

Antisense Oligonucleotide Therapy for the Nervous System: From Bench to Bedside with Emphasis on Pediatric Neurology

 , and
, and
Abstract
:1. Introduction
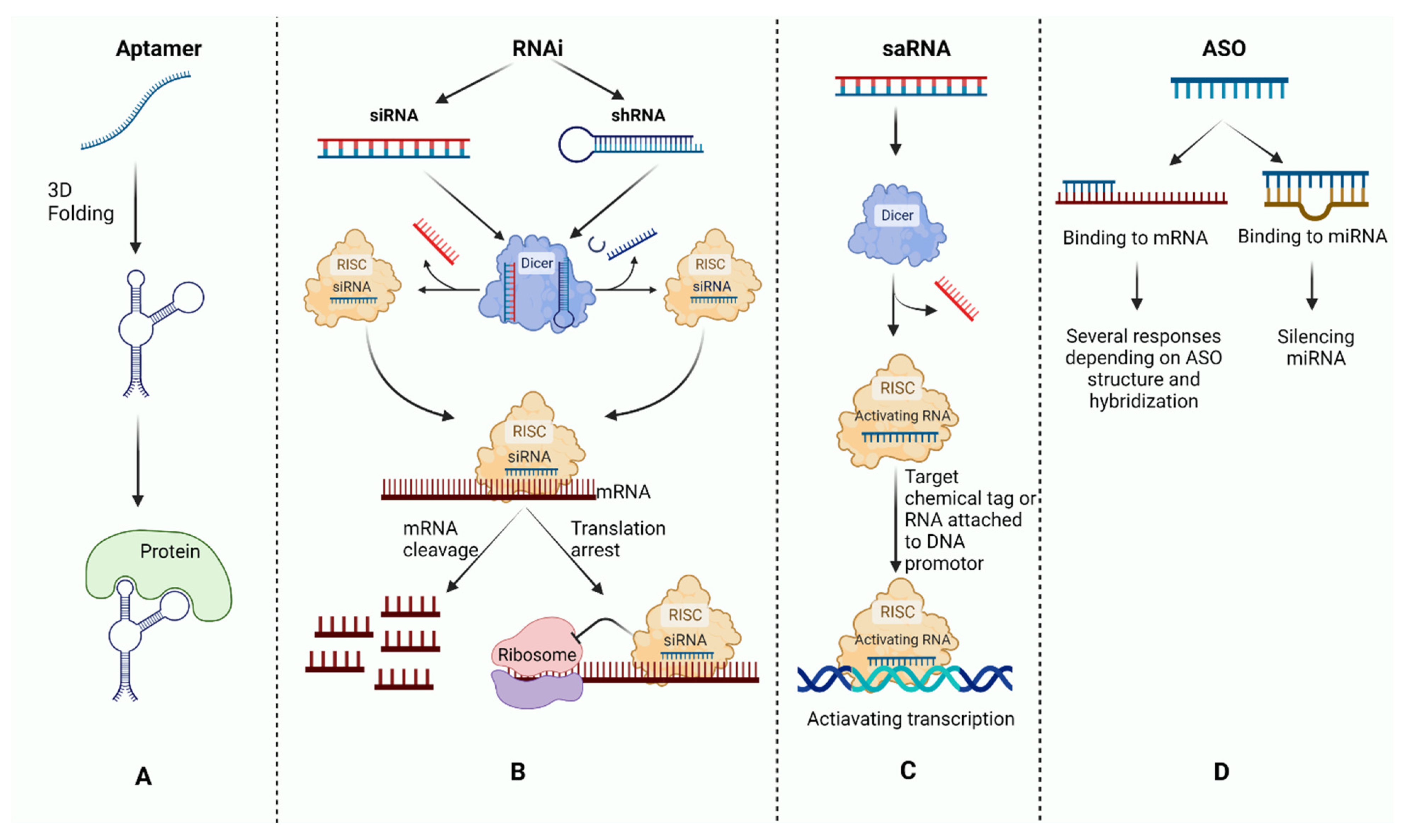
2. Antisense Oligonucleotide Mechanisms
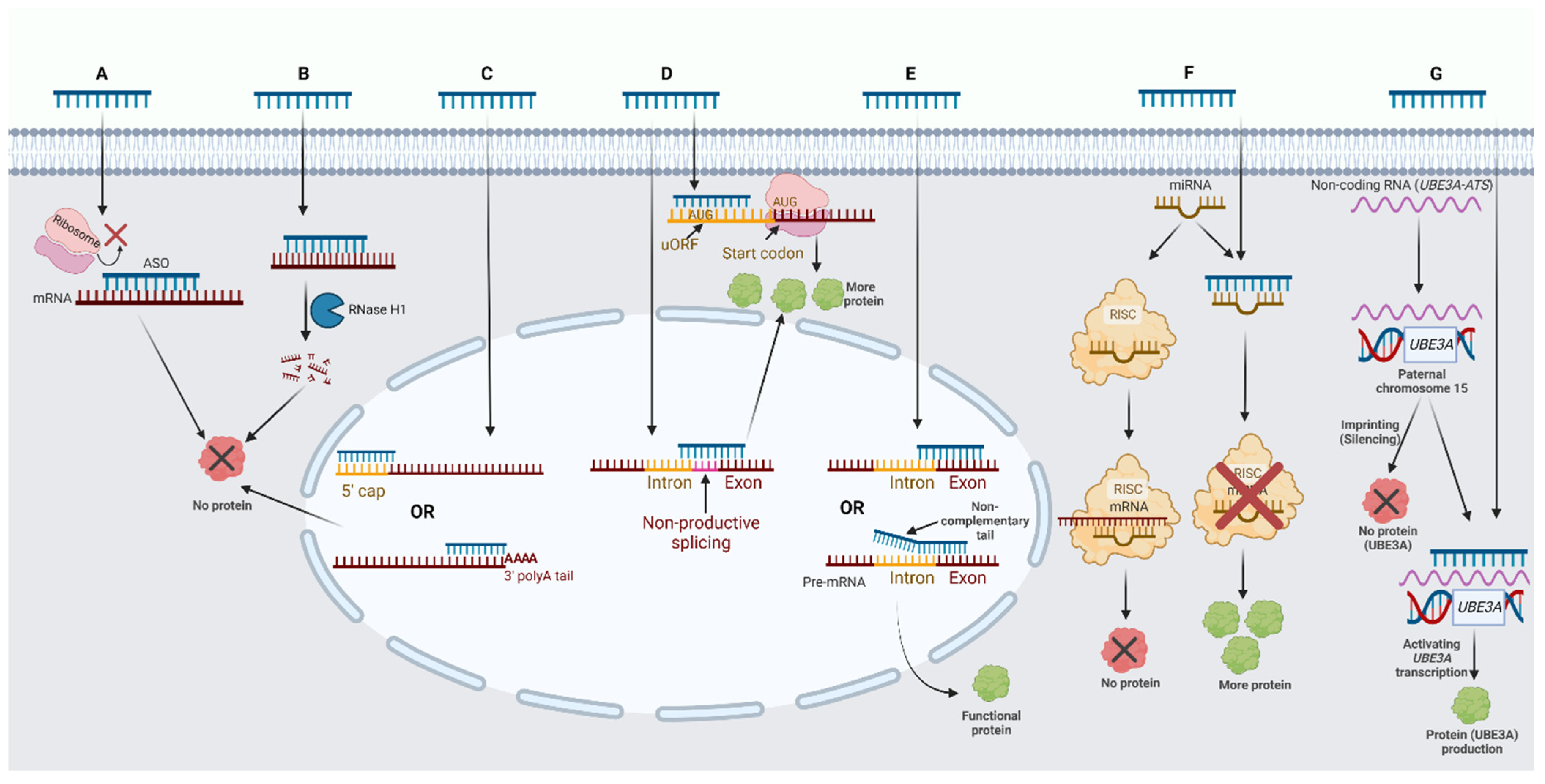
2.1. ASO Mechanisms of Action on Protein-Coding RNA
2.2. ASO Mechanisms of Action on Noncoding RNA
3. Design Considerations of Antisense Oligonucleotides
3.1. Accessibility of the Target Sequence That Binds the ASO
3.2. Stability of the ASO-RNA Duplex
3.3. Specificity of the ASO Target Sequence
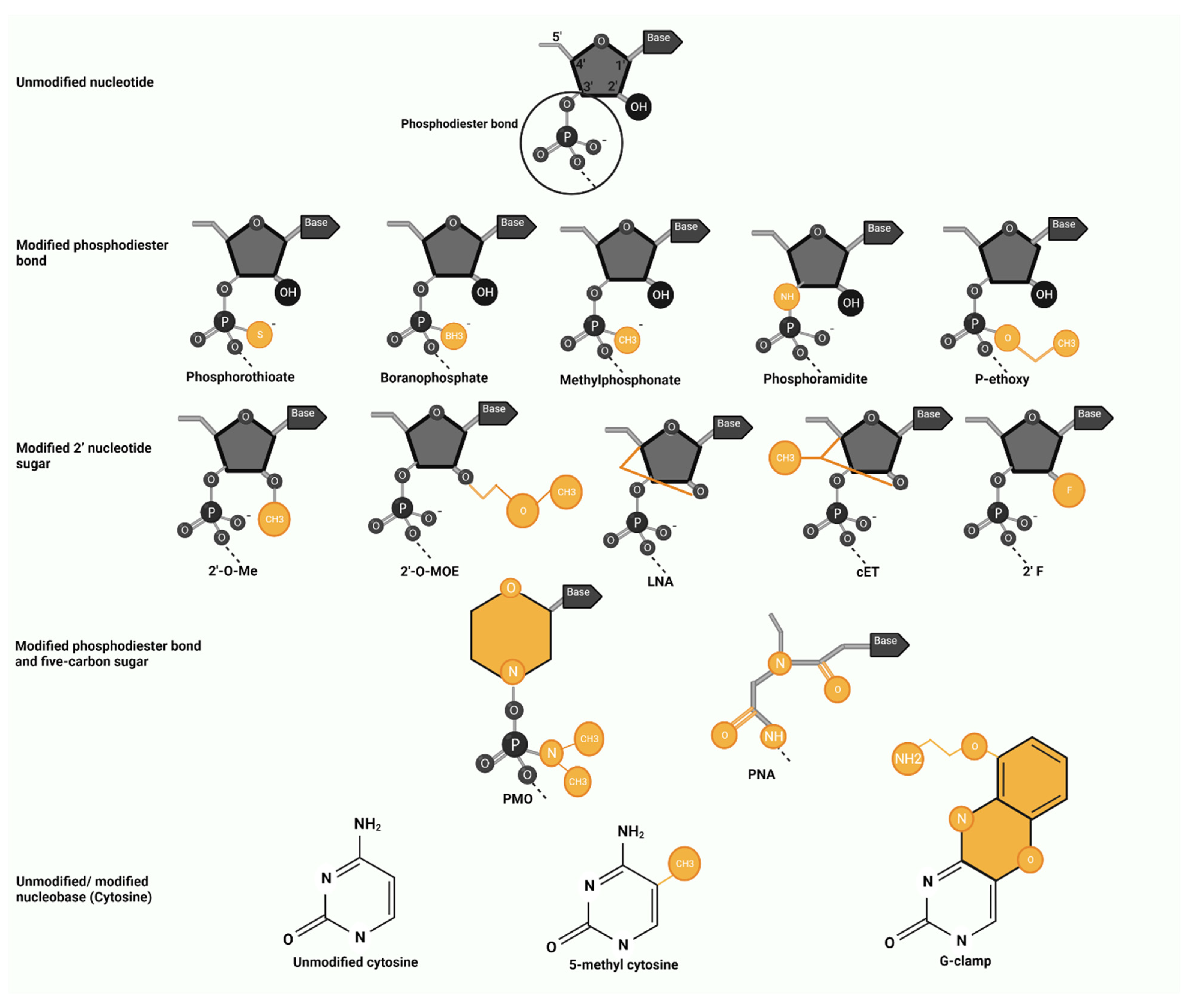
4. Antisense Oligonucleotide Modifications
4.1. Modified Phosphodiester Bond
4.2. Modified 2′ Nucleotide Sugar
4.3. Modified Phosphodiester Bond and Five-Carbon Sugar
4.4. Modified Nucleobase
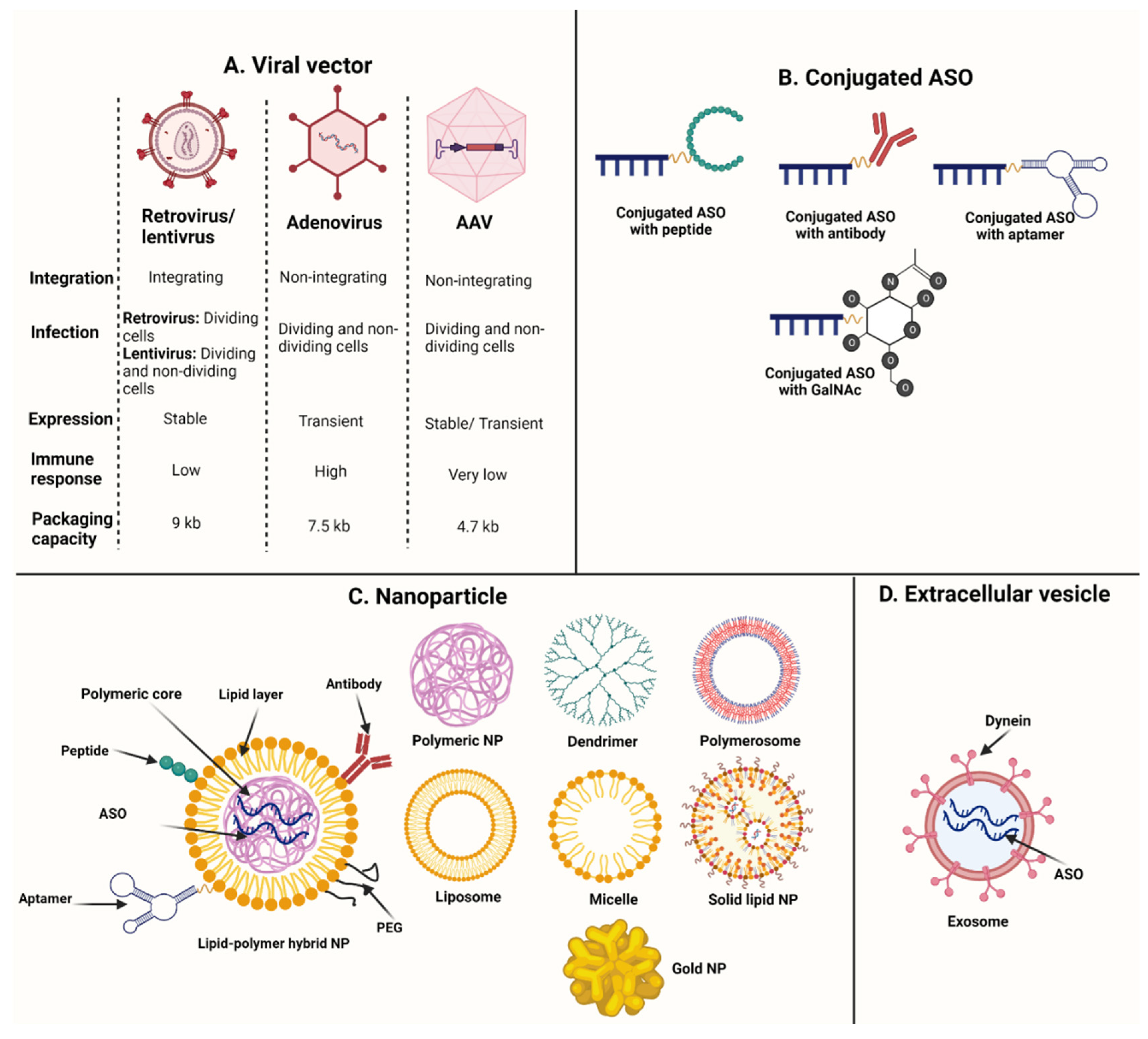
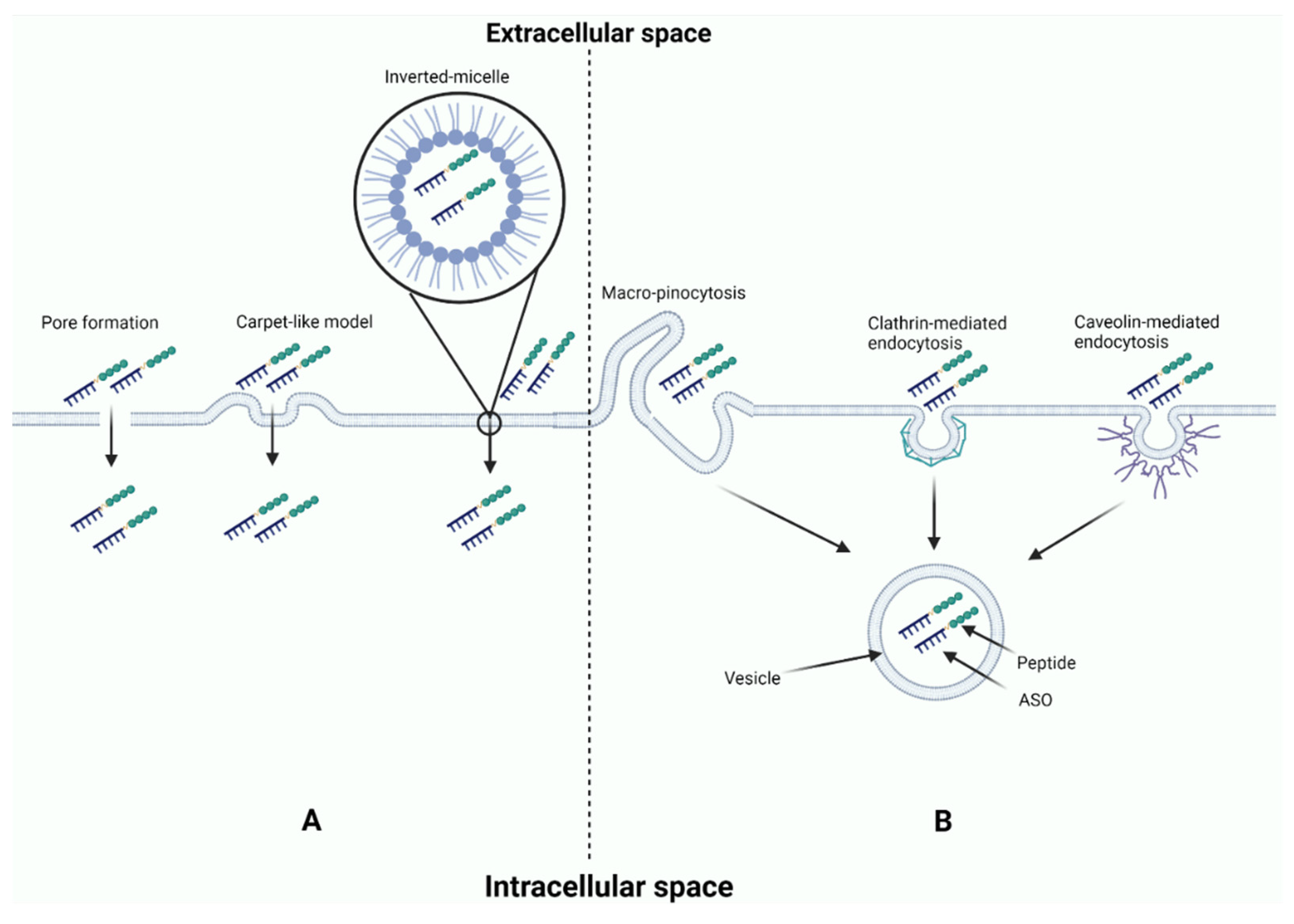
5. Antisense Oligonucleotide Delivery
5.1. Viral Vectors
5.2. Conjugated Peptides
5.3. Conjugated Antibodies
5.4. Other Conjugated Ligands
5.5. Nanoparticles
5.6. Extracellular Vesicles
6. Clinical Trials Using ASOs in Neurological Disorders
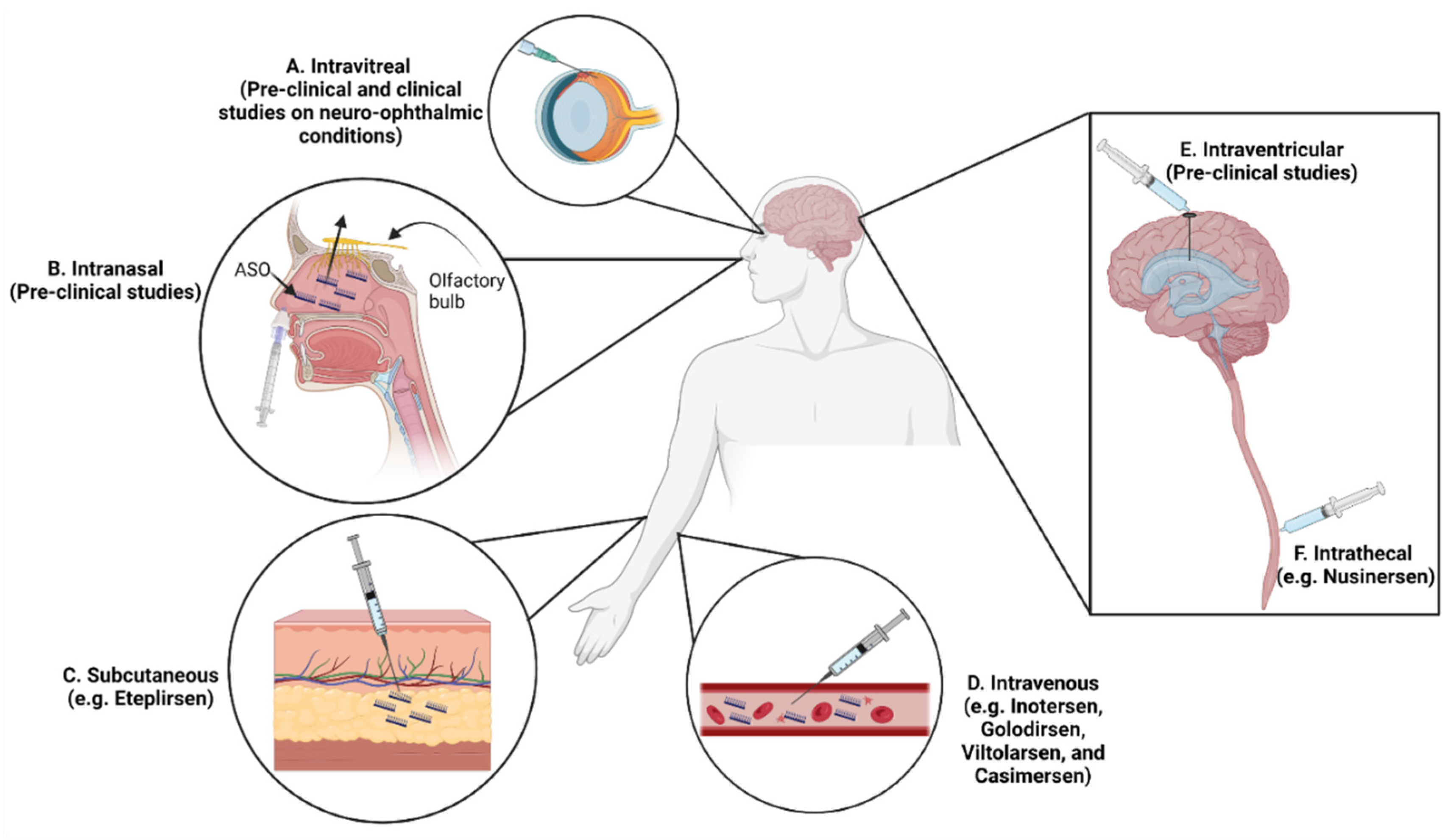
7. Routes of ASO Administration in Neurological Disorders
8. Neurological Disorders with FDA-Approved ASOs
8.1. Spinal Muscular Atrophy
8.2. Polyneuropathy Caused by Hereditary Transthyretin Amyloidosis
8.3. Duchenne Muscular Dystrophy
9. White Matter Disorders
9.1. Alexander Disease
9.2. Canavan Disease
9.3. Pelizaeus–Merzbacher Disease
9.4. Leukoencephalopathy with Brain Stem and Spinal Cord Involvement and Lactate Elevation
10. Neurodevelopmental Disorders
10.1. Angelman Syndrome
10.2. Dravet Syndrome
11. Antisense Oligonucleotide Adverse Events
12. Future Directions
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gragoudas, E.S.; Adamis, A.P.; Cunningham, E.T., Jr.; Feinsod, M.; Guyer, D.R. Pegaptanib for neovascular age-related macular degeneration. N. Engl. J. Med. 2004, 351, 2805–2816. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef]
- Askari, F.K.; McDonnell, W.M. Antisense-oligonucleotide therapy. N. Engl. J. Med. 1996, 334, 316–318. [Google Scholar] [CrossRef]
- Vitravene Study Group. A randomized controlled clinical trial of intravitreous fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus retinitis in patients with AIDS. Am. J. Ophthalmol. 2002, 133, 467–474. [Google Scholar]
- Thomas, G.S.; Cromwell, W.C.; Ali, S.; Chin, W.; Flaim, J.D.; Davidson, M. Mipomersen, an apolipoprotein B synthesis inhibitor, reduces atherogenic lipoproteins in patients with severe hypercholesterolemia at high cardiovascular risk: A randomized, double-blind, placebo-controlled trial. J. Am. Coll. Cardiol. 2013, 62, 2178–2184. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E.; Eteplirsen Study Group and Telethon Foundation DMD Italian Network. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef]
- Richardson, P.G.; Riches, M.L.; Kernan, N.A.; Brochstein, J.A.; Mineishi, S.; Termuhlen, A.M.; Arai, S.; Grupp, S.A.; Guinan, E.C.; Martin, P.L.; et al. Phase 3 trial of defibrotide for the treatment of severe veno-occlusive disease and multi-organ failure. Blood J. Am. Soc. Hematol. 2016, 127, 1656–1665. [Google Scholar] [CrossRef] [Green Version]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Servais, L.; Mercuri, E.; Straub, V.; Guglieri, M.; Seferian, A.M.; Scoto, M.; Leone, D.; Koenig, E.; Khan, N.; Dugar, A.; et al. Long-term safety and efficacy data of golodirsen in ambulatory patients with Duchenne muscular dystrophy amenable to exon 53 skipping: A first-in-human, multicenter, two-part, open-label, phase 1/2 trial. Nucleic Acid Ther. 2022, 32, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Komaki, H.; Nagata, T.; Saito, T.; Masuda, S.; Takeshita, E.; Sasaki, M.; Tachimori, H.; Nakamura, H.; Aoki, Y.; Takeda, S.I. Systemic administration of the antisense oligonucleotide NS-065/NCNP-01 for skipping of exon 53 in patients with Duchenne muscular dystrophy. Sci. Transl. Med. 2018, 10, eaan0713. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.R.; Kuntz, N.L.; Koenig, E.; East, L.; Upadhyay, S.; Han, B.; Shieh, P.B. Safety, tolerability, and pharmacokinetics of casimersen in patients with Duchenne muscular dystrophy amenable to exon 45 skipping: A randomized, double-blind, placebo-controlled, dose-titration trial. Muscle Nerve 2021, 64, 285–292. [Google Scholar] [CrossRef]
- Adams, D.; Tournev, I.L.; Taylor, M.S.; Coelho, T.; Planté-Bordeneuve, V.; Berk, J.L.; González-Duarte, A.; Gillmore, J.D.; Low, S.C.; Sekijima, Y.; et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: A randomized clinical trial. Amyloid 2022, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hulton, S.A.; Groothoff, J.W.; Frishberg, Y.; Koren, M.J.; Overcash, J.S.; Sellier-Leclerc, A.L.; Shasha-Lavsky, H.; Saland, J.M.; Hayes, W.; Magen, D.; et al. Randomized Clinical Trial on the Long-Term Efficacy and Safety of Lumasiran in Patients with Primary Hyperoxaluria Type 1. Kidney Int. Rep. 2022, 7, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Stoekenbroek, R.M.; Kallend, D.; Nishikido, T.; Leiter, L.A.; Landmesser, U.; Wright, R.S.; Wijngaard, P.L.; Kastelein, J.J. Effect of 1 or 2 doses of inclisiran on low-density lipoprotein cholesterol levels: One-year follow-up of the ORION-1 randomized clinical trial. JAMA Cardiol. 2019, 4, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.R.; Pottier, M.; Vekris, A.; Opolon, P.; Maksimenko, A.; Malvy, C. Comparison of antisense oligonucleotides and siRNAs in cell culture and in vivo. Biochem. Biophys. Res. Commun. 2002, 296, 1000–1004. [Google Scholar] [CrossRef]
- Miyagishi, M.; Hayashi, M.; Taira, K. Comparison of the suppressive effects of antisense oligonucleotides and siRNAs directed against the same targets in mammalian cells. Antisense Nucleic Acid Drug Dev. 2003, 13, 1–7. [Google Scholar] [CrossRef]
- Kretschmer-Kazemi Far, R.; Sczakiel, G. The activity of siRNA in mammalian cells is related to structural target accessibility: A comparison with antisense oligonucleotides. Nucleic Acids Res. 2003, 31, 4417–4424. [Google Scholar] [CrossRef] [Green Version]
- Vickers, T.A.; Koo, S.; Bennett, C.F.; Crooke, S.T.; Dean, N.M.; Baker, B.F. Efficient reduction of target RNAs by small interfering RNA and RNase H-dependent antisense agents: A comparative analysis. J. Biol. Chem. 2003, 278, 7108–7118. [Google Scholar] [CrossRef] [Green Version]
- Tallet-Lopez, B.; Aldaz-Carroll, L.; Chabas, S.; Dausse, E.; Staedel, C.; Toulmé, J.J. Antisense oligonucleotides targeted to the domain IIId of the hepatitis C virus IRES compete with 40S ribosomal subunit binding and prevent in vitro translation. Nucleic Acids Res. 2003, 31, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Walder, R.Y.; Walder, J.A. Role of RNase H in hybrid-arrested translation by antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1988, 85, 5011–5015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, B.F.; Lot, S.S.; Condon, T.P.; Cheng-Flournoy, S.; Lesnik, E.A.; Sasmor, H.M.; Bennett, C.F. 2′-O-(2-Methoxy) ethyl-modified anti-intercellular adhesion molecule 1 (ICAM-1) oligonucleotides selectively increase the ICAM-1 mRNA level and inhibit formation of the ICAM-1 translation initiation complex in human umbilical vein endothelial cells. J. Biol. Chem. 1997, 272, 11994–12000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchalot, A.; Horiot, C.; Lambert, J.M.; Carrion, C.; Oblet, C.; Pollet, J.; Cogné, M.; Moreau, J.; Laffleur, B.; Delpy, L. Targeting IgE polyadenylation signal with antisense oligonucleotides decreases IgE secretion and plasma cell viability. J. Allergy Clin. Immunol. 2022, 149, 1795–1801. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Shen, W.; Sun, H.; Migawa, M.T.; Vickers, T.A.; Crooke, S.T. Translation efficiency of mRNAs is increased by antisense oligonucleotides targeting upstream open reading frames. Nat. Biotechnol. 2016, 34, 875–880. [Google Scholar] [CrossRef]
- Liang, X.H.; Sun, H.; Shen, W.; Wang, S.; Yao, J.; Migawa, M.T.; Bui, H.H.; Damle, S.S.; Riney, S.; Graham, M.J.; et al. Antisense oligonucleotides targeting translation inhibitory elements in 5′ UTRs can selectively increase protein levels. Nucleic Acids Res. 2017, 45, 9528–9546. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Chen, C.; Christiansen, A.; Ji, S.; Lin, Q.; Anumonwo, C.; Liu, C.; Leiser, S.C.; Meena; Aznarez, I.; et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci. Transl. Med. 2020, 12, eaaz6100. [Google Scholar] [CrossRef]
- Al-Ali, R.; González-Sarmiento, R. Proximity of AUG sequences to initiation codon in genomic 5′ UTR regulates mammalian protein expression. Gene 2016, 594, 268–271. [Google Scholar] [CrossRef]
- Calvo, S.E.; Pagliarini, D.J.; Mootha, V.K. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. USA 2009, 106, 7507–7512. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.H.; Han, Z.; Jeon, H.Y.; Kach, J.; Jing, E.; Weyn-Vanhentenryck, S.; Downs, M.; Corrionero, A.; Oh, R.; Scharner, J.; et al. Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat. Commun. 2020, 11, 3501. [Google Scholar] [CrossRef]
- Climente-González, H.; Porta-Pardo, E.; Godzik, A.; Eyras, E. The functional impact of alternative splicing in cancer. Cell Rep. 2017, 20, 2215–2226. [Google Scholar] [CrossRef] [PubMed]
- Biamonti, G.; Amato, A.; Belloni, E.; Di Matteo, A.; Infantino, L.; Pradella, D.; Ghigna, C. Alternative splicing in Alzheimer’s disease. Aging Clin. Exp. Res. 2021, 33, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Slaugenhaupt, S.A.; Gusella, J.F. Familial dysautonomia. Curr. Opin. Genet. Dev. 2002, 12, 307–311. [Google Scholar] [CrossRef]
- Gold-von Simson, G.; Goldberg, J.D.; Rolnitzky, L.M.; Mull, J.; Leyne, M.; Voustianiouk, A.; Slaugenhaupt, S.A.; Axelrod, F.B. Kinetin in familial dysautonomia carriers: Implications for a new therapeutic strategy targeting mRNA splicing. Pediatr. Res. 2009, 65, 341–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porensky, P.N.; Burghes, A.H. Antisense oligonucleotides for the treatment of spinal muscular atrophy. Hum. Gene Ther. 2013, 24, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Skordis, L.A.; Dunckley, M.G.; Yue, B.; Eperon, I.C.; Muntoni, F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc. Natl. Acad. Sci. USA 2003, 100, 4114–4119. [Google Scholar] [CrossRef] [Green Version]
- van Berge, L.; Dooves, S.; van Berkel, C.G.; Polder, E.; van der Knaap, M.S.; Scheper, G.C. Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation is associated with cell-type-dependent splicing of mtAspRS mRNA. Biochem. J. 2012, 441, 955–962. [Google Scholar] [CrossRef]
- Maniataki, E.; Mourelatos, Z. A human, ATP-independent, RISC assembly machine fueled by pre-miRNA. Genes Dev. 2005, 19, 2979–2990. [Google Scholar] [CrossRef] [Green Version]
- Di Leva, G.; Croce, C.M. miRNA profiling of cancer. Curr. Opin. Genet. Dev. 2013, 23, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Reddy, K.B. MicroRNA (miRNA) in cancer. Cancer Cell Int. 2015, 15, 38. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yang, Y.B.; Zhang, X.H.; Yu, X.L.; Wang, Z.B.; Cheng, X.C. MicroRNA-21 gene and cancer. Med. Oncol. 2013, 30, 376. [Google Scholar] [CrossRef] [PubMed]
- Higgs, G.; Slack, F. The multiple roles of microRNA-155 in oncogenesis. J. Clin. Bioinform. 2013, 3, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Song, W.; Boulanger, J.; Xu, E.Y.; Wang, F.; Zhang, Y.; He, Q.; Wang, S.; Yang, L.; Pryce, C.; et al. Dysregulated expression of microRNA-21 and disease-related genes in human patients and in a mouse model of Alport syndrome. Hum. Gene Ther. 2019, 30, 865–881. [Google Scholar] [CrossRef] [PubMed]
- Ucar, A.; Gupta, S.K.; Fiedler, J.; Erikci, E.; Kardasinski, M.; Batkai, S.; Dangwal, S.; Kumarswamy, R.; Bang, C.; Holzmann, A.; et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 2012, 3, 1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aartsma-Rus, A.; Van Vliet, L.; Hirschi, M.; Janson, A.A.; Heemskerk, H.; De Winter, C.L.; De Kimpe, S.; Van Deutekom, J.C.; Ac’t Hoen, P.; van Ommen, G.J. Guidelines for antisense oligonucleotide design and insight into splice-modulating mechanisms. Mol. Ther. 2009, 17, 548–553. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef] [Green Version]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.; Hanson, J.; Paliwal, K.; Zhou, Y. RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning. Nat. Commun. 2019, 10, 5407. [Google Scholar] [CrossRef] [Green Version]
- Andronescu, M.; Aguirre-Hernandez, R.; Condon, A.; Hoos, H.H. RNAsoft: A suite of RNA secondary structure prediction and design software tools. Nucleic Acids Res. 2003, 31, 3416–3422. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.H.; Lim, S.; Wong, W.F. Antisense oligonucleotides: From design to therapeutic application. Clin. Exp. Pharmacol. Physiol. 2006, 33, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.S.; Britton, D.H.; Stone, B.A.; Behrens, D.L.; Leffet, L.M.; Hobbs, F.W.; Miller, J.A.; Trainor, G.L. Potent antisense oligonucleotides to the human multidrug resistance-1 mRNA are rationally selected by mapping RNA-accessible sites with oligonucleotide libraries. Nucleic Acids Res. 1996, 24, 1901–1907. [Google Scholar]
- Matveeva, O.V.; Tsodikov, A.D.; Giddings, M.; Freier, S.M.; Wyatt, J.R.; Spiridonov, A.A.; Shabalina, S.A.; Gesteland, R.F.; Atkins, J.F. Identification of sequence motifs in oligonucleotides whose presence is correlated with antisense activity. Nucleic Acids Res. 2000, 28, 2862–2865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35 (Suppl. S2), W43–W46. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36 (Suppl. S2), W5–W9. [Google Scholar] [CrossRef]
- Furdon, P.J.; Dominski, Z.; Kole, R. RNase H cleavage of RNA hybridized to oligonucleotides containing methylphosphonate, phosphorothioate and phosphodiester bonds. Nucleic Acids Res. 1989, 17, 9193–9204. [Google Scholar] [CrossRef] [Green Version]
- Eckstein, F. Phosphorothioate oligodeoxynucleotides: What is their origin and what is unique about them? Antisense Nucleic Acid Drug Dev. 2000, 10, 117–121. [Google Scholar] [CrossRef]
- Woolf, T.M.; Jennings, C.G.; Rebagliati, M.; Melton, D.A. The stability, toxicity and effectiveness of unmodified and phosphorothioate antisense oligodeoxynucleotides in Xenopus oocytes and embryos. Nucleic Acids Res. 1990, 18, 1763–1769. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.K.; Tewary, H.K.; Iversen, P.L. Characterization of binding sites, extent of binding, and drug interactions of oligonucleotides with albumin. Antisense Res. Dev. 1995, 5, 131–139. [Google Scholar] [CrossRef]
- Pohar, J.; Lainšček, D.; Kunšek, A.; Cajnko, M.M.; Jerala, R.; Benčina, M. Phosphodiester backbone of the CpG motif within immunostimulatory oligodeoxynucleotides augments activation of Toll-like receptor 9. Sci. Rep. 2017, 7, 14598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, A.A. A review of issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim. Et Biophys. Acta-Gene Struct. Expr. 2000, 1489, 69–84. [Google Scholar] [CrossRef]
- Rait, V.K.; Shaw, B.R. Boranophosphates support the RNase H cleavage of polyribonucleotides. Antisense Nucleic Acid Drug Dev. 1999, 9, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dobrikov, M.; Sergueev, D.; Ramsay Shaw, B. RNase H activation by stereoregular boranophosphate oligonucleotide. Nucl. Nucl. Nucleic Acids 2003, 22, 1151–1153. [Google Scholar] [CrossRef]
- Mignet, N.; Gryaznov, S.M. Zwitterionic oligodeoxyribonucleotide N3′→ P5′ phosphoramidates: Synthesis and properties. Nucleic Acids Res. 1998, 26, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Faria, M.; Spiller, D.G.; Dubertret, C.; Nelson, J.S.; White, M.R.; Scherman, D.; Hélène, C.; Giovannangeli, C. Phosphoramidate oligonucleotides as potent antisense molecules in cells and in vivo. Nat. Biotechnol. 2001, 19, 40–44. [Google Scholar] [CrossRef]
- Miroshnichenko, S.K.; Patutina, O.A.; Burakova, E.A.; Chelobanov, B.P.; Fokina, A.A.; Vlassov, V.V.; Altman, S.; Zenkova, M.A.; Stetsenko, D.A. Mesyl phosphoramidate antisense oligonucleotides as an alternative to phosphorothioates with improved biochemical and biological properties. Proc. Natl. Acad. Sci. USA 2019, 116, 1229–1234. [Google Scholar] [CrossRef] [Green Version]
- Skorski, T.; Perrotti, D.; Nieborowska-Skorska, M.; Gryaznov, S.; Calabretta, B. Antileukemia effect of c-myc N3′→ P5′ phosphoramidate antisense oligonucleotides in vivo. Proc. Natl. Acad. Sci. USA 1997, 94, 3966–3971. [Google Scholar] [CrossRef] [Green Version]
- Egli, M.; Minasov, G.; Tereshko, V.; Pallan, P.S.; Teplova, M.; Inamati, G.B.; Lesnik, E.A.; Owens, S.R.; Ross, B.S.; Prakash, T.P.; et al. Probing the influence of stereoelectronic effects on the biophysical properties of oligonucleotides: Comprehensive analysis of the RNA affinity, nuclease resistance, and crystal structure of ten 2 ‘-O-ribonucleic acid modifications. Biochemistry 2005, 44, 9045–9057. [Google Scholar] [CrossRef] [Green Version]
- Kurreck, J.; Wyszko, E.; Gillen, C.; Erdmann, V.A. Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res. 2002, 30, 1911–1918. [Google Scholar] [CrossRef] [Green Version]
- Pabon-Martinez, Y.V.; Xu, Y.; Villa, A.; Lundin, K.E.; Geny, S.; Nguyen, C.H.; Pedersen, E.B.; Jørgensen, P.T.; Wengel, J.; Nilsson, L.; et al. LNA effects on DNA binding and conformation: From single strand to duplex and triplex structures. Sci. Rep. 2017, 7, 11043. [Google Scholar] [CrossRef] [Green Version]
- Pandey, S.K.; Wheeler, T.M.; Justice, S.L.; Kim, A.; Younis, H.S.; Gattis, D.; Jauvin, D.; Puymirat, J.; Swayze, E.E.; Freier, S.M.; et al. Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1. J. Pharmacol. Exp. Ther. 2015, 355, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.P.; Stein, J.; Cai, Y.; Riemers, F.; Wexselblatt, E.; Wengel, J.; Tryfonidou, M.; Yayon, A.; Howard, K.A.; Creemers, L.B. Fibrin-hyaluronic acid hydrogel-based delivery of antisense oligonucleotides for ADAMTS5 inhibition in co-delivered and resident joint cells in osteoarthritis. J. Control. Release 2019, 294, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, K.T.; Pendergraff, H.M.; Deleavey, G.F.; Swayze, E.E.; Potier, P.; Randolph, J.; Roesch, E.B.; Chattopadhyaya, J.; Damha, M.J.; Bennett, C.F.; et al. Allele-selective inhibition of mutant huntingtin expression with antisense oligonucleotides targeting the expanded CAG repeat. Biochemistry 2010, 49, 10166–10178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.R.; Maruyama, R.; Echigoya, Y.; Nguyen, Q.; Zhang, A.; Khawaja, H.; Sen Chandra, S.; Jones, T.; Jones, P.; Chen, Y.W.; et al. Inhibition of DUX4 expression with antisense LNA gapmers as a therapy for facioscapulohumeral muscular dystrophy. Proc. Natl. Acad. Sci. USA 2020, 117, 16509–16515. [Google Scholar] [CrossRef]
- Kuespert, S.; Heydn, R.; Peters, S.; Wirkert, E.; Meyer, A.L.; Siebörger, M.; Johannesen, S.; Aigner, L.; Bogdahn, U.; Bruun, T.H. Antisense oligonucleotide in LNA-gapmer design targeting TGFBR2—A key single gene target for safe and effective inhibition of TGFβ signaling. Int. J. Mol. Sci. 2020, 21, 1952. [Google Scholar] [CrossRef] [Green Version]
- Seto, A.G.; Beatty, X.; Lynch, J.M.; Hermreck, M.; Tetzlaff, M.; Duvic, M.; Jackson, A.L. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br. J. Haematol. 2018, 183, 428–444. [Google Scholar] [CrossRef] [Green Version]
- Ottosen, S.; Parsley, T.B.; Yang, L.; Zeh, K.; van Doorn, L.J.; van der Veer, E.; Raney, A.K.; Hodges, M.R.; Patick, A.K. In vitro antiviral activity and preclinical and clinical resistance profile of miravirsen, a novel anti-hepatitis C virus therapeutic targeting the human factor miR-122. Antimicrob. Agents Chemother. 2015, 59, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Gomez, I.G.; MacKenna, D.A.; Johnson, B.G.; Kaimal, V.; Roach, A.M.; Ren, S.; Nakagawa, N.; Xin, C.; Newitt, R.; Pandya, S.; et al. Anti–microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Investig. 2015, 125, 141–156. [Google Scholar] [CrossRef]
- Zhang, S.; Cheng, Z.; Wang, Y.; Han, T. The risks of miRNA therapeutics: In a drug target perspective. Drug Des. Dev. Ther. 2021, 15, 721. [Google Scholar] [CrossRef]
- Damha, M.J.; Wilds, C.J.; Noronha, A.; Brukner, I.; Borkow, G.; Arion, D.; Parniak, M.A. Hybrids of RNA and arabinonucleic acids (ANA and 2 ‘F-ANA) are substrates of ribonuclease H. J. Am. Chem. Soc. 1998, 120, 12976–12977. [Google Scholar] [CrossRef]
- Takahashi, M.; Li, H.; Zhou, J.; Chomchan, P.; Aishwarya, V.; Damha, M.J.; Rossi, J.J. Dual mechanisms of action of self-delivering, anti-HIV-1 FANA oligonucleotides as a potential new approach to HIV therapy. Mol. Ther.-Nucleic Acids 2019, 17, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Harshe, R.P.; Xie, A.; Vuerich, M.; Frank, L.A.; Gromova, B.; Zhang, H.; Robles, R.J.; Mukherjee, S.; Csizmadia, E.; Kokkotou, E.; et al. Endogenous antisense RNA curbs CD39 expression in Crohn’s disease. Nat. Commun. 2020, 11, 5894. [Google Scholar] [CrossRef] [PubMed]
- Guimond, A.; Viau, E.; Aubé, P.; Renzi, P.M.; Paquet, L.; Ferrari, N. Advantageous toxicity profile of inhaled antisense oligonucleotides following chronic dosing in non-human primates. Pulm. Pharmacol. Ther. 2008, 21, 845–854. [Google Scholar] [CrossRef]
- Schmidt, K.; Weidmann, C.A.; Hilimire, T.A.; Yee, E.; Hatfield, B.M.; Schneekloth, J.S., Jr.; Weeks, K.M.; Novina, C.D. Targeting the oncogenic long non-coding RNA SLNCR1 by blocking its sequence-specific binding to the androgen receptor. Cell Rep. 2020, 30, 541–554. [Google Scholar] [CrossRef] [Green Version]
- Wuu, Y.R.; Hu, B.; Okunola, H.; Paul, A.M.; Blaber, E.A.; Cheng-Campbell, M.; Beheshti, A.; Grabham, P. LET-Dependent Low Dose and Synergistic Inhibition of Human Angiogenesis by Charged Particles: Validation of miRNAs that Drive Inhibition. IScience 2020, 23, 101771. [Google Scholar] [CrossRef]
- Pelisch, N.; Almanza, J.R.; Stehlik, K.E.; Aperi, B.V.; Kroner, A. Use of a self-delivering Anti-CCL3 FANA Oligonucleotide as an innovative approach to target inflammation after Spinal Cord Injury. eNeuro 2021, 8, ENEURO.0338-20.2021. [Google Scholar] [CrossRef]
- El-Khoury, R.; Damha, M.J. 2′-Fluoro-arabinonucleic Acid (FANA): A Versatile Tool for Probing Biomolecular Interactions. Acc. Chem. Res. 2021, 54, 2287–2297. [Google Scholar] [CrossRef]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense oligonucleotides: An emerging area in drug discovery and development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef]
- Castellanos, E.; Rosas, I.; Solanes, A.; Bielsa, I.; Lázaro, C.; Carrato, C.; Hostalot, C.; Prades, P.; Roca-Ribas, F.; Blanco, I.; et al. In vitro antisense therapeutics for a deep intronic mutation causing Neurofibromatosis type 2. Eur. J. Hum. Genet. 2013, 21, 769–773. [Google Scholar] [CrossRef]
- Aung-Htut, M.T.; McIntosh, C.S.; West, K.A.; Fletcher, S.; Wilton, S.D. In vitro validation of phosphorodiamidate morpholino oligomers. Molecules 2019, 24, 2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcclorey, G.; Moulton, H.M.; Iversen, P.L.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 2006, 13, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Hanvey, J.C.; Peffer, N.J.; Bisi, J.E.; Thomson, S.A.; Cadilla, R.; Josey, J.A.; Ricca, D.J.; Hassman, C.F.; Bonham, M.A.; Au, K.G.; et al. Antisense and antigene properties of peptide nucleic acids. Science 1992, 258, 1481–1485. [Google Scholar] [CrossRef]
- Nielsen, P.E. PNA technology. Mol. Biotechnol. 2004, 26, 233–248. [Google Scholar] [CrossRef]
- Knudsen, H.; Nielsen, P.E. Antisense properties of duplex-and triplex-forming PNAs. Nucleic Acids Res. 1996, 24, 494–500. [Google Scholar] [CrossRef] [Green Version]
- Bendifallah, N.; Rasmussen, F.W.; Zachar, V.; Ebbesen, P.; Nielsen, P.E.; Koppelhus, U. Evaluation of cell-penetrating peptides (CPPs) as vehicles for intracellular delivery of antisense peptide nucleic acid (PNA). Bioconjugate Chem. 2006, 17, 750–758. [Google Scholar] [CrossRef] [PubMed]
- El-Andaloussi, S.; Johansson, H.J.; Holm, T.; Langel, Ü. A novel cell-penetrating peptide, M918, for efficient delivery of proteins and peptide nucleic acids. Mol. Ther. 2007, 15, 1820–1826. [Google Scholar] [CrossRef]
- Sanghvi, Y.S.; Hoke, G.D.; Freier, S.M.; Zounes, M.C.; Gonzalez, C.; Cummins, L.; Sasmor, H.; Cook, P.D. Antisense oligodeoxynucleotides: Synthesis, biophysical and biological evaluation of oligodeoxynucleotides containing modified pyrimidines. Nucleic Acids Res. 1993, 21, 3197–3203. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, W.M.; Wolf, J.J.; Olson, P.; Grant, D.; Lin, K.Y.; Wagner, R.W.; Matteucci, M.D. A cytosine analog that confers enhanced potency to antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1999, 96, 3513–3518. [Google Scholar] [CrossRef] [Green Version]
- Ortega, J.A.; Blas, J.R.; Orozco, M.; Grandas, A.; Pedroso, E.; Robles, J. Binding affinities of oligonucleotides and PNAs containing phenoxazine and G-clamp cytosine analogues are unusually sequence-dependent. Org. Lett. 2007, 9, 4503–4506. [Google Scholar] [CrossRef]
- Wang, T.; Larcher, L.M.; Ma, L.; Veedu, R.N. Systematic screening of commonly used commercial transfection reagents towards efficient transfection of single-stranded oligonucleotides. Molecules 2018, 23, 2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viney, N.J.; Guo, S.; Tai, L.J.; Baker, B.F.; Aghajan, M.; Jung, S.W.; Yu, R.Z.; Booten, S.; Murray, H.; Machemer, T.; et al. Ligand conjugated antisense oligonucleotide for the treatment of transthyretin amyloidosis: Preclinical and phase 1 data. ESC Heart Fail. 2021, 8, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Ohanian, M.; Ashizawa, A.T.; Garcia-Manero, G.; Pemmaraju, N.; Kadia, T.; Jabbour, E.; Ravandi, F.; Borthakur, G.; Andreeff, M.; Konopleva, M.; et al. Liposomal Grb2 antisense oligodeoxynucleotide (BP1001) in patients with refractory or relapsed haematological malignancies: A single-centre, open-label, dose-escalation, phase 1/1b trial. Lancet Haematol. 2018, 5, e136–e146. [Google Scholar] [CrossRef]
- Bushman, F.D. Retroviral insertional mutagenesis in humans: Evidence for four genetic mechanisms promoting expansion of cell clones. Mol. Ther. 2020, 28, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.; Tisdale, J.F. A pause in gene therapy: Reflecting on the unique challenges of sickle cell disease. Mol. Ther. 2021, 29, 1355. [Google Scholar] [CrossRef] [PubMed]
- Servick, K. Gene Therapy Clinical Trial Halted as Cancer Risk Surfaces. Available online: https://www.science.org/content/article/gene-therapy-clinical-trial-halted-cancer-risk-surfaces (accessed on 15 July 2022).
- Chen, Y.H.; Keiser, M.S.; Davidson, B.L. Viral vectors for gene transfer. Curr. Protoc. Mouse Biol. 2018, 8, e58. [Google Scholar] [CrossRef] [PubMed]
- Schirmbeck, R.; Reimann, J.; Kochanek, S.; Kreppel, F. The immunogenicity of adenovirus vectors limits the multispecificity of CD8 T-cell responses to vector-encoded transgenic antigens. Mol. Ther. 2008, 16, 1609–1616. [Google Scholar] [CrossRef]
- Lai, Y.; Yue, Y.; Duan, D. Evidence for the failure of adeno-associated virus serotype 5 to package a viral genome ≥8.2 kb. Mol. Ther. 2010, 18, 75–79. [Google Scholar] [CrossRef]
- Ricobaraza, A.; Gonzalez-Aparicio, M.; Mora-Jimenez, L.; Lumbreras, S.; Hernandez-Alcoceba, R. High-capacity adenoviral vectors: Expanding the scope of gene therapy. Int. J. Mol. Sci. 2020, 21, 3643. [Google Scholar] [CrossRef]
- Gurumoorthy, N.; Nordin, F.; Tye, G.J.; Wan Kamarul Zaman, W.S.; Ng, M.H. Non-Integrating Lentiviral Vectors in Clinical Applications: A Glance Through. Biomedicines 2022, 10, 107. [Google Scholar] [CrossRef]
- Lu, D.; Yu, K.; Raizada, M.K. Retrovirus-mediated transfer of an angiotensin type I receptor (AT1-R) antisense sequence decreases AT1-Rs and angiotensin II action in astroglial and neuronal cells in primary cultures from the brain. Proc. Natl. Acad. Sci. USA 1995, 92, 1162–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, M.I. Antisense inhibition and adeno-associated viral vector delivery for reducing hypertension. Hypertension 1997, 29, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Imbert, M.; Dias-Florencio, G.; Goyenvalle, A. Viral vector-mediated antisense therapy for genetic diseases. Genes 2017, 8, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geib, T.; Hertel, K.J. Restoration of full-length SMN promoted by adenoviral vectors expressing RNA antisense oligonucleotides embedded in U7 snRNAs. PLoS ONE 2009, 4, e8204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquis, J.; Meyer, K.; Angehrn, L.; Kämpfer, S.S.; Rothen-Rutishauser, B.; Schümperli, D. Spinal muscular atrophy: SMN2 pre-mRNA splicing corrected by a U7 snRNA derivative carrying a splicing enhancer sequence. Mol. Ther. 2007, 15, 1479–1486. [Google Scholar] [CrossRef]
- Goyenvalle, A.; Wright, J.; Babbs, A.; Wilkins, V.; Garcia, L.; Davies, K.E. Engineering multiple U7snRNA constructs to induce single and multiexon-skipping for Duchenne muscular dystrophy. Mol. Ther. 2012, 20, 1212–1221. [Google Scholar] [CrossRef] [Green Version]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Ter-Avetisyan, G.; Tünnemann, G.; Nowak, D.; Nitschke, M.; Herrmann, A.; Drab, M.; Cardoso, M.C. Cell entry of arginine-rich peptides is independent of endocytosis. J. Biol. Chem. 2009, 284, 3370–3378. [Google Scholar] [CrossRef] [Green Version]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogs with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef]
- Shin, M.C.; Zhang, J.; Min, K.A.; Lee, K.; Byun, Y.; David, A.E.; He, H.; Yang, V.C. Cell-penetrating peptides: Achievements and challenges in application for cancer treatment. J. Biomed. Mater. Res. Part A Off. J. Soc. Biomater. Jpn. Soc. Biomater. Aust. Soc. Biomater. Korean Soc. Biomater. 2014, 102, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, K.; Yoneyama, S.; Murase, O.; Miyajima, K. Transbilayer transport of ions and lipids coupled with mastoparan × translocation. Biochemistry 1996, 35, 8450–8456. [Google Scholar] [CrossRef]
- Derossi, D.; Chassaing, G.; Prochiantz, A. Trojan peptides: The penetratin system for intracellular delivery. Trends Cell Biol. 1998, 8, 84–87. [Google Scholar] [CrossRef]
- Jones, A.T. Macropinocytosis: Searching for an endocytic identity and role in the uptake of cell penetrating peptides. J. Cell. Mol. Med. 2007, 11, 670–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, Y.; Takeuchi, T.; Kuwata, K.; Chiba, J.; Hatanaka, Y.; Nakase, I.; Futaki, S. Syndecan-4 is a receptor for clathrin-mediated endocytosis of arginine-rich cell-penetrating peptides. Bioconjugate Chem. 2016, 27, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.N.; Zaro, J.L.; Shen, W.C. Cell penetrating peptides: Intracellular pathways and pharmaceutical perspectives. Pharm. Res. 2007, 24, 1977–1992. [Google Scholar] [CrossRef]
- Rhee, M.; Davis, P. Mechanism of uptake of C105Y; a novel cell-penetrating peptide. J. Biol. Chem. 2006, 281, 1233–1240. [Google Scholar] [CrossRef] [Green Version]
- Walker, I.; Irwin, W.J.; Akhtar, S. Improved cellular delivery of antisense oligonucleotides using transferrin receptor antibody-oligonucleotide conjugates. Pharm. Res. 1995, 12, 1548–1553. [Google Scholar] [CrossRef]
- Arnold, A.E.; Malek-Adamian, E.; Le, P.U.; Meng, A.; Martínez-Montero, S.; Petrecca, K.; Damha, M.J.; Shoichet, M.S. Antibody-antisense oligonucleotide conjugate downregulates a key gene in glioblastoma stem cells. Mol. Ther.-Nucleic Acids 2018, 11, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Satake, N.; Duong, C.; Yoshida, S.; Oestergaard, M.; Chen, C.; Peralta, R.; Guo, S.; Seth, P.P.; Li, Y.; Beckett, L.; et al. Novel targeted therapy for Precursor B-Cell acute Lymphoblastic Leukemia: Anti-CD22 antibody-MXD3 antisense Oligonucleotide Conjugate. Mol. Med. 2016, 22, 632–642. [Google Scholar] [CrossRef] [Green Version]
- Vorobyeva, M.; Vorobjev, P.; Venyaminova, A. Multivalent aptamers: Versatile tools for diagnostic and therapeutic applications. Molecules 2016, 21, 1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.; Okuda, T.; Kasahara, Y.; Obika, S. Base-modified aptamers obtained by cell-internalization SELEX facilitate cellular uptake of an antisense oligonucleotide. Mol. Ther.-Nucleic Acids 2021, 23, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Graham, M.J.; Yu, J.; Carty, R.; Low, A.; Chappell, A.; Schmidt, K.; Zhao, C.; Aghajan, M.; Murray, H.F.; et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014, 42, 8796–8807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcón-Arís, D.; Recasens, A.; Galofré, M.; Carballo-Carbajal, I.; Zacchi, N.; Ruiz-Bronchal, E.; Pavia-Collado, R.; Chica, R.; Ferrés-Coy, A.; Santos, M.; et al. Selective α-synuclein knockdown in monoamine neurons by intranasal oligonucleotide delivery: Potential therapy for Parkinson’s disease. Mol. Ther. 2018, 26, 550–567. [Google Scholar] [CrossRef] [Green Version]
- Doane, T.; Burda, C. Nanoparticle mediated non-covalent drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 607–621. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.K.; Ghosh, P.; Pagliuca, C.; Zhu, Z.J.; Menichetti, S.; Rotello, V.M. Entrapment of hydrophobic drugs in nanoparticle monolayers with efficient release into cancer cells. J. Am. Chem. Soc. 2009, 131, 1360–1361. [Google Scholar] [CrossRef] [Green Version]
- Manju, S.; Sreenivasan, K. Enhanced drug loading on magnetic nanoparticles by layer-by-layer assembly using drug conjugates: Blood compatibility evaluation and targeted drug delivery in cancer cells. Langmuir 2011, 27, 14489–14496. [Google Scholar] [CrossRef]
- Kim, B.S.; Park, S.W.; Hammond, P.T. Hydrogen-bonding layer-by-layer-assembled biodegradable polymeric micelles as drug delivery vehicles from surfaces. ACS Nano 2008, 2, 386–392. [Google Scholar] [CrossRef]
- Kester, M.; Heakal, Y.; Fox, T.; Sharma, A.; Robertson, G.P.; Morgan, T.T.; Altinoglu, E.I.; Tabakovic, A.; Parette, M.R.; Rouse, S.M.; et al. Calcium phosphate nanocomposite particles for in vitro imaging and encapsulated chemotherapeutic drug delivery to cancer cells. Nano Lett. 2008, 8, 4116–4121. [Google Scholar] [CrossRef] [Green Version]
- Lambert, G.; Fattal, E.; Couvreur, P. Nanoparticulate systems for the delivery of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2001, 47, 99–112. [Google Scholar] [CrossRef]
- Mendonça, M.C.; Kont, A.; Aburto, M.R.; Cryan, J.F.; O’Driscoll, C.M. Advances in the design of (nano) formulations for delivery of antisense oligonucleotides and small interfering RNA: Focus on the central nervous system. Mol. Pharm. 2021, 18, 1491–1506. [Google Scholar] [CrossRef] [PubMed]
- Min, H.S.; Kim, H.J.; Naito, M.; Ogura, S.; Toh, K.; Hayashi, K.; Kim, B.S.; Fukushima, S.; Anraku, Y.; Miyata, K.; et al. Systemic brain delivery of antisense oligonucleotides across the blood–brain barrier with a glucose-coated polymeric nanocarrier. Angew. Chem. Int. Ed. 2020, 59, 8173–8180. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Moss, K.H.; Popova, P.; Hadrup, S.R.; Astakhova, K.; Taskova, M. Lipid nanoparticles for delivery of therapeutic RNA oligonucleotides. Mol. Pharm. 2019, 16, 2265–2277. [Google Scholar] [CrossRef]
- Sheng, Y.; Chang, L.; Kuang, T.; Hu, J. PEG/heparin-decorated lipid–polymer hybrid nanoparticles for long-circulating drug delivery. RSC Adv. 2016, 6, 23279–23287. [Google Scholar] [CrossRef]
- Wu, R.; Zhang, Z.; Wang, B.; Chen, G.; Zhang, Y.; Deng, H.; Tang, Z.; Mao, J.; Wang, L. Combination chemotherapy of lung cancer–co-delivery of docetaxel prodrug and cisplatin using aptamer-decorated lipid–polymer hybrid nanoparticles. Drug Des. Dev. Ther. 2020, 14, 2249. [Google Scholar] [CrossRef] [PubMed]
- Thanki, K.; Papai, S.; Lokras, A.; Rose, F.; Falkenberg, E.; Franzyk, H.; Foged, C. Application of a quality-by-design approach to optimise lipid-polymer hybrid nanoparticles loaded with a splice-correction antisense oligonucleotide: Maximising loading and intracellular delivery. Pharm. Res. 2019, 36, 37. [Google Scholar] [CrossRef] [PubMed]
- Küçüktürkmen, B.; Devrim, B.; Saka, O.M.; Yilmaz, Ş.; Arsoy, T.; Bozkir, A. Co-delivery of pemetrexed and miR-21 antisense oligonucleotide by lipid-polymer hybrid nanoparticles and effects on glioblastoma cells. Drug Dev. Ind. Pharm. 2017, 43, 12–21. [Google Scholar] [CrossRef]
- Chen, L.; Watson, C.; Morsch, M.; Cole, N.J.; Chung, R.S.; Saunders, D.N.; Yerbury, J.J.; Vine, K.L. Improving the delivery of SOD1 antisense oligonucleotides to motor neurons using calcium phosphate-lipid nanoparticles. Front. Neurosci. 2017, 11, 476. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Qian, L.; Ge, J.; Fu, J.; Yuan, P.; Yao, S.C.; Yao, S.Q. Cell-penetrating poly (disulfide) assisted intracellular delivery of mesoporous silica nanoparticles for inhibition of miR-21 function and detection of subsequent therapeutic effects. Angew. Chem. Int. Ed. 2016, 55, 9272–9276. [Google Scholar] [CrossRef]
- Yoshida, S.; Duong, C.; Oestergaard, M.; Fazio, M.; Chen, C.; Peralta, R.; Guo, S.; Seth, P.P.; Li, Y.; Beckett, L.; et al. MXD3 antisense oligonucleotide with superparamagnetic iron oxide nanoparticles: A new targeted approach for neuroblastoma. Nanomed. Nanotechnol. Biol. Med. 2020, 24, 102127. [Google Scholar] [CrossRef] [PubMed]
- Beha, M.J.; Ryu, J.S.; Kim, Y.S.; Chung, H.J. Delivery of antisense oligonucleotides using multi-layer coated gold nanoparticles to methicillin-resistant S. aureus for combinatorial treatment. Mater. Sci. Eng. C 2021, 126, 112167. [Google Scholar] [CrossRef] [PubMed]
- Falzarano, M.S.; Passarelli, C.; Ferlini, A. Nanoparticle delivery of antisense oligonucleotides and their application in the exon skipping strategy for Duchenne muscular dystrophy. Nucleic Acid Ther. 2014, 24, 87–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tkach, M.; Théry, C. Communication by extracellular vesicles: Where we are and where we need to go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Margolis, L.; Sadovsky, Y. The biology of extracellular vesicles: The known unknowns. PLoS Biol. 2019, 17, e3000363. [Google Scholar] [CrossRef]
- Usman, W.M.; Pham, T.C.; Kwok, Y.Y.; Vu, L.T.; Ma, V.; Peng, B.; Chan, Y.S.; Wei, L.; Chin, S.M.; Azad, A.; et al. Efficient RNA drug delivery using red blood cell extracellular vesicles. Nat. Commun. 2018, 9, 2359. [Google Scholar] [CrossRef] [Green Version]
- Adamus, T.; Hung, C.Y.; Yu, C.; Kang, E.; Hammad, M.; Flores, L.; Nechaev, S.; Zhang, Q.; Gonzaga, J.M.; Muthaiyah, K.; et al. Glioma-targeted delivery of exosome-encapsulated antisense oligonucleotides using neural stem cells. Mol. Ther.-Nucleic Acids 2022, 27, 611–620. [Google Scholar] [CrossRef]
- Yang, J.; Luo, S.; Zhang, J.; Yu, T.; Fu, Z.; Zheng, Y.; Xu, X.; Liu, C.; Fan, M.; Zhang, Z. Exosome-mediated delivery of antisense oligonucleotides targeting α-synuclein ameliorates the pathology in a mouse model of Parkinson’s disease. Neurobiol. Dis. 2021, 148, 105218. [Google Scholar] [CrossRef]
- Tang, M.; Chen, Y.; Li, B.; Sugimoto, H.; Yang, S.; Yang, C.; LeBleu, V.S.; McAndrews, K.M.; Kalluri, R. Therapeutic targeting of STAT3 with small interference RNAs and antisense oligonucleotides embedded exosomes in liver fibrosis. FASEB J. 2021, 35, e21557. [Google Scholar] [CrossRef]
- Gao, X.; Ran, N.; Dong, X.; Zuo, B.; Yang, R.; Zhou, Q.; Moulton, H.M.; Seow, Y.; Yin, H. Anchor peptide captures, targets, and loads exosomes of diverse origins for diagnostics and therapy. Sci. Transl. Med. 2018, 10, eaat0195. [Google Scholar] [CrossRef] [Green Version]
- Didiot, M.C.; Hall, L.M.; Coles, A.H.; Haraszti, R.A.; Godinho, B.M.; Chase, K.; Sapp, E.; Ly, S.; Alterman, J.F.; Hassler, M.R.; et al. Exosome-mediated delivery of hydrophobically modified siRNA for huntingtin mRNA silencing. Mol. Ther. 2016, 24, 1836–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Limmroth, V.; Barkhof, F.; Desem, N.; Diamond, M.P.; Tachas, G.; ATL1102 Study Group. CD49d antisense drug ATL1102 reduces disease activity in patients with relapsing-remitting MS. Neurology 2014, 83, 1780–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, K.J.; Witchell, D.R.; Graham, M.J.; Koo, S.; Butler, M.; Condon, T.P. Antisense oligonucleotide blockade of alpha 4 integrin prevents and reverses clinical symptoms in murine experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 160, 12–24. [Google Scholar] [CrossRef]
- Woodcock, I.R.; Tachas, G.; Desmin, N.; Houweling, P.J.; Yiu, E.; Kean, M.; Emmanuel, J.; Kennedy, R.; Carroll, K.; de Valle, K.; et al. A Phase 2 open-label study to determine the safety and efficacy of weekly dosing of ATL1102 in patients with non-ambulatory Duchenne muscular dystrophy. medRxiv 2022. [Google Scholar] [CrossRef]
- Astra Zeneca. Eplontersen Met Co-Primary and Secondary Endpoints in Interim Analysis of the NEURO-TTRansform Phase III Trial for Hereditary Transthyretin-Mediated Amyloid Polyneuropathy (ATTRv-PN). Available online: https://www.astrazeneca.com/media-centre/press-releases/2022/eplontersen-phase-iii-trial-met-co-primary-endpoints.html (accessed on 15 July 2022).
- Kim, J.; Hu, C.; Moufawad El Achkar, C.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-customized oligonucleotide therapy for a rare genetic disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef]
- Benedict, C.; Frey, W.H., II; Schiöth, H.B.; Schultes, B.; Born, J.; Hallschmid, M. Intranasal insulin as a therapeutic option in the treatment of cognitive impairments. Exp. Gerontol. 2011, 46, 112–115. [Google Scholar] [CrossRef]
- Wu, H.; Zhou, Y.; Wang, Y.; Tong, L.; Wang, F.; Song, S.; Xu, L.; Liu, B.; Yan, H.; Sun, Z. Current state and future directions of intranasal delivery route for central nervous system disorders: A scientometric and visualization analysis. Front. Pharmacol. 2021, 12, 717192. [Google Scholar] [CrossRef]
- Zhang, Y.T.; He, K.J.; Zhang, J.B.; Ma, Q.H.; Wang, F.; Liu, C.F. Advances in intranasal application of stem cells in the treatment of central nervous system diseases. Stem Cell Res. Ther. 2021, 12, 210. [Google Scholar] [CrossRef]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H., II. Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 10962–10967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugarman, E.A.; Nagan, N.; Zhu, H.; Akmaev, V.R.; Zhou, Z.; Rohlfs, E.M.; Flynn, K.; Hendrickson, B.C.; Scholl, T.; Sirko-Osadsa, D.A.; et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of> 72 400 specimens. Eur. J. Hum. Genet. 2012, 20, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Leonard, D.G.; Rennert, H.; Ewens, W.J.; Wilson, R.B. Genetic risk assessment in carrier testing for spinal muscular atrophy. Am. J. Med. Genet. 2002, 110, 301–307. [Google Scholar] [CrossRef]
- Emery, A.E. Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef]
- Hayashi, M.; Araki, S.; Arai, N.; Kumada, S.; Itoh, M.; Tamagawa, K.; Oda, M.; Morimatsu, Y. Oxidative stress and disturbed glutamate transport in spinal muscular atrophy. Brain Dev. 2002, 24, 770–775. [Google Scholar] [CrossRef]
- Schrank, B.; Götz, R.; Gunnersen, J.M.; Ure, J.M.; Toyka, K.V.; Smith, A.G.; Sendtner, M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. USA 1997, 94, 9920–9925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keil, J.M.; Seo, J.; Howell, M.D.; Hsu, W.H.; Singh, R.N.; Di Donato, C.J. A short antisense oligonucleotide ameliorates symptoms of severe mouse models of spinal muscular atrophy. Mol. Ther.-Nucleic Acids 2014, 3, e174. [Google Scholar] [CrossRef]
- Nizzardo, M.; Simone, C.; Dametti, S.; Salani, S.; Ulzi, G.; Pagliarani, S.; Rizzo, F.; Frattini, E.; Pagani, F.; Bresolin, N.; et al. Spinal muscular atrophy phenotype is ameliorated in human motor neurons by SMN increase via different novel RNA therapeutic approaches. Sci. Rep. 2015, 5, 11746. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Moshe-Lilie, O.; Visser, A.; Chahin, N.; Ragole, T.; Dimitrova, D.; Karam, C. Nusinersen in adult patients with spinal muscular atrophy: Observations from a single center. Neurology 2020, 95, e413–e416. [Google Scholar] [CrossRef]
- Hagenacker, T.; Wurster, C.D.; Günther, R.; Schreiber-Katz, O.; Osmanovic, A.; Petri, S.; Weiler, M.; Ziegler, A.; Kuttler, J.; Koch, J.C.; et al. Nusinersen in adults with 5q spinal muscular atrophy: A non-interventional, multicentre, observational cohort study. Lancet Neurol. 2020, 19, 317–325. [Google Scholar] [CrossRef]
- Maggi, L.; Bello, L.; Bonanno, S.; Govoni, A.; Caponnetto, C.; Passamano, L.; Grandis, M.; Trojsi, F.; Cerri, F.; Ferraro, M.; et al. Nusinersen safety and effects on motor function in adult spinal muscular atrophy type 2 and 3. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Luigetti, M.; Romano, A.; Di Paolantonio, A.; Bisogni, G.; Sabatelli, M. Diagnosis and treatment of hereditary transthyretin amyloidosis (hATTR) polyneuropathy: Current perspectives on improving patient care. Ther. Clin. Risk Manag. 2020, 16, 109–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoy, S.M. Patisiran: First global approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef]
- Keam, S.J. Vutrisiran: First Approval. Drugs 2022, 82, 1419–1425. [Google Scholar] [CrossRef]
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef]
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; De Kock, S.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef] [Green Version]
- Duan, D.; Goemans, N.; Takeda, S.I.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Prim. 2021, 7, 13. [Google Scholar] [CrossRef]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Koo, T.; Wood, M.J. Clinical trials using antisense oligonucleotides in duchenne muscular dystrophy. Hum. Gene Ther. 2013, 24, 479–488. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; Van Deutekom, J.; van Ommen, G.J.; Den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Kinane, T.B.; Mayer, O.H.; Duda, P.W.; Lowes, L.P.; Moody, S.L.; Mendell, J.R. Long-term pulmonary function in Duchenne muscular dystrophy: Comparison of eteplirsen-treated patients to natural history. J. Neuromuscul. Dis. 2018, 5, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Brogna, C.; Coratti, G.; Pane, M.; Ricotti, V.; Messina, S.; D’Amico, A.; Bruno, C.; Vita, G.; Berardinelli, A.; Mazzone, E.; et al. Long-term natural history data in Duchenne muscular dystrophy ambulant patients with mutations amenable to skip exons 44, 45, 51 and 53. PLoS ONE 2019, 14, e0218683. [Google Scholar]
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020, 94, e2270–e2282. [Google Scholar] [CrossRef] [Green Version]
- Komaki, H.; Takeshima, Y.; Matsumura, T.; Ozasa, S.; Funato, M.; Takeshita, E.; Iwata, Y.; Yajima, H.; Egawa, Y.; Toramoto, T.; et al. Viltolarsen in Japanese Duchenne muscular dystrophy patients: A phase 1/2 study. Ann. Clin. Transl. Neurol. 2020, 7, 2393–2408. [Google Scholar] [CrossRef]
- Clemens, P.R.; Rao, V.K.; Connolly, A.M.; Harper, A.D.; Mah, J.K.; Smith, E.C.; McDonald, C.M.; Zaidman, C.M.; Morgenroth, L.P.; Osaki, H.; et al. Safety, tolerability, and efficacy of viltolarsen in boys with duchenne muscular dystrophy amenable to exon 53 skipping: A phase 2 randomized clinical trial. JAMA Neurol. 2020, 77, 982–991. [Google Scholar] [CrossRef]
- Clemens, P.R.; Rao, V.K.; Connolly, A.M.; Harper, A.D.; Mah, J.K.; McDonald, C.M.; Smith, E.C.; Zaidman, C.M.; Nakagawa, T.; Hoffman, E.P.; et al. Long-Term Functional Efficacy and Safety of Viltolarsen in Patients with Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2022, 9, 493–501. [Google Scholar] [CrossRef]
- USA FDA. Amondys 45 (Casimersen) Injection, for Intravenous Use: US Prescribing Information 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213026lbl.pdf (accessed on 12 August 2022).
- Evers, M.M.; Toonen, L.J.; van Roon-Mom, W.M. Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv. Drug Deliv. Rev. 2015, 87, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.F.; Krainer, A.R.; Cleveland, D.W. Antisense oligonucleotide therapies for neurodegenerative diseases. Annu. Rev. Neurosci. 2019, 42, 385. [Google Scholar] [CrossRef]
- Leavitt, B.R.; Tabrizi, S.J. Antisense oligonucleotides for neurodegeneration. Science 2020, 367, 1428–1429. [Google Scholar] [CrossRef]
- Silva, A.C.; Lobo, D.D.; Martins, I.M.; Lopes, S.M.; Henriques, C.; Duarte, S.P.; Dodart, J.C.; Nobre, R.J.; Pereira de Almeida, L. Antisense oligonucleotide therapeutics in neurodegenerative diseases: The case of polyglutamine disorders. Brain 2020, 143, 407–429. [Google Scholar] [CrossRef] [PubMed]
- Wurster, C.D.; Ludolph, A.C. Antisense oligonucleotides in neurological disorders. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418776932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet de Courssou, J.B.; Durr, A.; Adams, D.; Corvol, J.C.; Mariani, L.L. Antisense therapies in neurological diseases. Brain 2022, 145, 816–831. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.A.; Pelfrey, C.M.; Tranquill, L.R.; Maloni, H.; McFarland, H.F. VLA-4 expression on peripheral blood lymphocytes is downregulated after treatment of multiple sclerosis with interferon beta. Neurology 1997, 49, 1111–1116. [Google Scholar] [CrossRef]
- De Andrés, C.; Teijeiro, R.; Alonso, B.; Sánchez-Madrid, F.; Martínez, M.L.; De Villoria, J.G.; Fernández-Cruz, E.; Sánchez-Ramón, S. Long-term decrease in VLA-4 expression and functional impairment of dendritic cells during natalizumab therapy in patients with multiple sclerosis. PLoS ONE 2012, 7, e34103. [Google Scholar] [CrossRef] [Green Version]
- Schwab, N.; Schneider-Hohendorf, T.; Wiendl, H. Therapeutic uses of anti-α4-integrin (anti-VLA-4) antibodies in multiple sclerosis. Int. Immunol. 2015, 27, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Soilu-Hänninen, M.; Epa, R.; Shipham, K.; Butzkueven, H.; Bucci, T.; Barrett, G.; Bartlett, P.F.; Kilpatrick, T.J. Treatment of experimental autoimmune encephalomyelitis with antisense oligonucleotides against the low affinity neurotrophin receptor. J. Neurosci. Res. 2000, 59, 712–721. [Google Scholar] [CrossRef]
- Vaknin-Dembinsky, A.; Balashov, K.; Weiner, H.L. IL-23 is increased in dendritic cells in multiple sclerosis and down-regulation of IL-23 by antisense oligos increases dendritic cell IL-10 production. J. Immunol. 2006, 176, 7768–7774. [Google Scholar] [CrossRef] [Green Version]
- Heim, P.; Claussen, M.; Hoffmann, B.; Conzelmann, E.; Gärtner, J.; Harzer, K.; Hunneman, D.H.; Köhler, W.; Kurlemann, G.; Kohlschütter, A. Leukodystrophy incidence in Germany. Am. J. Med. Genet. 1997, 71, 475–478. [Google Scholar] [CrossRef]
- Vanderver, A.; Hussey, H.; Schmidt, J.L.; Pastor, W.; Hoffman, H.J. Relative incidence of inherited white matter disorders in childhood to acquired pediatric demyelinating disorders. Semin. Pediatr. Neurol. 2012, 19, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Bonkowsky, J.L.; Nelson, C.; Kingston, J.L.; Filloux, F.M.; Mundorff, M.B.; Srivastava, R. The burden of inherited leukodystrophies in children. Neurology 2010, 75, 718–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soderholm, H.E.; Chapin, A.B.; Bayrak-Toydemir, P.; Bonkowsky, J.L. Elevated leukodystrophy incidence predicted from genomics databases. Pediatr. Neurol. 2020, 111, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.L.; Powers, B.; Mazur, C.; Kim, A.; Wheeler, S.; Hung, G.; Swayze, E.; Messing, A. Antisense suppression of glial fibrillary acidic protein as a treatment for Alexander disease. Ann. Neurol. 2018, 83, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Elitt, M.S.; Barbar, L.; Shick, H.E.; Powers, B.E.; Maeno-Hikichi, Y.; Madhavan, M.; Allan, K.C.; Nawash, B.S.; Gevorgyan, A.S.; Hung, S.; et al. Suppression of proteolipid protein rescues Pelizaeus–Merzbacher disease. Nature 2020, 585, 397–403. [Google Scholar] [CrossRef]
- Hull, V.; Wang, Y.; Burns, T.; Zhang, S.; Sternbach, S.; McDonough, J.; Guo, F.; Pleasure, D. Antisense oligonucleotide reverses leukodystrophy in Canavan disease mice. Ann. Neurol. 2020, 87, 480–485. [Google Scholar] [CrossRef]
- Alexander, W.S. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain 1949, 72, 373–381. [Google Scholar] [CrossRef]
- Yoshida, T.; Sasaki, M.; Yoshida, M.; Namekawa, M.; Okamoto, Y.; Tsujino, S.; Sasayama, H.; Mizuta, I.; Nakagawa, M. Nationwide survey of Alexander disease in Japan and proposed new guidelines for diagnosis. J. Neurol. 2011, 258, 1998–2008. [Google Scholar] [CrossRef]
- Springer, S.; Erlewein, R.; Naegele, T.; Becker, I.; Auer, D.; Grodd, W.; Krägeloh-Mann, I. Alexander disease-classification revisited and isolation of a neonatal form. Neuropediatrics 2000, 31, 86–92. [Google Scholar] [CrossRef]
- Srivastava, S.; Naidu, S. Alexander disease. In GeneReviews [Internet]; Pagon, R.A., Adam, M.P., Ardinger, H.H., Eds.; University of Washington: Seattle, WA, USA, 1993–2022; Last update: 12 November 2020. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1172 (accessed on 16 September 2022).
- Prust, M.; Wang, J.; Morizono, H.; Messing, A.; Brenner, M.; Gordon, E.; Hartka, T.; Sokohl, A.; Schiffmann, R.; Gordish-Dressman, H.; et al. GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology 2011, 77, 1287–1294. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, R.A.; Brenner, M.; Goldman, J.E.; Messing, A. GFAP and its role in Alexander disease. Exp. Cell Res. 2007, 313, 2077–2087. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Johnson, A.B.; Salomons, G.; Goldman, J.E.; Naidu, S.; Quinlan, R.; Cree, B.; Ruyle, S.Z.; Banwell, B.; D’Hooghe, M.; et al. Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann. Neurol. 2005, 57, 310–326. [Google Scholar] [CrossRef] [PubMed]
- Gomi, H.; Yokoyama, T.; Fujimoto, K.; Ikeda, T.; Katoh, A.; Itoh, T.; Itohara, S. Mice devoid of the glial fibrillary acidic protein develop normally and are susceptible to scrapie prions. Neuron 1995, 14, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Hagemann, T.L.; Connor, J.X.; Messing, A. Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress response. J. Neurosci. 2006, 26, 11162–11173. [Google Scholar] [CrossRef] [Green Version]
- Jany, P.L.; Hagemann, T.L.; Messing, A. GFAP expression as an indicator of disease severity in mouse models of Alexander disease. ASN Neuro 2013, 5, e00109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemann, T.L.; Powers, B.; Lin, N.H.; Mohamed, A.F.; Dague, K.L.; Hannah, S.C.; Bachmann, G.; Mazur, C.; Rigo, F.; Olsen, A.L.; et al. Antisense therapy in a rat model of Alexander disease reverses GFAP pathology, white matter deficits, and motor impairment. Sci. Transl. Med. 2021, 13, eabg4711. [Google Scholar] [CrossRef] [PubMed]
- Globus, J.H.; Strauss, I. Progressive degenerative subcortical encephalopathy (Schilder’s disease). Arch. Neurol. Psychiatry 1928, 20, 1190–1228. [Google Scholar] [CrossRef]
- Kronn, D.; Oddoux, C.; Phillips, J.; Ostrer, H. Prevalence of Canavan disease heterozygotes in the New York metropolitan Ashkenazi Jewish population. Am. J. Hum. Genet. 1995, 57, 1250. [Google Scholar]
- Feigenbaum, A.; Moore, R.; Clarke, J.; Hewson, S.; Chitayat, D.; Ray, P.N.; Stockley, T.L. Canavan disease: Carrier-frequency determination in the Ashkenazi Jewish population and development of a novel molecular diagnostic assay. Am. J. Med. Genet. Part A 2004, 124, 142–147. [Google Scholar] [CrossRef]
- Fares, F.; Badarneh, K.; Abosaleh, M.; Harari-Shaham, A.; Diukman, R.; David, M. Carrier frequency of autosomal-recessive disorders in the Ashkenazi Jewish population: Should the rationale for mutation choice for screening be reevaluated? Prenat. Diagn. 2008, 28, 236–241. [Google Scholar] [CrossRef]
- Wei, H.; Moffett, J.R.; Amanat, M.; Fatemi, A.; Tsukamoto, T.; Namboodiri, A.M.; Slusher, B.S. The pathogenesis of, and pharmacological treatment for, Canavan disease. Drug Discov. Today 2022, 27, 2467–2483. [Google Scholar] [CrossRef]
- Matalon, D.; Matalon, K.M.; Matalon, R. Canavan disease. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 6th ed.; Rosenberg, R.N., Pascual, J.M., Eds.; Academic Press: London, UK, 2020; pp. 909–916. [Google Scholar]
- Wang, Y.; Hull, V.; Sternbach, S.; Popovich, B.; Burns, T.; McDonough, J.; Guo, F.; Pleasure, D. Ablating the Transporter Sodium-Dependent Dicarboxylate Transporter 3 Prevents Leukodystrophy in Canavan Disease Mice. Ann. Neurol. 2021, 90, 845–850. [Google Scholar] [CrossRef]
- Pederzolli, C.D.; Mescka, C.P.; Scapin, F.; Rockenbach, F.J.; Sgaravatti, Â.M.; Sgarbi, M.B.; Wyse, A.T.; Wannmacher, C.M.; Wajner, M.; Dutra-Filho, C.S. N-acetylaspartic acid promotes oxidative stress in cerebral cortex of rats. Int. J. Dev. Neurosci. 2007, 25, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Surendran, S.; Bhatnagar, M. Upregulation of N-acetylaspartic acid induces oxidative stress to contribute in disease pathophysiology. Int. J. Neurosci. 2011, 121, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Assadi, M.; Janson, C.; Wang, D.J.; Goldfarb, O.; Suri, N.; Bilaniuk, L.; Leone, P. Lithium citrate reduces excessive intra-cerebral N-acetyl aspartate in Canavan disease. Eur. J. Paediatr. Neurol. 2010, 14, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Baslow, M.H.; Kitada, K.; Suckow, R.F.; Hungund, B.L.; Serikawa, T. The effects of lithium chloride and other substances on levels of brain N-acetyl-L-aspartic acid in Canavan disease-like rats. Neurochem. Res. 2002, 27, 403–406. [Google Scholar] [CrossRef]
- O’Donnell, T.; Rotzinger, S.; Nakashima, T.T.; Hanstock, C.C.; Ulrich, M.; Silverstone, P.H. Chronic lithium and sodium valproate both decrease the concentration of myo-inositol and increase the concentration of inositol monophosphates in rat brain. Brain Res. 2000, 880, 84–91. [Google Scholar] [CrossRef]
- Numata, Y.; Gotoh, L.; Iwaki, A.; Kurosawa, K.; Takanashi, J.I.; Deguchi, K.; Yamamoto, T.; Osaka, H.; Inoue, K. Epidemiological, clinical, and genetic landscapes of hypomyelinating leukodystrophies. J. Neurol. 2014, 261, 752–758. [Google Scholar] [CrossRef]
- Pelizaeus, F. Über eine eigenartige familiäre Entwicklungshemmung vornehmlich auf motorischem Gebiet. Arch. Für Psychiatr. Und Nervenkrankh. 1899, 31, 100–104. [Google Scholar]
- Merzbacher, L. Eine eigenartige familiär-hereditäre Erkrankungsform (Aplasia axialis extracorticalis congenita). Z. Für Die Gesamte Neurol. Und Psychiatr. 1910, 3, 1–138. [Google Scholar] [CrossRef] [Green Version]
- Seitelberger, F.; Urbanits, S.; Nave, K.A. Pelizaeus–Merzbacher disease. In Handbook of Clinical Neurology; Vinken, P., Bruyn, G., Eds.; Elsevier: Amsterdam, The Netherlands, 1996; pp. 551–579. [Google Scholar]
- Osório, M.J.; Goldman, S.A. Neurogenetics of Pelizaeus–Merzbacher disease. Handb. Clin. Neurol. 2018, 148, 701–722. [Google Scholar]
- Klugmann, M.; Schwab, M.H.; Pühlhofer, A.; Schneider, A.; Zimmermann, F.; Griffiths, I.R.; Nave, K.A. Assembly of CNS myelin in the absence of proteolipid protein. Neuron 1997, 18, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Van Der Knaap, M.S.; Van Der Voorn, P.; Barkhof, F.; Van Coster, R.; Krägeloh-Mann, I.; Feigenbaum, A.; Blaser, S.; Vles, J.S.; Rieckmann, P.; Pouwels, P.J. A new leukoencephalopathy with brainstem and spinal cord involvement and high lactate. Ann. Neurol. 2003, 53, 252–258. [Google Scholar] [CrossRef]
- Isohanni, P.; Linnankivi, T.; Buzkova, J.; Lönnqvist, T.; Pihko, H.; Valanne, L.; Tienari, P.J.; Elovaara, I.; Pirttilä, T.; Reunanen, M.; et al. DARS2 mutations in mitochondrial leucoencephalopathy and multiple sclerosis. J. Med. Genet. 2010, 47, 66–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aradjanski, M.; Dogan, S.A.; Lotter, S.; Wang, S.; Hermans, S.; Wibom, R.; Rugarli, E.; Trifunovic, A. DARS2 protects against neuroinflammation and apoptotic neuronal loss, but is dispensable for myelin producing cells. Hum. Mol. Genet. 2017, 26, 4181–4189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fine, A.S.; Nemeth, C.L.; Kaufman, M.L.; Fatemi, A. Mitochondrial aminoacyl-tRNA synthetase disorders: An emerging group of developmental disorders of myelination. J. Neurodev. Disord. 2019, 11, 29. [Google Scholar] [CrossRef]
- Williams, L.A.; Gerber, D.J.; Elder, A.; Tseng, W.C.; Baru, V.; Delaney-Busch, N.; Ambrosi, C.; Mahimkar, G.; Joshi, V.; Shah, H.; et al. Developing antisense oligonucleotides for a TECPR2 mutation-induced, ultra-rare neurological disorder using patient-derived cellular models. Mol. Ther.-Nucleic Acids 2022, 29, 189–203. [Google Scholar] [CrossRef]
- Synofzik, M.; van Roon-Mom, W.M.; Marckmann, G.; van Duyvenvoorde, H.A.; Graessner, H.; Schüle, R.; Aartsma-Rus, A.; behalf of the 1M1M consortium. Preparing n-of-1 antisense oligonucleotide treatments for rare neurological diseases in Europe: Genetic, regulatory, and ethical perspectives. Nucleic Acid Ther. 2022, 32, 83–94. [Google Scholar] [CrossRef]
- Angelman, H. ‘Puppet’children a report on three cases. Dev. Med. Child Neurol. 1965, 7, 681–688. [Google Scholar] [CrossRef]
- Williams, C.A.; Beaudet, A.L.; Clayton-Smith, J.; Knoll, J.H.; Kyllerman, M.; Laan, L.A.; Magenis, R.E.; Moncla, A.; Schinzel, A.A.; Summers, J.A.; et al. Angelman syndrome 2005: Updated consensus for diagnostic criteria. Am. J. Med. Genet. Part A 2006, 140, 413–418. [Google Scholar] [CrossRef]
- Margolis, S.S.; Sell, G.L.; Zbinden, M.A.; Bird, L.M. Angelman syndrome. Neurotherapeutics 2015, 12, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Person, R.E.; Beaudet, A.L. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum. Mol. Genet. 2012, 21, 3001–3012. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Ward, A.J.; Chun, S.; Bennett, C.F.; Beaudet, A.L.; Rigo, F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 2015, 518, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milazzo, C.; Mientjes, E.J.; Wallaard, I.; Rasmussen, S.V.; Erichsen, K.D.; Kakunuri, T.; van der Sman, A.E.; Kremer, T.; Miller, M.T.; Hoener, M.C.; et al. Antisense oligonucleotide treatment rescues UBE3A expression and multiple phenotypes of an Angelman syndrome mouse model. JCI Insight 2021, 6, e145991. [Google Scholar] [CrossRef]
- Sonzogni, M.; Hakonen, J.; Bernabé Kleijn, M.; Silva-Santos, S.; Judson, M.C.; Philpot, B.D.; van Woerden, G.M.; Elgersma, Y. Delayed loss of UBE3A reduces the expression of Angelman syndrome-associated phenotypes. Mol. Autism 2019, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Elgersma, Y.; Sonzogni, M. UBE3A reinstatement as a disease-modifying therapy for Angelman syndrome. Dev. Med. Child Neurol. 2021, 63, 802–807. [Google Scholar] [CrossRef]
- Dravet, C. Les epilepsies graves de l’enfant. Vie Med. 1978, 8, 543–548. [Google Scholar]
- Dravet, C. The core Dravet syndrome phenotype. Epilepsia 2011, 52, 3–9. [Google Scholar] [CrossRef]
- Inoue, Y.; Ohtsuka, Y.; Oguni, H.; Tohyama, J.; Baba, H.; Fukushima, K.; Ohtani, H.; Takahashi, Y.; Ikeda, S. Stiripentol open study in Japanese patients with Dravet syndrome. Epilepsia 2009, 50, 2362–2368. [Google Scholar] [CrossRef]
- Nabbout, R.; Mistry, A.; Zuberi, S.; Villeneuve, N.; Gil-Nagel, A.; Sanchez-Carpintero, R.; Stephani, U.; Laux, L.; Wirrell, E.; Knupp, K.; et al. Fenfluramine for treatment-resistant seizures in patients with Dravet syndrome receiving stiripentol-inclusive regimens: A randomized clinical trial. JAMA Neurol. 2020, 77, 300–308. [Google Scholar] [CrossRef] [Green Version]
- Devinsky, O.; Cross, J.H.; Laux, L.; Marsh, E.; Miller, I.; Nabbout, R.; Scheffer, I.E.; Thiele, E.A.; Wright, S. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N. Engl. J. Med. 2017, 376, 2011–2020. [Google Scholar] [CrossRef] [Green Version]
- Marini, C.; Scheffer, I.E.; Nabbout, R.; Suls, A.; De Jonghe, P.; Zara, F.; Guerrini, R. The genetics of Dravet syndrome. Epilepsia 2011, 52, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jancovski, N.; Jafar-Nejad, P.; Burbano, L.E.; Rollo, B.; Richards, K.; Drew, L.; Sedo, A.; Heighway, J.; Pachernegg, S.; et al. Antisense oligonucleotide therapy reduces seizures and extends life span in an SCN2A gain-of-function epilepsy model. J. Clin. Investig. 2021, 131, e152079. [Google Scholar] [CrossRef] [PubMed]
- Lenk, G.M.; Jafar-Nejad, P.; Hill, S.F.; Huffman, L.D.; Smolen, C.E.; Wagnon, J.L.; Petit, H.; Yu, W.; Ziobro, J.; Bhatia, K.; et al. Scn8a antisense oligonucleotide is protective in mouse models of SCN8A encephalopathy and Dravet syndrome. Ann. Neurol. 2020, 87, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhamadani, F.; Zhang, K.; Parikh, R.; Wu, H.; Rasmussen, T.P.; Bahal, R.; Zhong, X.B.; Manautou, J.E. Adverse Drug Reactions and Toxicity of the Food and Drug Administration–Approved Antisense Oligonucleotide Drugs. Drug Metab. Dispos. 2022, 50, 879–887. [Google Scholar] [CrossRef]
- Kumar, V.B.; Farr, S.A.; Flood, J.F.; Kamlesh, V.; Franko, M.; Banks, W.A.; Morley, J.E. Site-directed antisense oligonucleotide decreases the expression of amyloid precursor protein and reverses deficits in learning and memory in aged SAMP8 mice. Peptides 2000, 21, 1769–1775. [Google Scholar] [CrossRef]
- Poon, H.F.; Joshi, G.; Sultana, R.; Farr, S.A.; Banks, W.A.; Morley, J.E.; Calabrese, V.; Butterfield, D.A. Antisense directed at the Aβ region of APP decreases brain oxidative markers in aged senescence accelerated mice. Brain Res. 2004, 1018, 86–96. [Google Scholar] [CrossRef]
- Chang, J.L.; Hinrich, A.J.; Roman, B.; Norrbom, M.; Rigo, F.; Marr, R.A.; Norstrom, E.M.; Hastings, M.L. Targeting amyloid-β precursor protein, APP; splicing with antisense oligonucleotides reduces toxic amyloid-β production. Mol. Ther. 2018, 26, 1539–1551. [Google Scholar] [CrossRef]
- Zhao, H.T.; Damle, S.; Ikeda-Lee, K.; Kuntz, S.; Li, J.; Mohan, A.; Kim, A.; Hung, G.; Scheideler, M.A.; Scherer, S.S.; et al. PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models. J. Clin. Investig. 2018, 128, 359–368. [Google Scholar] [CrossRef]
- Ansseau, E.; Vanderplanck, C.; Wauters, A.; Harper, S.Q.; Coppée, F.; Belayew, A. Antisense oligonucleotides used to target the DUX4 mRNA as therapeutic approaches in faciosscapulohumeral muscular dystrophy (FSHD). Genes 2017, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.C.; King, O.D.; Zhang, Y.; Clayton, N.P.; Spencer, C.; Wentworth, B.M.; Emerson, C.P., Jr.; Wagner, K.R. Morpholino-mediated knockdown of DUX4 toward facioscapulohumeral muscular dystrophy therapeutics. Mol. Ther. 2016, 24, 1405–1411. [Google Scholar] [CrossRef] [Green Version]
- Derbis, M.; Kul, E.; Niewiadomska, D.; Sekrecki, M.; Piasecka, A.; Taylor, K.; Hukema, R.K.; Stork, O.; Sobczak, K. Short antisense oligonucleotides alleviate the pleiotropic toxicity of RNA harboring expanded CGG repeats. Nat. Commun. 2021, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Borgonetti, V.; Galeotti, N. Intranasal delivery of an antisense oligonucleotide to the RNA-binding protein HuR relieves nerve injury-induced neuropathic pain. Pain 2021, 162, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Shohami, E.; Kaufer, D.; Chen, Y.; Seidman, S.; Cohen, O.; Ginzberg, D.; Melamed-Book, N.; Yirmiya, R.; Soreq, H. Antisense prevention of neuronal damages following head injury in mice. J. Mol. Med. 2000, 78, 228–236. [Google Scholar] [CrossRef]
- Ghirnikar, R.S.; Lee, Y.L.; Li, J.D.; Eng, L.F. Chemokine inhibition in rat stab wound brain injury using antisense oligodeoxynucleotides. Neurosci. Lett. 1998, 247, 21–24. [Google Scholar] [CrossRef]
- Lu, K.T.; Huang, T.C.; Tsai, Y.H.; Yang, Y.L. Transient receptor potential vanilloid type 4 channels mediate Na-K-Cl-co-transporter-induced brain edema after traumatic brain injury. J. Neurochem. 2017, 140, 718–727. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug (Year of Approval) | Indication | Mechanism | Design |
|---|---|---|---|
| Fomivirsen (1998) | Retinitis induced by CMV in people with AIDS [5] | Inhibitor of the major IE2 proteins synthesis in CMV (RNase H-inducing) | Phosphorothioate nucleotides 5′-G-C-G-T-T-T-G-C-T-C-T-T-C-T-T-C-T-T-G-C-G-3′ |
| Mipomersen (2013) | Reducing LDL cholesterol concentrations in individuals with homozygous familial hypercholesterolemia [6] | Inhibitor of apolipoprotein B 100 synthesis in liver (RNase H-inducing) | 2′-O-methoxyethyl and phosphorothioate nucleotides 5′-mG-mC*-mC*-mU*-mC*-A-G-T-C*-T-G-C*-T-T-C*-mG-mC*-mA-mC*-mC*-3′ |
| Eteplirsen (2016) | Treatment of Duchenne muscular dystrophy [7] | Inducing the production of functional dystrophin through specific skipping of exon 51 in defective DMD variants (Splicing modulation) | phosphorodiamidate morpholino nucleotides 5′-C-T-C-C-A-A-C-A-T-C-A-A-G-G-A-A-G-A-T-G-G-C-A-T-T-T-C-T-A-G-3′ |
| Defibrotide (2016) | Treatment of severe hepatic veno-occlusive disease [8] | It has antithrombotic, profibrinolytic, anti-ischemic, and antiadhesive features that hydrolyze clots but no specific mechanism was found | It has a natural origin achieved from polymerization of porcine intestinal mucosal DNA. It is composed of a single and double-stranded phosphodiester mixture with a length of about 50 nucleotides |
| Nusinersen (2016) | Treatment of spinal muscular atrophy [9] | Inducing the inclusion of exon 7 in SMN2 gene by binding to intronic splicing silencer to produce full-length SMN protein (Splicing modulation) | 2′-O-methoxyethyl and phosphorothioate nucleotides mU*-mC*-mA-mC*-mU*-mU*-mU*-mC*-mA-mU*-mA-mA-mU*-G-mC*-mU*-mG-mG |
| Inotersen (2018) | Treatment of hereditary transthyretin amyloidosis [10] | Inhibitor of transthyretin synthesis by binding to 3′ untranslated region of TTR mRNA (RNase H-inducing) | 2′-O-methoxyethyl and phosphorothioate nucleotides 5′-mT-mC*-mT-mT-mG-G-T-T-A-C*-A-T-G-A-A-mA-mT-mC*-mC*-mC* |
| Golodirsen (2019) | Treatment of Duchenne muscular dystrophy [11] | Inducing the production of functional dystrophin through specific skipping of exon 53 in defective DMD variants (Splicing modulation) | phosphorodiamidate morpholino nucleotides 5′-G-T-T-G-C-C-T-C-C-G-G-T-T-C-T-G-A-A-G-G-T-G-T-T-C-3′ |
| Viltolarsen (2020) | Treatment of Duchenne muscular dystrophy [12] | Inducing the production of functional dystrophin through specific skipping of exon 53 in defective DMD variants (Splicing modulation) | phosphorodiamidate morpholino nucleotides 5′-C-C-T-C-C-G-G-T-T-C-T-G-A-A-G-G-T-G-T-T-C-3′ |
| Casimersen (2021) | Treatment of Duchenne muscular dystrophy [13] | Inducing the production of functional dystrophin through specific skipping of exon 45 in defective DMD variants (Splicing modulation) | phosphorodiamidate morpholino nucleotides 5′-C-A-A-T-G-C-C-A-T-C-C-T-G-G-A-G-T-T-C-C-T-G-3′ |
| Drug | Disease | Target mRNA | Suggested Dose | Delivery Route | Adverse Events * |
|---|---|---|---|---|---|
| Nusinersen (Spinraza) | Spinal muscular atrophy | SMN2 to increase exon 7 inclusion | 12 mg Treatment includes four loading doses. The first three doses are injected at 14-day intervals and the fourth dose should be injected 30 days after the third dose. The drug is then administered every four months. | IT | Nephrotoxicity, coagulation defects, thrombocytopenia, and IT-related adverse events including headaches, fever, and meningitis |
| Inotersen (Tegsedi) | Polyneuropathy caused by hereditary transthyretin amyloidosis | TTR to prevent protein synthesis | 300 mg Once per week | SC | Injection site reaction, headaches, fever, nausea, decreased appetite, liver inflammation, and nephrotoxicity are common adverse events. One patient who received inotersen in a clinical trial died after intracranial hemorrhage due to thrombocytopenia |
| Eteplirsen (Exondys 51) | Duchenne muscular dystrophy | DMD to skip exon 51 | 30 mg/kg Once per week as 35- to 60-min infusion | IV | Hypersensitivity reaction (e.g., bronchospasm, chest pain, cough, tachycardia, and urticaria), headaches, nausea, vomiting, upper respiratory tract infection, nasopharyngitis, balance disorder, contact dermatitis, and arthralgia |
| Golodirsen (Vyondys 53) | Duchenne muscular dystrophy | DMD to skip exon 53 | 30 mg/kg Once per week as 35- to 60-min infusion | IV | Hypersensitivity reaction, headaches, nausea, vomiting, cough, fever, abdominal pain, nasopharyngitis, falls, and nephrotoxicity |
| Viltolarsen (Viltepso) | Duchenne muscular dystrophy | DMD to skip exon 53 | 80 mg/kg Once per week as 60-min infusion | IV | Upper respiratory tract infection, injection site reaction, cough, fever, contusion, arthralgia, diarrhea, nausea, vomiting, abdominal pain, reduced ejection fraction, urticarial, and nephrotoxicity |
| Casimersen (Amondys 45) | Duchenne muscular dystrophy | DMD to skip exon 45 | 30 mg/kg Once per week as 35- to 60-min infusion | IV | Upper respiratory tract infection, cough, fever, headaches, arthralgia, pain in mouth and throat, and nephrotoxicity |
| Identifier | Disease | Drug | Phase | Delivery Route | Notes |
|---|---|---|---|---|---|
| NCT04849741 | Alexander disease | ASO inhibiting GFAP synthesis (ION373) | I–III | IT | The drug or placebo will be injected for a 60-week period. All participants will then receive ION373 for a 60-week open-label treatment period. |
| NCT03186989 | Alzheimer’s disease and frontotemporal dementia | ASO inhibiting tau protein synthesis (IONIS-MAPTRx) | I/II | IT | The study will include two parts. In part 1, the ASO or placebo will be administered at 4-week intervals over the course of a 13-week treatment period for four dose levels. In part 2, ASO will be administered at quarterly intervals for 48 weeks. |
| NCT04931862 | Amyotrophic lateral sclerosis or frontotemporal dementia with documented mutation of G4C2 repeat expansion in the first intronic region of the C9orf72 gene | ASO targeting C9orf72 mRNA to selectively reduce C9orf72-repeat-containing transcripts (WVE-004) | I/II | IT | Different doses of WVE-004 or artificial cerebrospinal fluid (placebo) will be administered |
| NCT04768972 | Amyotrophic lateral sclerosis with FUS mutation | ASO inhibiting FUS protein synthesis (ION363) | III | IT | The study will include two parts. Part 1 will consist of participants that will be randomized in a 2:1 ratio to receive a multidose regimen of ION363 or placebo for a period of 61 weeks, followed by Part 2, in which participants will receive ION363 for a period of 85 weeks. |
| NCT04494256 | Amyotrophic lateral sclerosis with no mutation in SOD1 or FUS gene | ASO inhibiting ataxin-2 synthesis (BIIB105) | I | IT | The study will include three loading doses for three days, followed by two or five maintenance doses. |
| NCT04856982 | Amyotrophic lateral sclerosis with SOD1 mutation | ASO inhibiting SOD1 protein synthesis (Tofersen) | III | IT | The drug will be administered on days 1, 15, 29, and every 28 days thereafter for up to 2 years. In the next part, participants who received placebo before, will receive tofersen 100 mg on days 1, 15, 29, and every 28 days thereafter for up to 2 years. Participants who received tofersen before, will receive tofersen 100 mg on days 1, 29, and every 28 days thereafter for up to 2 years, with a dose of placebo on day 15 to maintain the study blind |
| NCT04259281 | Angelman Syndrome | ASO inhibiting UBE3A antisense transcript to increase paternal UBE3A expression (GTX-102) | I/II | IT | Treatment will include 2 mg, 3.3 mg, or 5 mg for 3–4 monthly doses followed by a quarterly maintenance regimen |
| NCT04428281 | Angelman Syndrome | ASO inhibiting UBE3A antisense transcript to increase paternal UBE3A expression (RO7248824) | I | IT | The drug will be administered in different dose levels over a period of 8 weeks, with a minimum of approximately 4 weeks between each dose administration. In the long-term extension part RO7248824 will be administered up to 10 doses in selected dose levels with a minimum of approximately 16 weeks between each dose administration. |
| NCT05127226 | Angelman Syndrome | ASO inhibiting UBE3A antisense transcript to increase paternal UBE3A expression (ION582) | I/II | IT | ION582 will be administered in different doses over a period of 13 weeks, with a minimum of approximately 4 weeks between each dose administration. |
| NCT04123626 | Autosomal dominant retinitis pigmentosa due to the P23H mutation in the RHO gene | ASO inhibiting P23H protein expression while preserving expression of the wild type rhodopsin protein (QR-1123) | I/II | IVT | The study will comprise up to 8 single dose and repeat dose cohorts. In the single dose, patients will receive a single injection. In the repeat dose cohorts, subjects will be randomized to receive either a unilateral injection of every 3 months or a unilateral sham-procedure every 3 months |
| NCT04442295 | Dravet syndrome | ASO targeting nonproductive alternative splicing in SCN1A mRNA to upregulate Nav1.1 (STK001) | I/II | IT | In one group single ascending doses including 10 mg, 20 mg, 30 mg, and 45 mg will be administered, and in another group, multiple ascending doses including 20 mg, 30 mg, and 45 mg will be administered |
| NCT04906460 | Duchenne muscular dystrophy | Exon-skipping ASO to promote skipping over exon 53 in DMD pre-mRNA (WVE-N531) | I/II | IV | Up to 4 dose levels of WVE-N531 will be administered at least 4 weeks apart |
| NCT04004065 | Duchenne muscular dystrophy | Peptide-conjugated eteplirsen to promote skipping over exon 51 in DMD pre-mRNA (vesleteplirsen or SRP-5051) | II | IV | Participants will receive escalating dose levels of treatment, every 4 weeks, for up to 75 weeks during Part A of the study. Once the doses have been selected for Part B, all participants who have completed Part A will transition to Part B. In this part participants will receive treatment at the doses selected based on data from Part A every 4 weeks, for up to 2 years. |
| NCT05524883 | Duchenne muscular dystrophy | Exon-skipping ASO to promote skipping over exon 51 in DMD pre-mRNA (DYNE-251) | I/II | IV | DYNE-251 or placebo will be administered 6 times over 24 weeks. Then, DYNE-251 will be administered up to 30 times over 120 weeks in all participants |
| NCT05032196 | Early Huntington’s disease | Allele selective ASO targeting single nucleotide polymorphism (SNP)-3 to reduce mutant HTT synthesis (WVE-003) | I/II | IT | Injection of 4 doses of WVE-003 or placebo to find out the safety and pharmacokinetics of the drug |
| NCT03761849 | Huntington’s disease | Allele nonselective ASO lowering mutant and wild-type HTT synthesis (RO7234292) | III | IT | The drug will be administered every 8 or 16 weeks to compare its safety and efficacy to placebo that will be injected every 8 weeks |
| NCT04855045 | Leber congenital amaurosis 10 due to the c.2991+1655A>G | ASO targeting mutated CEP290 pre-mRNA to redirect normal splicing (Sepofarsen) | II/III | IVT | The study will consist of two parts: an open-label dose escalation part with three planned dose groups, followed by a double-masked randomized part. Patients will receive an initial loading dose, followed by maintenance doses every 6 months |
| NCT03913143 | Leber congenital amaurosis 10 due to the c.2991+1655A>G | ASO targeting mutated CEP290 pre-mRNA to redirect normal splicing (Sepofarsen) | II/III | IVT | Treatment will include an initial loading dose, followed by maintenance doses at month 3 and every 6 months |
| NCT04165486 | Multiple system atrophy | ASO inhibiting alpha-synuclein synthesis (ION464) | I | IT | The study will include two parts. Part 1 of the study will consist a screening period of up to 6 weeks, a treatment period of 12 weeks, and a follow-up period of 24 weeks. The study duration for each participant in part 2 will be approximately 96 weeks, which will consist a 72-week treatment period and a 24-week follow-up period. |
| NCT05481879 | Myotonic dystrophy type I | ASO inhibiting DM1 protein kinase synthesis (DYNE-101) | I/II | IV | The study will include screening period (up to 6 weeks), a multiple-ascending dose placebo-controlled period (24 weeks), a treatment period (24 weeks) and a long-term extension period (96 weeks). |
| NCT04485949 | Newly diagnosed glioblastoma multiforme | IGV-001, containing autologous glioblastoma cells treated with antisense oligonucleotide (IMV-001) targeting IGF-1R, encapsulated in biodiffusion chambers; followed by radiotherapy and temozolomide | II | Abdominal rectus sheath | Biodiffusion chambers including IGV-001 or placebo will be implanted and then, remove at approximately 48 h after implantation. After 6 weeks of ASO therapy, participants will receive radiotherapy per institutional standards for 5 days per week along with temozolomide 75 mg/m2 orally, once daily for up to 12 weeks followed by temozolomide 150 to 200 mg/m2, orally, on Days 1 to 5 of each 28-day cycle for up to 6 cycles |
| NCT03976349 | Parkinson’s disease | ASO inhibiting LRRK2 synthesis (ION859) | I | IT | Study will have two parts. In part A, Participants will receive a single injection in different doses, and in part B, Participants will receive a single injection in different doses on multiple days |
| NCT04539041 | Progressive supranuclear palsy | ASO targeting MPAT mRNA to reduce tau expression (NIO752) | I | IT | Drug in different doses or placebo will be administered in 6 cohorts over 3 months |
| NCT05160558 | Spinocerebellar Ataxia 3 | ASO inhibiting ATXN3 protein (BIIB132) | I | IT | The study will consist of five cohorts to administer drug or placebo every four weeks for up to 85 days |
| NCT05085964 | USH2A-associated retinitis pigmentosa | ASO targeting USH2A mRNA to skip exon 13 (Ultevursen or QR-421a) | II | IVT | This is an extension of a completed phase I/II trial (Stellar study with NCT03780257 identifier). The QR-421a Injection will be in one or both eyes of all participants and repeat every 6 months for an anticipated period of 24 months (Helia study) |
| NCT05176717 | USH2A-associated retinitis pigmentosa | ASO targeting USH2A mRNA to skip exon 13 (Ultevursen or QR-421a) | II/III | IVT | Patients with early to moderate vision loss will receive loading dose of 180 µg (group 1) or 60 µg (group 2) and maintenance dose of 60 µg. The third group receives sham-procedure (Celeste study) |
| NCT05158296 | USH2A-associated retinitis pigmentosa | ASO targeting USH2A mRNA to skip exon 13 (Ultevursen or QR-421a) | II/III | IVT | Patients with advanced vision loss will receive loading dose of 180 µg (group 1) or 60 µg (group 2) and maintenance dose of 60 µg. The third group receives sham-procedure (Sirius study) |
| Authors | Disease | Mechanism | Study Setting (Route of Administration/Type of Animals or Cells) |
|---|---|---|---|
| Kumar et al. [271]; Poon et al. [272]; Chang et al. [273] | Alzheimer’s disease | Reducing the levels of toxic amyloid-β precursor protein | In vivo ICV in mice |
| Zhao et al. [274] | Charcot–Marie–Tooth type 1A | Reducing the levels of PMP22 mRNA | In vivo SC in mice |
| Chang et al. [273] | Down syndrome | Reducing the levels of toxic amyloid-β precursor protein | In vitro Patient-derived fibroblasts |
| Ansseau et al. [275]; Chen et al. [276]; Lim et al. [75] | Facioscapulohumeral muscular dystrophy (FSHD) | Reducing the levels of DUX4 mRNA | Ansseau et al. and Lim et al.: In vivo intramuscular injection in mice Chen et al.: In vitro patient-derived myotubes In vivo Electroporation of ASO into FSHD patient muscle xenografts in mice |
| Derbis et al. [277] | Fragile X-associated tremor/ataxia syndrome | Targeting the expanded CGG repeat in FMR1 mRNA | In vitro Patient-derived fibroblasts In vivo ICV in mice |
| Gagnon et al. [74] | Huntington’s disease | Targeting the expanded CAG repeat in HTT mRNA | In vitro Patient-derived fibroblasts |
| Borgonetti and Galeotti [278] | Neuropathic pain | Reducing the levels of HuR mRNA | In vivo Intranasal or IT injection in mice |
| Alarcón-Arís et al. [135] | Parkinson’s disease | Reducing the levels of alpha-synuclein | In vivo Intranasal in mice |
| Pelisch et al. [87] | Spinal cord injury | Reducing the levels of CCL3 chemokine to inhibit inflammation | In vitro Bone marrow-derived macrophages of mice In vivo IT in mice |
| Shohami et al. [279]; Ghirinkar et al. [280]; Lu et al. [281] | Traumatic brain injury | Shohami et al.: Reducing the levels of acetylcholinesterase Ghirinkar et al.: Reducing the levels of monocyte chemoattractant protein 1 Lu et al.: Reducing the levels of transient receptor potential vanilloid type 4 | In vivo Shohami et al.: ICV in mice Ghirinkar et al.: Infusion into the injured area in brain of rats Lu et al.: ICV in rats |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amanat, M.; Nemeth, C.L.; Fine, A.S.; Leung, D.G.; Fatemi, A. Antisense Oligonucleotide Therapy for the Nervous System: From Bench to Bedside with Emphasis on Pediatric Neurology. Pharmaceutics 2022, 14, 2389. https://doi.org/10.3390/pharmaceutics14112389
Amanat M, Nemeth CL, Fine AS, Leung DG, Fatemi A. Antisense Oligonucleotide Therapy for the Nervous System: From Bench to Bedside with Emphasis on Pediatric Neurology. Pharmaceutics. 2022; 14(11):2389. https://doi.org/10.3390/pharmaceutics14112389
Chicago/Turabian StyleAmanat, Man, Christina L. Nemeth, Amena Smith Fine, Doris G. Leung, and Ali Fatemi. 2022. "Antisense Oligonucleotide Therapy for the Nervous System: From Bench to Bedside with Emphasis on Pediatric Neurology" Pharmaceutics 14, no. 11: 2389. https://doi.org/10.3390/pharmaceutics14112389