stability and magneto-structural properties. J. Magn. Magn. Mater. 2015, 384, 335–343. [Google Scholar] [CrossRef]
- Frenkel, J.; Doefman, J. Spontaneous and Induced Magnetisation in Ferromagnetic Bodies. Nature 1930, 126, 274–275. [Google Scholar] [CrossRef]
- Néel, L. Théorie du traînage magnétique des substances massives dans le domaine de Rayleigh. J. Phys. Radium 1950, 11, 49–61. [Google Scholar] [CrossRef]
- Bean, C.P.; Livingston, J.D. Superparamagnetism. J. Appl. Phys. 1959, 30, S120–S129. [Google Scholar] [CrossRef]
- Savliwala, S.; Chiu-Lam, A.; Unni, M.; Rivera-Rodriguez, A.; Fuller, E.; Sen, K.; Threadcraft, K.; Rinaldi, C. Chapter 13-Magnetic nanoparticles. In Nanoparticles for Biomedical Applications; Elsevier: Amsterdam, The Netherlands, 2020; pp. 195–221. [Google Scholar]
- Weissleder, R.; Hahn, P.F.; Stark, D.D.; Elizondo, G.; Saini, S.; Todd, L.E.; Wittenberg, J.; Ferrucci, J.T. Superparamagnetic iron oxide: Enhanced detection of focal splenic tumors with MR imaging. Radiology 1988, 169, 399–403. [Google Scholar] [CrossRef]
- Weissleder, R.; Elizondo, G.; Wittenberg, J.; Rabito, C.A.; Bengele, H.H.; Josephson, L. Ultrasmall superparamagnetic iron oxide: Characterization of a new class of contrast agents for MR imaging. Radiology 1990, 175, 489–493. [Google Scholar] [CrossRef]
- Stark, D.D.; Weissleder, R.; Elizondo, G.; Hahn, P.F.; Saini, S.; Todd, L.E.; Wittenberg, J.; Ferrucci, J.T. Superparamagnetic iron oxide: Clinical application as a contrast agent for MR imaging of the liver. Radiology 1988, 168, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.; Stark, D.D.; Hahn, P.F.; Wittenberg, J.; Brady, T.J.; Ferrucci, J.T., Jr. Ferrite particles: A superparamagnetic MR contrast agent for the reticuloendothelial system. Radiology 1987, 162, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.; Stark, D.D.; Hahn, P.F.; Bousquet, J.C.; Introcasso, J.; Wittenberg, J.; Brady, T.J.; Ferrucci, J.T., Jr. Ferrite particles: A superparamagnetic MR contrast agent for enhanced detection of liver carcinoma. Radiology 1987, 162, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Zoghbi, S.S.; Gore, J.C. Regional differences in rat brain displayed by fast MRI with superparamagnetic contrast agents. Magn. Reson. Imaging 1988, 6, 611–615. [Google Scholar] [CrossRef]
- Hervault, A.; Thanh, N.T.K. Magnetic nanoparticle-based therapeutic agents for thermo-chemotherapy treatment of cancer. Nanoscale 2014, 6, 11553–11573. [Google Scholar] [CrossRef] [Green Version]
- Antonelli, A.; Magnani, M. SPIO nanoparticles and magnetic erythrocytes as contrast agents for biomedical and diagnostic applications. J. Magn. Magn. Mater. 2022, 541, 168520. [Google Scholar] [CrossRef]
- Bjørnerud, A.; Johansson, L. The utility of superparamagnetic contrast agents in MRI: Theoretical consideration and applications in the cardiovascular system: Superparamagnetic contrast agents. NMR Biomed. 2004, 17, 465–477. [Google Scholar] [CrossRef]
- Koenig, S.H.; Kellar, K.E. Theory of 1/T1 and 1/T2 NMRD profiles of solutions of magnetic nanoparticles. Magn. Reson. Med. 1995, 34, 227–233. [Google Scholar] [CrossRef]
- Bae, K.H.; Kim, Y.B.; Lee, Y.; Hwang, J.; Park, H.; Park, T.G. Bioinspired Synthesis and Characterization of Gadolinium-Labeled Magnetite Nanoparticles for Dual Contrast T1- and T2-Weighted Magnetic Resonance Imaging. Bioconjugate Chem. 2010, 21, 505–512. [Google Scholar] [CrossRef]
- Kellar, K.E.; Fujii, D.K.; Gunther, W.H.H.; Briley-Saebo, K.; Bjornerud, A.; Spiller, M.; Koenig, S.H. NC100150 injection, a preparation of optimized iron oxide nanoparticles for positive-contrast MR angiography. J. Magn. Reson. Imaging 2000, 11, 488–494. [Google Scholar] [CrossRef]
- Alipour, A.; Soran-Erdem, Z.; Utkur, M.; Sharma, V.K.; Algin, O.; Saritas, E.U.; Demir, H.V. A new class of cubic SPIONs as a dual-mode T1 and T2 contrast agent for MRI. Magn. Reson. Imaging 2018, 49, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Hahn, P.F.; Stark, D.D.; Lewis, J.M.; Saini, S.; Elizondo, G.; Weissleder, R.; Fretz, C.J.; Ferrucci, J.T. First clinical trial of a new superparamagnetic iron oxide for use as an oral gastrointestinal contrast agent in MR imaging. Radiology 1990, 175, 695–700. [Google Scholar] [CrossRef]
- Neuwelt, A.; Sidhu, N.; Hu, C.-A.A.; Mlady, G.; Eberhardt, S.C.; Sillerud, L.O. Iron-Based Superparamagnetic Nanoparticle Contrast Agents for MRI of Infection and Inflammation. Am. J. Roentgenol. 2015, 204, W302–W313. [Google Scholar] [CrossRef] [Green Version]
- Kaim, A.H.; Jundt, G.; Wischer, T.; O’Reilly, T.; Fröhlich, J.; von Schulthess, G.K.; Allegrini, P.R. Functional-Morphologic MR Imaging with Ultrasmall Superparamagnetic Particles of Iron Oxide in Acute and Chronic Soft-Tissue Infection: Study in Rats. Radiology 2003, 227, 169–174. [Google Scholar] [CrossRef]
- Lutz, A.M.; Weishaupt, D.; Persohn, E.; Goepfert, K.; Froehlich, J.; Sasse, B.; Gottschalk, J.; Marincek, B.; Kaim, A.H. Imaging of Macrophages in Soft-Tissue Infection in Rats: Relationship between Ultrasmall Superparamagnetic Iron Oxide Dose and MR Signal Characteristics. Radiology 2005, 234, 765–775. [Google Scholar] [CrossRef]
- Stoll, G.; Bendszus, M. New approaches to neuroimaging of central nervous system inflammation. Curr. Opin. Neurol. 2010, 23, 282–286. [Google Scholar] [CrossRef]
- Sillerud, L.O.; Solberg, N.O.; Chamberlain, R.; Orlando, R.A.; Heidrich, J.E.; Brown, D.C.; Brady, C.I.; Vander Jagt, T.A.; Garwood, M.; Vander Jagt, D.L. SPION-Enhanced Magnetic Resonance Imaging of Alzheimer’s Disease Plaques in AβPP/PS-1 Transgenic Mouse Brain. JAD 2013, 34, 349–365. [Google Scholar] [CrossRef] [Green Version]
- Ruehm, S.G.; Corot, C.; Vogt, P.; Kolb, S.; Debatin, J.F. Magnetic Resonance Imaging of Atherosclerotic Plaque With Ultrasmall Superparamagnetic Particles of Iron Oxide in Hyperlipidemic Rabbits. Circulation 2001, 103, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Harnan, S.E.; Cooper, K.L.; Meng, Y.; Ward, S.E.; Fitzgerald, P.; Papaioannou, D.; Ingram, C.; Lorenz, E.; Wilkinson, I.D.; Wyld, L. Magnetic resonance for assessment of axillary lymph node status in early breast cancer: A systematic review and meta-analysis. Eur. J. Surg. Oncol. 2011, 37, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Weissleder, R.; Elizondo, G.; Wittenberg, J.; Lee, A.S.; Josephson, L.; Brady, T.J. Ultrasmall superparamagnetic iron oxide: An intravenous contrast agent for assessing lymph nodes with MR imaging. Radiology 1990, 175, 494–498. [Google Scholar] [CrossRef]
- Chambon, C.; Clement, O.; Le Blanche, A.; Schouman-Claeys, E.; Frija, G. Superparamagnetic iron oxides as positive MR contrast agents: In vitro and in vivo evidence. Magn. Reson. Imaging 1993, 11, 509–519. [Google Scholar] [CrossRef]
- Serkova, N.J. Nanoparticle-Based Magnetic Resonance Imaging on Tumor-Associated Macrophages and Inflammation. Front. Immunol. 2017, 8, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storey, P.; Lim, R.P.; Chandarana, H.; Rosenkrantz, A.B.; Kim, D.; Stoffel, D.R.; Lee, V.S. MRI assessment of hepatic iron clearance rates after USPIO administration in healthy adults. Investig. Radiol. 2012, 47, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, S.A.; Thurman, J.M. Molecular imaging of autoimmune diseases and inflammation. Mol. Imaging 2012, 11, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissleder, R. Molecular Imaging: Principles and Practice; People’s Medical Publishing House-USA: Shelton, CT, USA, 2010. [Google Scholar]
- Peiravi, M.; Eslami, H.; Ansari, M.; Zare-Zardini, H. Magnetic hyperthermia: Potentials and limitations. J. Indian Chem. Soc. 2022, 99, 100269. [Google Scholar] [CrossRef]
- Yan, B.; Liu, C.; Wang, S.; Li, H.; Jiao, J.; Lee, W.S.V.; Zhang, S.; Hou, Y.; Hou, Y.; Ma, X.; et al. Magnetic hyperthermia induces effective and genuine immunogenic tumor cell death with respect to exogenous heating. J. Mater. Chem. B 2022, 10, 5364–5374. [Google Scholar] [CrossRef]
- Deatsch, A.E.; Evans, B.A. Heating efficiency in magnetic nanoparticle hyperthermia. J. Magn. Magn. Mater. 2014, 354, 163–172. [Google Scholar] [CrossRef]
- Jiao, W.; Zhang, T.; Peng, M.; Yi, J.; He, Y.; Fan, H. Design of Magnetic Nanoplatforms for Cancer Theranostics. Biosensors 2022, 12, 38. [Google Scholar] [CrossRef]
- Palanisamy, S.; Wang, Y.M. Superparamagnetic iron oxide nanoparticulate system: Synthesis, targeting, drug delivery and therapy in cancer. Dalton Trans. 2019, 48, 9490–9515. [Google Scholar] [CrossRef]
- Samrot, A.V.; Sahithya, C.S.; Selvarani, A.J.; Purayil, S.K.; Ponnaiah, P. A review on synthesis, characterization and potential biological applications of superparamagnetic iron oxide nanoparticles. Curr. Res. Green Sustain. Chem. 2021, 4, 100042. [Google Scholar] [CrossRef]
- Berret, J.F.; Graillot, A. Versatile Coating Platform for Metal Oxide Nanoparticles: Applications to Materials and Biological Science. Langmuir 2022, 38, 5323–5338. [Google Scholar] [CrossRef]
- Nuzhina, J.V.; Shtil, A.A.; Prilepskii, A.Y.; Vinogradov, V.V. Preclinical Evaluation and Clinical Translation of Magnetite-Based Nanomedicines. J. Drug Deliv. Sci. Technol. 2019, 54, 101282. [Google Scholar] [CrossRef]
- Wlodarczyk, A.; Gorgon, S.; Radon, A.; Bajdak-Rusinek, K. Magnetite Nanoparticles in Magnetic Hyperthermia and Cancer Therapies: Challenges and Perspectives. Nanomaterials 2022, 12, 1807. [Google Scholar] [CrossRef]
- Rubia-Rodriguez, I.; Santana-Otero, A.; Spassov, S.; Tombacz, E.; Johansson, C.; De La Presa, P.; Teran, F.J.; Del Puerto Morales, M.; Veintemillas-Verdaguer, S.; Thanh, N.T.K.; et al. Whither Magnetic Hyperthermia? A Tentative Roadmap. Materials 2021, 14, 706. [Google Scholar] [CrossRef]
- Tong, S.; Hou, S.; Zheng, Z.; Zhou, J.; Bao, G. Coating optimization of superparamagnetic iron oxide nanoparticles for high T2 relaxivity. Nano Lett. 2010, 10, 4607–4613. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.L.; Fan, H.M.; Yi, J.B.; Yang, Y.; Choo, E.S.G.; Xue, J.M.; Fan, D.D.; Ding, J. Optimization of surface coating on Fe3O4 nanoparticles for high performance magnetic hyperthermia agents. J. Mater. Chem. 2012, 22, 8235. [Google Scholar] [CrossRef]
- Egea-Benavente, D.; Ovejero, J.G.; Morales, M.D.P.; Barber, D.F. Understanding MNPs Behaviour in Response to AMF in Biological Milieus and the Effects at the Cellular Level: Implications for a Rational Design That Drives Magnetic Hyperthermia Therapy toward Clinical Implementation. Cancers 2021, 13, 4583. [Google Scholar] [CrossRef]
- Guardia, P.; Di Corato, R.; Lartigue, L.; Wilhelm, C.; Espinosa, A.; Garcia-Hernandez, M.; Gazeau, F.; Manna, L.; Pellegrino, T. Water-soluble iron oxide nanocubes with high values of specific absorption rate for cancer cell hyperthermia treatment. ACS Nano 2012, 6, 3080–3091. [Google Scholar] [CrossRef]
- Mai, B.T.; Balakrishnan, P.B.; Barthel, M.J.; Piccardi, F.; Niculaes, D.; Marinaro, F.; Fernandes, S.; Curcio, A.; Kakwere, H.; Autret, G.; et al. Thermoresponsive Iron Oxide Nanocubes for an Effective Clinical Translation of Magnetic Hyperthermia and Heat-Mediated Chemotherapy. ACS Appl. Mater. Interfaces 2019, 11, 5727–5739. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.; Revia, R.A.; Zhang, M. Iron oxide nanoparticles for immune cell labeling and cancer immunotherapy. Nanoscale Horiz. 2021, 6, 696–717. [Google Scholar] [CrossRef]
- Persano, S.; Das, P.; Pellegrino, T. Magnetic Nanostructures as Emerging Therapeutic Tools to Boost Anti-Tumour Immunity. Cancers 2021, 13, 2735. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, X.; Cheng, Y.; Guo, X.; Yuan, W. Iron Oxide Nanoparticles-Based Vaccine Delivery for Cancer Treatment. Mol. Pharm. 2018, 15, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Ortega, L.; Rojas, J.M.; Marcos, A.; Portilla, Y.; Stein, J.V.; Barber, D.F. T cells loaded with magnetic nanoparticles are retained in peripheral lymph nodes by the application of a magnetic field. J. Nanobiotechnology 2019, 17, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karoon Kiani, F.; Izadi, S.; Ansari Dezfouli, E.; Ebrahimi, F.; Mohammadi, M.; Chalajour, H.; Mortazavi Bulus, M.; Nasr Esfahani, M.; Karpisheh, V.; Mahmoud Salehi Khesht, A.; et al. Simultaneous silencing of the A2aR and PD-1 immune checkpoints by siRNA-loaded nanoparticles enhances the immunotherapeutic potential of dendritic cell vaccine in tumor experimental models. Life Sci. 2022, 288, 120166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cao, S.; Liang, S.; Tan, C.H.; Luo, B.; Xu, X.; Saw, P.E. Differently Charged Super-Paramagnetic Iron Oxide Nanoparticles Preferentially Induced M1-Like Phenotype of Macrophages. Front. Bioeng. Biotechnol. 2020, 8, 537. [Google Scholar] [CrossRef]
- Wang, W.; Li, F.; Li, S.; Hu, Y.; Xu, M.; Zhang, Y.; Khan, M.I.; Wang, S.; Wu, M.; Ding, W.; et al. M2 macrophage-targeted iron oxide nanoparticles for magnetic resonance image-guided magnetic hyperthermia therapy. J. Mater. Sci. Technol. 2021, 81, 77–87. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, X.; Yan, B.; Wen, N.; Lee, W.S.V.; Liang, X.J.; Liu, X. Enhancement of CD8(+) T-Cell-Mediated Tumor Immunotherapy via Magnetic Hyperthermia. ChemMedChem 2022, 17, e202100656. [Google Scholar] [CrossRef]
- Chang, D.; Lim, M.; Goos, J.; Qiao, R.; Ng, Y.Y.; Mansfeld, F.M.; Jackson, M.; Davis, T.P.; Kavallaris, M. Biologically Targeted Magnetic Hyperthermia: Potential and Limitations. Front. Pharmacol. 2018, 9, 831. [Google Scholar] [CrossRef] [Green Version]
- Pucci, C.; Degl’Innocenti, A.; Belenli Gumus, M.; Ciofani, G. Superparamagnetic iron oxide nanoparticles for magnetic hyperthermia: Recent advancements, molecular effects, and future directions in the omics era. Biomater. Sci. 2022, 10, 2103–2121. [Google Scholar] [CrossRef]
- Imashiro, C.; Takeshita, H.; Morikura, T.; Miyata, S.; Takemura, K.; Komotori, J. Development of accurate temperature regulation culture system with metallic culture vessel demonstrates different thermal cytotoxicity in cancer and normal cells. Sci. Rep. 2021, 11, 21466. [Google Scholar] [CrossRef]
- Kanamori, T.; Miyazaki, N.; Aoki, S.; Ito, K.; Hisaka, A.; Hatakeyama, H. Investigation of energy metabolic dynamism in hyperthermia-resistant ovarian and uterine cancer cells under heat stress. Sci. Rep. 2021, 11, 14726. [Google Scholar] [CrossRef]
- van der Zee, J. Heating the patient: A promising approach? Ann. Oncol. 2002, 13, 1173–1184. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Fiorentini, G.; Szasz, A.M.; Szigeti, G.; Szasz, A.; Minnaar, C.A. Quo Vadis Oncological Hyperthermia (2020)? Front. Oncol. 2020, 10, 1690. [Google Scholar] [CrossRef]
- Yagawa, Y.; Tanigawa, K.; Kobayashi, Y.; Yamamoto, M. Cancer immunity and therapy using hyperthermia with immunotherapy, radiotherapy, chemotherapy, and surgery. J. Cancer Metastasis Treat. 2017, 3, 218–230. [Google Scholar] [CrossRef]
- Adnan, A.; Munoz, N.M.; Prakash, P.; Habibollahi, P.; Cressman, E.N.K.; Sheth, R.A. Hyperthermia and Tumor Immunity. Cancers 2021, 13, 2507. [Google Scholar] [CrossRef]
- Carter, T.J.; Agliardi, G.; Lin, F.Y.; Ellis, M.; Jones, C.; Robson, M.; Richard-Londt, A.; Southern, P.; Lythgoe, M.; Zaw Thin, M.; et al. Potential of Magnetic Hyperthermia to Stimulate Localized Immune Activation. Small 2021, 17, e2005241. [Google Scholar] [CrossRef]
- Maier-Hauff, K.; Rothe, R.; Scholz, R.; Gneveckow, U.; Wust, P.; Thiesen, B.; Feussner, A.; von Deimling, A.; Waldoefner, N.; Felix, R.; et al. Intracranial thermotherapy using magnetic nanoparticles combined with external beam radiotherapy: Results of a feasibility study on patients with glioblastoma multiforme. J. Neurooncol. 2007, 81, 53–60. [Google Scholar] [CrossRef]
- Johannsen, M.; Gneveckow, U.; Thiesen, B.; Taymoorian, K.; Cho, C.H.; Waldofner, N.; Scholz, R.; Jordan, A.; Loening, S.A.; Wust, P. Thermotherapy of prostate cancer using magnetic nanoparticles: Feasibility, imaging, and three-dimensional temperature distribution. Eur. Urol. 2007, 52, 1653–1661. [Google Scholar] [CrossRef]
- Wust, P.; Gneveckow, U.; Johannsen, M.; Bohmer, D.; Henkel, T.; Kahmann, F.; Sehouli, J.; Felix, R.; Ricke, J.; Jordan, A. Magnetic nanoparticles for interstitial thermotherapy--feasibility, tolerance and achieved temperatures. Int. J. Hyperth. 2006, 22, 673–685. [Google Scholar] [CrossRef]
- Maier-Hauff, K.; Ulrich, F.; Nestler, D.; Niehoff, H.; Wust, P.; Thiesen, B.; Orawa, H.; Budach, V.; Jordan, A. Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme. J. Neurooncol. 2011, 103, 317–324. [Google Scholar] [CrossRef]
- Persano, S.; Vicini, F.; Poggi, A.; Fernandez, J.L.C.; Rizzo, G.M.R.; Gavilan, H.; Silvestri, N.; Pellegrino, T. Elucidating the Innate Immunological Effects of Mild Magnetic Hyperthermia on U87 Human Glioblastoma Cells: An In Vitro Study. Pharmaceutics 2021, 13, 1668. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gruosso, T.; Zuo, D.; Omeroglu, A.; Meterissian, S.; Guiot, M.C.; Salazar, A.; Park, M.; Levine, H. Infiltration of CD8(+) T cells into tumor cell clusters in triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3678–3687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covarrubias, G.; Lorkowski, M.E.; Sims, H.M.; Loutrianakis, G.; Rahmy, A.; Cha, A.; Abenojar, E.; Wickramasinghe, S.; Moon, T.J.; Samia, A.C.S.; et al. Hyperthermia-mediated changes in the tumor immune microenvironment using iron oxide nanoparticles. Nanoscale Adv. 2021, 3, 5890–5899. [Google Scholar] [CrossRef] [PubMed]
- Forte, D.; Barone, M.; Palandri, F.; Catani, L. The “Vesicular Intelligence” Strategy of Blood Cancers. Genes 2021, 12, 416. [Google Scholar] [CrossRef] [PubMed]
- Saggioro, M.; D’Angelo, E.; Bisogno, G.; Agostini, M.; Pozzobon, M. Carcinoma and Sarcoma Microenvironment at a Glance: Where We Are. Front. Oncol. 2020, 10, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Huang, Q.; Deng, Y.; Guo, F.; Wang, G.; Liu, M. Crosstalk Between the Tumor Microenvironment and Cancer Cells: A Promising Predictive Biomarker for Immune Checkpoint Inhibitors. Front. Cell Dev. Biol. 2021, 9, 738373. [Google Scholar] [CrossRef]
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef]
- Liao, D.; Johnson, R.S. Hypoxia: A key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007, 26, 281–290. [Google Scholar] [CrossRef]
- Mulens-Arias, V.; Rojas, J.M.; Barber, D.F. The Use of Iron Oxide Nanoparticles to Reprogram Macrophage Responses and the Immunological Tumor Microenvironment. Front. Immunol. 2021, 12, 693709. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Komohara, Y.; Fujiwara, Y.; Ohnishi, K.; Takeya, M. Tumor-associated macrophages: Potential therapeutic targets for anti-cancer therapy. Adv. Drug Deliv. Rev. 2016, 99, 180–185. [Google Scholar] [CrossRef]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Pittet, M.J.; Michielin, O.; Migliorini, D. Clinical relevance of tumour-associated macrophages. Nat. Rev. Clin. Oncol. 2022, 19, 402–421. [Google Scholar] [CrossRef]
- Roszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediators Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [Green Version]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Rayahin, J.E.; Gemeinhart, R.A. Activation of Macrophages in Response to Biomaterials. Results Probl. Cell Differ. 2017, 62, 317–351. [Google Scholar] [CrossRef]
- Galdiero, M.R.; Biswas, S.K.; Mantovani, A. Polarized Activation of Macrophages. In Macrophages: Biology and Role in the Pathology of Diseases; Biswas, S.K., Mantovani, A., Eds.; Springer: New York, NY, USA, 2014; pp. 37–57. [Google Scholar]
- Yu, T.; Gan, S.; Zhu, Q.; Dai, D.; Li, N.; Wang, H.; Chen, X.; Hou, D.; Wang, Y.; Pan, Q.; et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat. Commun. 2019, 10, 4353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchacher, T.; Ohradanova-Repic, A.; Stockinger, H.; Fischer, M.B.; Weber, V. M2 Polarization of Human Macrophages Favors Survival of the Intracellular Pathogen Chlamydia pneumoniae. PLoS ONE 2015, 10, e0143593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dalen, F.J.; van Stevendaal, M.; Fennemann, F.L.; Verdoes, M.; Ilina, O. Molecular Repolarisation of Tumour-Associated Macrophages. Molecules 2018, 24, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado Jde, D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef] [Green Version]
- Vogel, D.Y.; Glim, J.E.; Stavenuiter, A.W.; Breur, M.; Heijnen, P.; Amor, S.; Dijkstra, C.D.; Beelen, R.H. Human macrophage polarization in vitro: Maturation and activation methods compared. Immunobiology 2014, 219, 695–703. [Google Scholar] [CrossRef]
- Nascimento Da Conceicao, V.; Sun, Y.; Ramachandran, K.; Chauhan, A.; Raveendran, A.; Venkatesan, M.; DeKumar, B.; Maity, S.; Vishnu, N.; Kotsakis, G.A.; et al. Resolving macrophage polarization through distinct Ca(2+) entry channel that maintains intracellular signaling and mitochondrial bioenergetics. iScience 2021, 24, 103339. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Sierra-Filardi, E.; Nieto, C.; Dominguez-Soto, A.; Barroso, R.; Sanchez-Mateos, P.; Puig-Kroger, A.; Lopez-Bravo, M.; Joven, J.; Ardavin, C.; Rodriguez-Fernandez, J.L.; et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: Identification of CCL2/CCR2-dependent gene expression profile. J. Immunol. 2014, 192, 3858–3867. [Google Scholar] [CrossRef]
- Sica, A.; Erreni, M.; Allavena, P.; Porta, C. Macrophage polarization in pathology. Cell Mol. Life Sci. 2015, 72, 4111–4126. [Google Scholar] [CrossRef]
- Sen, P.; Kemppainen, E.; Oresic, M. Perspectives on Systems Modeling of Human Peripheral Blood Mononuclear Cells. Front Mol. Biosci. 2017, 4, 96. [Google Scholar] [CrossRef] [Green Version]
- Italiani, P.; Boraschi, D. Development and Functional Differentiation of Tissue-Resident Versus Monocyte-Derived Macrophages in Inflammatory Reactions. Results Probl Cell Differ 2017, 62, 23–43. [Google Scholar] [CrossRef]
- Isidro, R.A.; Appleyard, C.B. Colonic macrophage polarization in homeostasis, inflammation, and cancer. Am. J. Physiol. Gastrointest Liver Physiol. 2016, 311, G59–G73. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Helming, L.; Milde, R.; Varin, A.; Melgert, B.N.; Draijer, C.; Thomas, B.; Fabbri, M.; Crawshaw, A.; Ho, L.P.; et al. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: Similarities and differences. Blood 2013, 121, e57–e69. [Google Scholar] [CrossRef]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef]
- Polasky, C.; Studt, T.; Steuer, A.K.; Loyal, K.; Ludtke-Buzug, K.; Bruchhage, K.L.; Pries, R. Impact of Superparamagnetic Iron Oxide Nanoparticles on THP-1 Monocytes and Monocyte-Derived Macrophages. Front. Mol. Biosci. 2022, 9, 811116. [Google Scholar] [CrossRef]
- Wu, Q.; Miao, T.; Feng, T.; Yang, C.; Guo, Y.; Li, H. Dextrancoated superparamagnetic iron oxide nanoparticles activate the MAPK pathway in human primary monocyte cells. Mol. Med. Rep. 2018, 18, 564–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonnissen, D.; Qu, Y.; Langer, K.; Ozturk, C.; Zhao, Y.; Chen, C.; Seebohm, G.; Dufer, M.; Fuchs, H.; Galla, H.J.; et al. Comparison of cellular effects of starch-coated SPIONs and poly(lactic-co-glycolic acid) matrix nanoparticles on human monocytes. Int. J. Nanomed. 2016, 11, 5221–5236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharkey, J.; Starkey Lewis, P.J.; Barrow, M.; Alwahsh, S.M.; Noble, J.; Livingstone, E.; Lennen, R.J.; Jansen, M.A.; Carrion, J.G.; Liptrott, N.; et al. Functionalized superparamagnetic iron oxide nanoparticles provide highly efficient iron-labeling in macrophages for magnetic resonance-based detection in vivo. Cytotherapy 2017, 19, 555–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulens-Arias, V.; Rojas, J.M.; Perez-Yague, S.; Morales, M.P.; Barber, D.F. Polyethylenimine-coated SPIONs trigger macrophage activation through TLR-4 signaling and ROS production and modulate podosome dynamics. Biomaterials 2015, 52, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Liu, L.; Zhu, W.; Li, D.; Yang, L.; Duan, J.; Cai, Z.; Nie, Y.; Zhang, Y.; Gong, Q.; et al. Iron oxide nanoparticles promote macrophage autophagy and inflammatory response through activation of toll-like Receptor-4 signaling. Biomaterials 2019, 203, 23–30. [Google Scholar] [CrossRef]
- Rojas, J.M.; Sanz-Ortega, L.; Mulens-Arias, V.; Gutierrez, L.; Perez-Yague, S.; Barber, D.F. Superparamagnetic iron oxide nanoparticle uptake alters M2 macrophage phenotype, iron metabolism, migration and invasion. Nanomedicine 2016, 12, 1127–1138. [Google Scholar] [CrossRef]
- Laskar, A.; Eilertsen, J.; Li, W.; Yuan, X.M. SPION primes THP1 derived M2 macrophages towards M1-like macrophages. Biochem. Biophys. Res. Commun. 2013, 441, 737–742. [Google Scholar] [CrossRef]
- Yu, M.; Huang, S.; Yu, K.J.; Clyne, A.M. Dextran and polymer polyethylene glycol (PEG) coating reduce both 5 and 30 nm iron oxide nanoparticle cytotoxicity in 2D and 3D cell culture. Int. J. Mol. Sci. 2012, 13, 5554–5570. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Hu, Y.; Wang, J.; Gao, X.; Qian, X.; Tang, M. Superparamagnetic Iron Oxide Nanoparticles: Cytotoxicity, Metabolism, and Cellular Behavior in Biomedicine Applications. Int. J. Nanomed. 2021, 16, 6097–6113. [Google Scholar] [CrossRef]
- Kodali, V.; Littke, M.H.; Tilton, S.C.; Teeguarden, J.G.; Shi, L.; Frevert, C.W.; Wang, W.; Pounds, J.G.; Thrall, B.D. Dysregulation of macrophage activation profiles by engineered nanoparticles. ACS Nano 2013, 7, 6997–7010. [Google Scholar] [CrossRef]
- Robert, J. Textbook of Cell Signalling in Cancer: An Educational Approach; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Heinsbroek, S.E.; Gordon, S. The role of macrophages in inflammatory bowel diseases. Expert Rev. Mol. Med. 2009, 11, e14. [Google Scholar] [CrossRef]
- Lim, S.G.; Kim, J.K.; Suk, K.; Lee, W.H. Crosstalk between signals initiated from TLR4 and cell surface BAFF results in synergistic induction of proinflammatory mediators in THP-1 cells. Sci. Rep. 2017, 7, 45826. [Google Scholar] [CrossRef]
- Broz, P.; Monack, D.M. Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 2013, 13, 551–565. [Google Scholar] [CrossRef]
- Chen, H.; Li, P.; Yin, Y.; Cai, X.; Huang, Z.; Chen, J.; Dong, L.; Zhang, J. The promotion of type 1 T helper cell responses to cationic polymers in vivo via toll-like receptor-4 mediated IL-12 secretion. Biomaterials 2010, 31, 8172–8180. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef] [Green Version]
- Siegrist, S.; Kettiger, H.; Fasler-Kan, E.; Huwyler, J. Selective stimulation of the JAK/STAT signaling pathway by silica nanoparticles in human endothelial cells. Toxicol. Vitr. 2017, 42, 308–318. [Google Scholar] [CrossRef]
- Nascimento, C.S.; Alves, E.A.R.; de Melo, C.P.; Correa-Oliveira, R.; Calzavara-Silva, C.E. Immunotherapy for cancer: Effects of iron oxide nanoparticles on polarization of tumor-associated macrophages. Nanomedicine 2021, 16, 2633–2650. [Google Scholar] [CrossRef]
- Sukhbaatar, N.; Weichhart, T. Iron Regulation: Macrophages in Control. Pharmaceuticals 2018, 11, 137. [Google Scholar] [CrossRef]
- Jennifer, B.; Berg, V.; Modak, M.; Puck, A.; Seyerl-Jiresch, M.; Kunig, S.; Zlabinger, G.J.; Steinberger, P.; Chou, J.; Geha, R.S.; et al. Transferrin receptor 1 is a cellular receptor for human heme-albumin. Commun. Biol. 2020, 3, 621. [Google Scholar] [CrossRef]
- Nelson, N.R.; Port, J.D.; Pandey, M.K. Use of Superparamagnetic Iron Oxide Nanoparticles (SPIONs) via Multiple Imaging Modalities and Modifications to Reduce Cytotoxicity: An Educational Review. JNT 2020, 1, 8. [Google Scholar] [CrossRef]
- Nairz, M.; Theurl, I.; Swirski, F.K.; Weiss, G. “Pumping iron”-how macrophages handle iron at the systemic, microenvironmental, and cellular levels. Pflugers Arch. 2017, 469, 397–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaneda, O.A.; Lee, S.C.; Ho, C.T.; Huang, T.C. Macrophages in oxidative stress and models to evaluate the antioxidant function of dietary natural compounds. J. Food Drug Anal. 2017, 25, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canton, M.; Sanchez-Rodriguez, R.; Spera, I.; Venegas, F.C.; Favia, M.; Viola, A.; Castegna, A. Reactive Oxygen Species in Macrophages: Sources and Targets. Front. Immunol. 2021, 12, 734229. [Google Scholar] [CrossRef]
- Freeman, S.A.; Grinstein, S. Phagocytosis: Receptors, signal integration, and the cytoskeleton. Immunol. Rev. 2014, 262, 193–215. [Google Scholar] [CrossRef]
- Tertrais, M.; Bigot, C.; Martin, E.; Poincloux, R.; Labrousse, A.; Maridonneau-Parini, I. Phagocytosis is coupled to the formation of phagosome-associated podosomes and a transient disruption of podosomes in human macrophages. Eur. J. Cell Biol. 2021, 100, 151161. [Google Scholar] [CrossRef]
- Wheeler, K.C.; Jena, M.K.; Pradhan, B.S.; Nayak, N.; Das, S.; Hsu, C.D.; Wheeler, D.S.; Chen, K.; Nayak, N.R. VEGF may contribute to macrophage recruitment and M2 polarization in the decidua. PLoS ONE 2018, 13, e0191040. [Google Scholar] [CrossRef]
- Richter, M.; Piwocka, O.; Musielak, M.; Piotrowski, I.; Suchorska, W.M.; Trzeciak, T. From Donor to the Lab: A Fascinating Journey of Primary Cell Lines. Front. Cell Dev. Biol. 2021, 9, 711381. [Google Scholar] [CrossRef]





| Type | Spontaneous Magnetization | Description |
|---|---|---|
| Diamagnetism | No | Electron magnetic moment compensation. Magnetic interactions within atoms. No exchange magnetic interaction between atoms and molecules. Weakly repelled by magnetic fields. |
Paramagnetism | No | Presence of unpaired electrons in the electronic configuration. Weakly attracted by magnetic fields. |
Antiferromagnetism | No | Antiparallel ordered magnetic moments. Canting of magnetic moments leading to the appearance of small net magnetization along the direction of the applied magnetic field. |
Ferrimagnetism | Yes | Antiparallel unbalanced magnetic moments. Small net magnetic moment at zero applied magnetic field. |
Ferromagnetism | Yes | Parallel magnetic moments. Strong net magnetic moment at zero applied magnetic field. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dias, A.M.M.; Courteau, A.; Bellaye, P.-S.; Kohli, E.; Oudot, A.; Doulain, P.-E.; Petitot, C.; Walker, P.-M.; Decréau, R.; Collin, B. Superparamagnetic Iron Oxide Nanoparticles for Immunotherapy of Cancers through Macrophages and Magnetic Hyperthermia. Pharmaceutics 2022, 14, 2388. https://doi.org/10.3390/pharmaceutics14112388
Dias AMM, Courteau A, Bellaye P-S, Kohli E, Oudot A, Doulain P-E, Petitot C, Walker P-M, Decréau R, Collin B. Superparamagnetic Iron Oxide Nanoparticles for Immunotherapy of Cancers through Macrophages and Magnetic Hyperthermia. Pharmaceutics. 2022; 14(11):2388. https://doi.org/10.3390/pharmaceutics14112388
Chicago/Turabian StyleDias, Alexandre M. M., Alan Courteau, Pierre-Simon Bellaye, Evelyne Kohli, Alexandra Oudot, Pierre-Emmanuel Doulain, Camille Petitot, Paul-Michael Walker, Richard Decréau, and Bertrand Collin. 2022. "Superparamagnetic Iron Oxide Nanoparticles for Immunotherapy of Cancers through Macrophages and Magnetic Hyperthermia" Pharmaceutics 14, no. 11: 2388. https://doi.org/10.3390/pharmaceutics14112388