
An Overview on Immunogenic Cell Death in Cancer Biology and Therapy

 , ,
, ,  and
and
Abstract
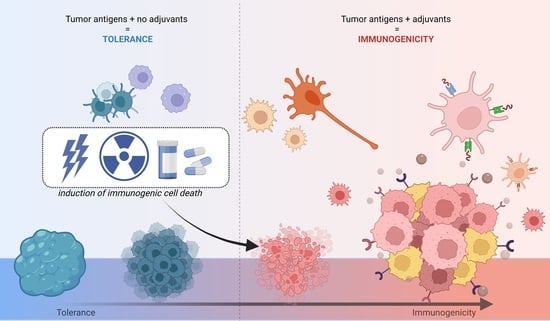
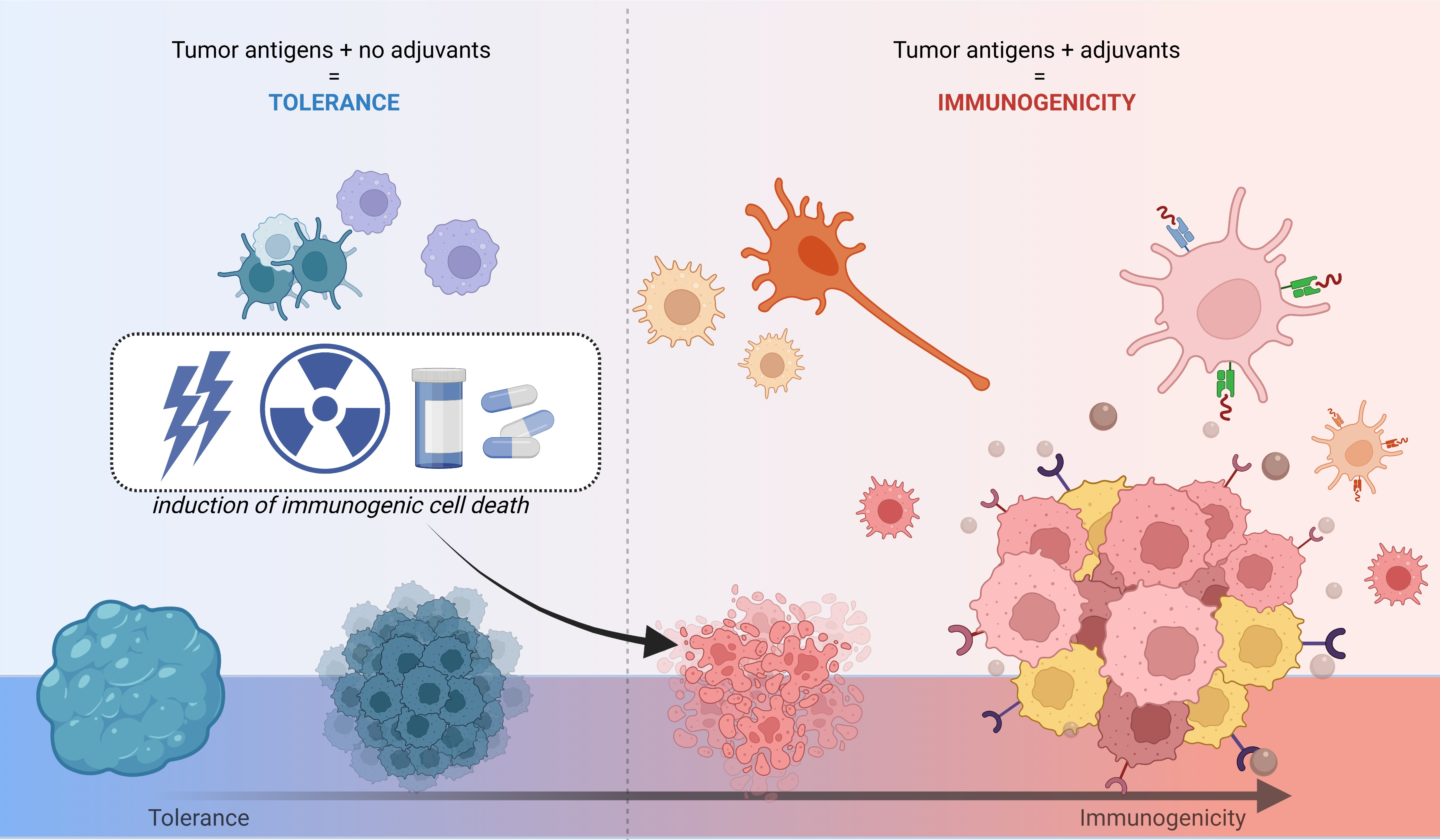
:
1. Introduction
2. A Historical Overview on Cancer Immunotherapy
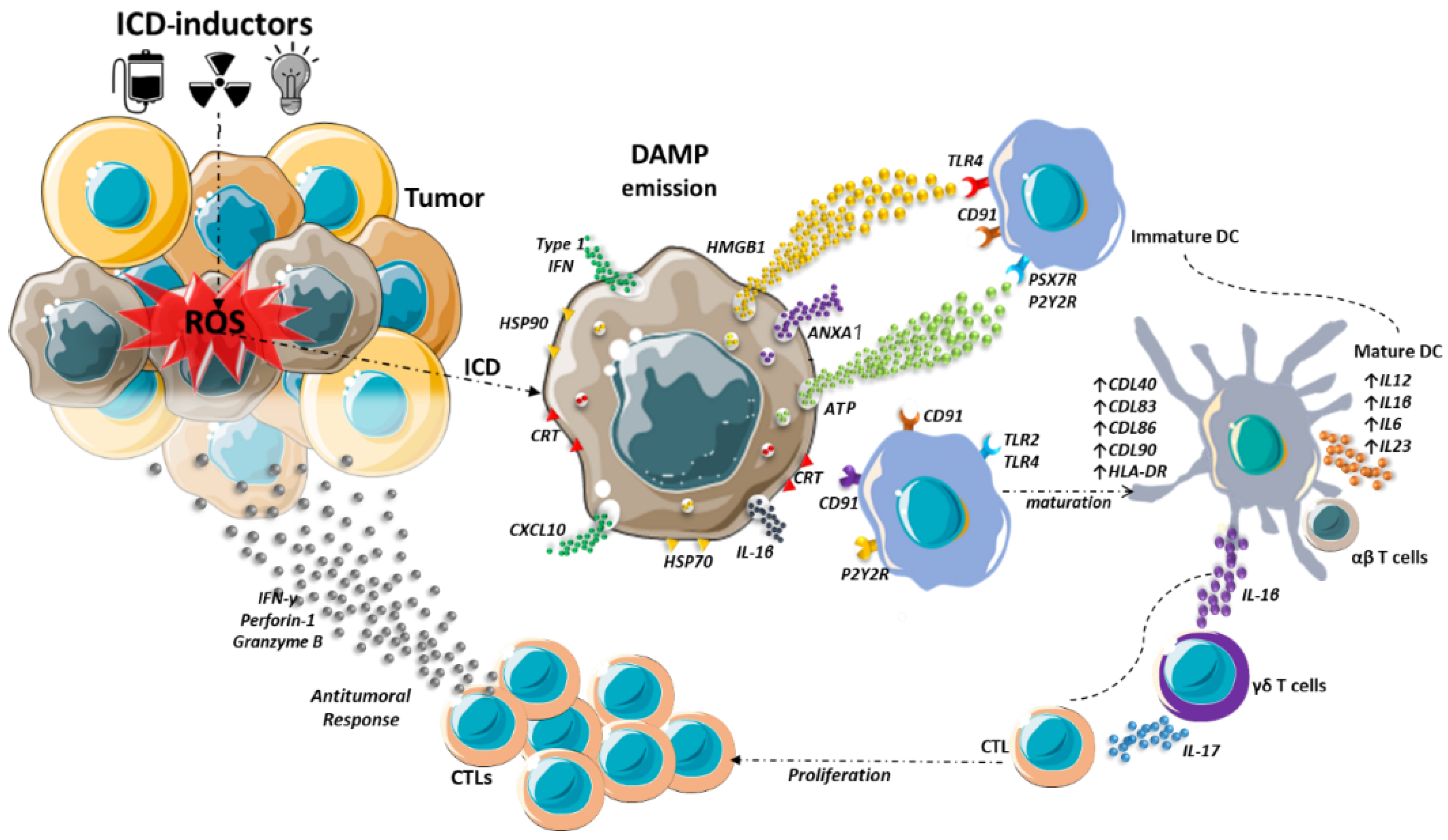
3. Immunogenic Cell Death
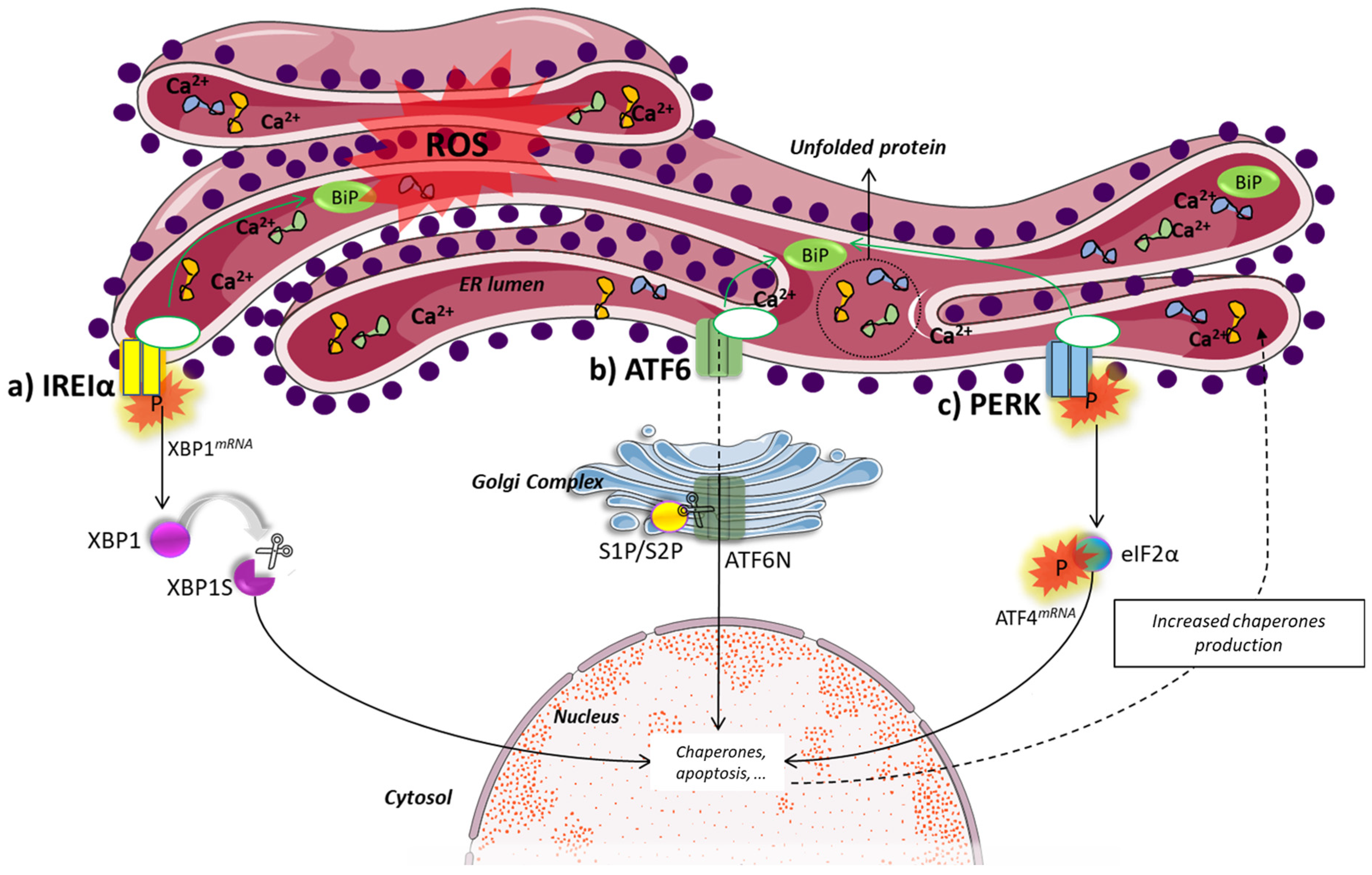
4. Endoplasmic Reticulum Stress
5. Damage-Associated Molecular Patterns
6. ICD and DAMPs in Cancer Therapy
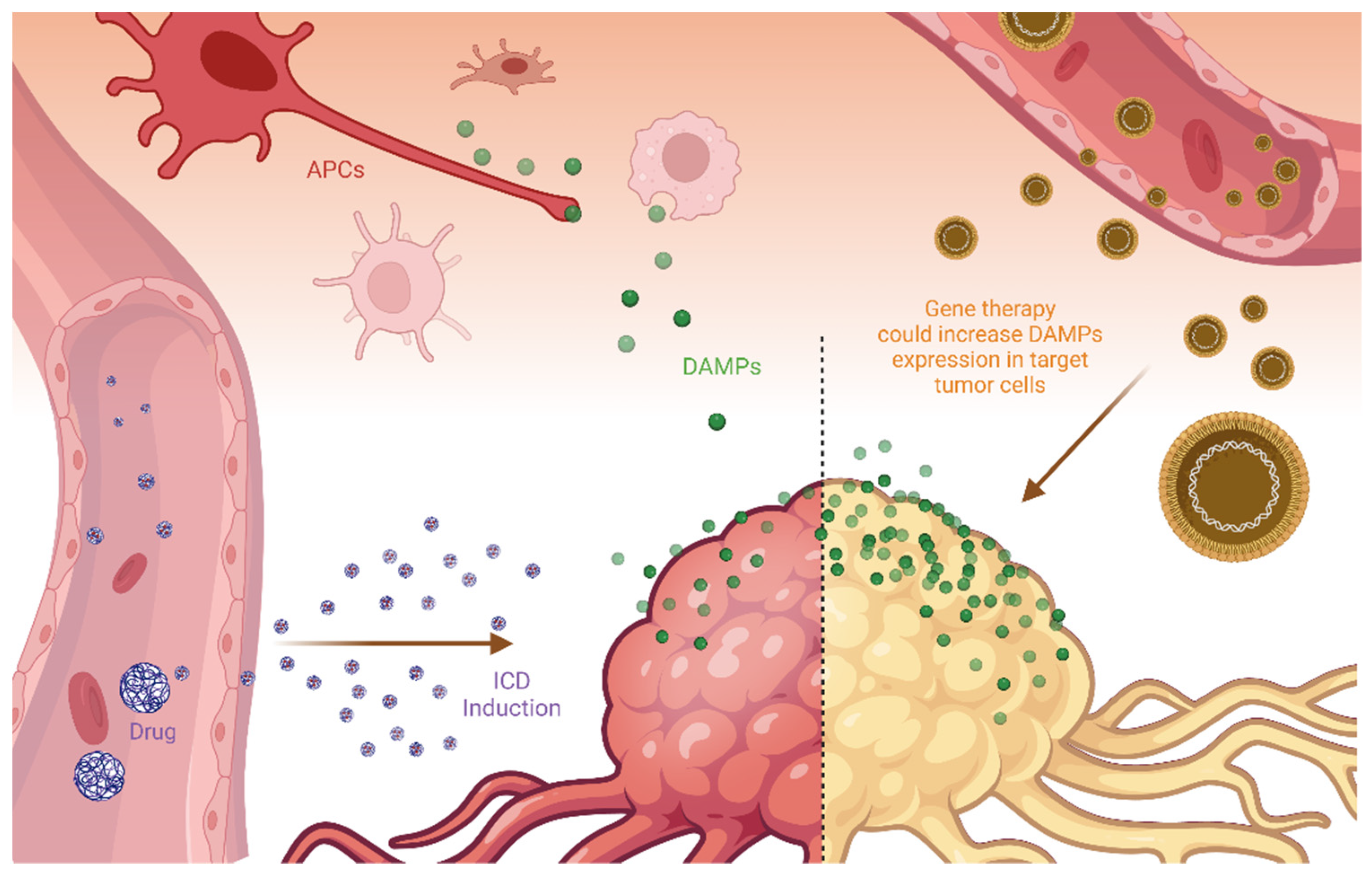
7. Delivery of DAMPs and ICD-Inducers to Tumor Tissues
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.N.; Kishton, R.J.; Restifo, N.P. Developing neoantigen-targeted T cell–based treatments for solid tumors. Nat. Med. 2019, 25, 1488–1499. [Google Scholar] [CrossRef]
- Weinberg, R.A. The Biology of Cancer, 2nd ed.; Garland Science, Taylor & Francis Group, LLC: New York, NY, USA, 2013. [Google Scholar]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Busch, W. Aus der Sitzung der medicinischen Section vom 13 November 1867. Berl. Klin. Wochenschr. 1868, 5, 137. [Google Scholar]
- Fehleisen, F. Die Aetiologie des Erysipels; Theodor Fischer: Berlin, Germany, 1883. [Google Scholar]
- Bruns, P. Die Heilwirkung des Erysipels auf Geschwülste. Bruns’ Beiträge zur klinischen Chirurgie. 1887. Available online: https://books.google.com.br/books?id=0Gw1AQAAMAAJ&printsec=frontcover&source=gbs_ge_summary_r&cad=0#v=onepage&q&f=false (accessed on 5 May 2022).
- Coley, W.B., II. Contribution to the Knowledge of Sarcoma. Ann. Surg. 1891, 14, 199–220. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 2006, 26, 154–158. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1888599/ (accessed on 6 May 2022). [PubMed]
- Oiseth, S.J.; Aziz, M.S. Cancer immunotherapy: A brief review of the history, possibilities, and challenges ahead. J. Cancer Metastasis Treat. 2017, 3, 250. [Google Scholar] [CrossRef]
- Codman, E. Symposium on the Treatment of Primary Malignant Bone Tumors: The Memorial Hospital Conference on the Treatment of Bone Sarcoma. Am. J. Surg. 1935, 27, 3–6. [Google Scholar] [CrossRef]
- Ehrlich, P. “Über den Jetzigen Stand der Karzinomforschung” Beiträge zur Experimentellen. Pathologie und Chemotherapie. 1909. Available online: https://curiosity.lib.harvard.edu/contagion/catalog/36-990061083080203941 (accessed on 5 May 2022).
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Burnet, M. Cancer—A biological approach. I. The processes of control. Br. Med. J. 1957, 1, 779–786. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Pernot, S.; Terme, M.; Voron, T.; Colussi, O.; Marcheteau, E.; Tartour, E.; Taieb, J. Colorectal cancer and immunity: What we know and perspectives. World J. Gastroenterol. 2014, 20, 3738–3750. [Google Scholar] [CrossRef]
- Sanchez-Castañón, M.; Er, T.; Bujanda, L.; Herreros-Villanueva, M. Immunotherapy in colorectal cancer: What have we learned so far? Clin. Chim. Acta 2016, 460, 78–87. [Google Scholar] [CrossRef]
- Dobosz, P.; Dzieciątkowski, T. The Intriguing History of Cancer Immunotherapy. Front. Immunol. 2019, 10, 2965. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.F.A.P.; Mitchell, G.F.; Weiss, N.S. Cellular Basis of the Immunological Defects in Thymectomized Mice. Nature 1967, 214, 992–997. [Google Scholar] [CrossRef]
- Steinman, R.M.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice: I. morphology, quantitation, tissue distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef]
- Kiessling, R.; Klein, E.; Wigzell, H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 1975, 5, 112–117. [Google Scholar] [CrossRef]
- Yousefi, H.; Yuan, J.; Keshavarz-Fathi, M.; Murphy, J.F.; Rezaei, N. Immunotherapy of cancers comes of age. Expert Rev. Clin. Immunol. 2017, 13, 1001–1015. [Google Scholar] [CrossRef]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Krombach, J.; Hennel, R.; Brix, N.; Orth, M.; Schoetz, U.; Ernst, A.; Schuster, J.; Zuchtriegel, G.; Reichel, C.A.; Bierschenk, S.; et al. Priming anti-tumor immunity by radiotherapy: Dying tumor cell-derived DAMPs trigger endothelial cell activation and recruitment of myeloid cells. OncoImmunology 2018, 8, e1523097. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.C.; Júnior, W.T.D.S.; Mundim, T.; Vale, C.L.C.; de Oliveira, J.V.; Ganassin, R.; Pacheco, T.J.A.; Morais, J.A.V.; Longo, J.P.F.; Azevedo, R.B.; et al. Induction of Immunogenic Cell Death by Photodynamic Therapy Mediated by Aluminum-Phthalocyanine in Nanoemulsion. Pharmaceutics 2022, 14, 196. [Google Scholar] [CrossRef]
- Morais, J.A.V.; Almeida, L.R.; Rodrigues, M.C.; Azevedo, R.B.; Muehlmann, L.A. The induction of immunogenic cell death by photodynamic therapy in B16F10 cells in vitro is effected by the concentration of the photosensitizer. Photodiagn. Photodyn. Ther. 2021, 35, 102392. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [Green Version]
- Minton, K. DAMP-driven metabolic adaptation. Nat. Rev. Immunol. 2019, 20, 1. [Google Scholar] [CrossRef] [Green Version]
- Collett, G.P.; Redman, C.W.; Sargent, I.L.; Vatish, M. Endoplasmic reticulum stress stimulates the release of extracellular vesicles carrying danger-associated molecular pattern (DAMP) molecules. Oncotarget 2018, 9, 6707–6717. [Google Scholar] [CrossRef] [Green Version]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2019, 20, 95–112. [Google Scholar] [CrossRef]
- Serrano-del Valle, A.; Anel, A.; Naval, J.; Marzo, I. Immunogenic cell death and immunotherapy of multiple myeloma. Front. Cell Dev. Biol. 2019, 7, 50. [Google Scholar] [CrossRef]
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 2022, 23, 487–500. [Google Scholar] [CrossRef]
- Baracco, E.E.; Stoll, G.; Van Endert, P.; Zitvogel, L.; Vacchelli, E.; Kroemer, G. Contribution of annexin A1 to anticancer immunosurveillance. OncoImmunology 2019, 8, e1647760. [Google Scholar] [CrossRef] [Green Version]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2006, 13, 54–61. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Liu, Y.-H.; Fang, Z.-S.; Lin, C.-L.; Lin, J.-C.; Yao, B.-Y.; Hu, C.-M.J. Synthetic Immunogenic Cell Death Mediated by Intracellular Delivery of STING Agonist Nanoshells Enhances Anticancer Chemo-immunotherapy. Nano Lett. 2020, 20, 2246–2256. [Google Scholar] [CrossRef]
- Jafari, S.; Lavasanifar, A.; Hejazi, M.S.; Maleki-Dizaji, N.; Mesgari, M.; Molavi, O. STAT3 inhibitory stattic enhances immunogenic cell death induced by chemotherapy in cancer cells. DARU J. Pharm. Sci. 2020, 28, 159–169. [Google Scholar] [CrossRef]
- Vanmeerbeek, I.; Sprooten, J.; De Ruysscher, D.; Tejpar, S.; Vandenberghe, P.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. OncoImmunology 2020, 9, 1703449. [Google Scholar] [CrossRef] [Green Version]
- Doix, B.; Trempolec, N.; Riant, O.; Feron, O. Low Photosensitizer Dose and Early Radiotherapy Enhance Antitumor Immune Response of Photodynamic Therapy-Based Dendritic Cell Vaccination. Front. Oncol. 2019, 9, 811. [Google Scholar] [CrossRef]
- Rapoport, B.L.; Anderson, R. Realizing the Clinical Potential of Immunogenic Cell Death in Cancer Chemotherapy and Radiotherapy. Int. J. Mol. Sci. 2019, 20, 959. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Yang, J.; Luo, L.; Jiang, M.; Qin, B.; Yin, H.; Zhu, C.; Yuan, X.; Zhang, J.; Luo, Z.; et al. Targeting photodynamic and photothermal therapy to the endoplasmic reticulum enhances immunogenic cancer cell death. Nat. Commun. 2019, 10, 3349. [Google Scholar] [CrossRef] [Green Version]
- Turubanova, V.D.; Balalaeva, I.V.; Mishchenko, T.A.; Catanzaro, E.; Alzeibak, R.; Peskova, N.N.; Efimova, I.; Bachert, C.; Mitroshina, E.V.; Krysko, O.; et al. Immunogenic cell death induced by a new photodynamic therapy based on photosens and photodithazine. J. Immunother. Cancer 2019, 7, 350. [Google Scholar] [CrossRef]
- He, H.; Liu, L.; Liang, R.; Zhou, H.; Pan, H.; Zhang, S.; Cai, L. Tumor-targeted nanoplatform for in situ oxygenation-boosted immunogenic phototherapy of colorectal cancer. Acta Biomater. 2020, 104, 188–197. [Google Scholar] [CrossRef]
- Deng, H.; Zhou, Z.; Yang, W.; Lin, L.-S.; Wang, S.; Niu, G.; Song, J.; Chen, X. Endoplasmic Reticulum Targeting to Amplify Immunogenic Cell Death for Cancer Immunotherapy. Nano Lett. 2020, 20, 1928–1933. [Google Scholar] [CrossRef]
- Ganassin, R.; Oliveira, G.R.T.; da Rocha, M.C.O.; Morais, J.A.V.; Rodrigues, M.C.; Motta, F.N.; Azevedo, R.B.; Muehlmann, L.A. Curcumin induces immunogenic cell death in murine colorectal carcinoma CT26 cells. Pharmacol. Res.-Mod. Chin. Med. 2022, 2, 100046. [Google Scholar] [CrossRef]
- Dulloo, I.; Atakpa-Adaji, P.; Yeh, Y.-C.; Levet, C.; Muliyil, S.; Lu, F.; Taylor, C.W.; Freeman, M. iRhom pseudoproteases regulate ER stress-induced cell death through IP3 receptors and BCL-2. Nat. Commun. 2022, 13, 1257. [Google Scholar] [CrossRef]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2015, 283, 2640–2652. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 Controls Diverse Cell Type- and Condition-Specific Transcriptional Regulatory Networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, A.-Z.; Chen, S.-T.; Chen, L.-S.; Guo, F.-Q. SPL6 represses signalling outputs of ER stress in control of panicle cell death in rice. Nat. Plants 2018, 4, 280–288. [Google Scholar] [CrossRef]
- Shibusawa, R.; Yamada, E.; Okada, S.; Nakajima, Y.; Bastie, C.C.; Maeshima, A.; Kaira, K.; Yamada, M. Dapagliflozin rescues endoplasmic reticulum stress-mediated cell death. Sci. Rep. 2019, 9, 9887. [Google Scholar] [CrossRef]
- Bezu, L.; Sauvat, A.; Humeau, J.; Leduc, M.; Kepp, O.; Kroemer, G. eIF2α phosphorylation: A hallmark of immunogenic cell death. OncoImmunology 2018, 7, e1431089. [Google Scholar] [CrossRef] [Green Version]
- Anspach, L.; Tsaryk, R.; Seidmann, L.; Unger, R.E.; Jayasinghe, C.; Simiantonaki, N.; Kirkpatrick, C.J.; Pröls, F. Function and mutual interaction of BiP-, PERK-, and IRE1α-dependent signalling pathways in vascular tumours. J. Pathol. 2020, 251, 123–134. [Google Scholar] [CrossRef]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 Activated by Proteolysis Binds in the Presence of NF-Y (CBF) Directly to the cis -Acting Element Responsible for the Mammalian Unfolded Protein Response. Mol. Cell. Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef] [Green Version]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Klymenko, O.; Huehn, M.; Wilhelm, J.; Wasnick, R.; Shalashova, I.; Ruppert, C.; Henneke, I.; Hezel, S.; Guenther, K.; Mahavadi, P.; et al. Regulation and role of the ER stress transcription factor CHOP in alveolar epithelial type-II cells. Klin. Wochenschr. 2019, 97, 973–990. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Niemeijer, M.; van de Water, B.; Beltman, J.B. ATF6 Is a Critical Determinant of CHOP Dynamics during the Unfolded Protein Response. iScience 2020, 23, 100860. [Google Scholar] [CrossRef] [Green Version]
- Sheng, X.; Nenseth, H.Z.; Qu, S.; Kuzu, O.F.; Frahnow, T.; Simon, L.; Greene, S.; Zeng, Q.; Fazli, L.; Rennie, P.S.; et al. IRE1α-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat. Commun. 2019, 10, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Fang, H.; Wu, Q.; Wang, X.; Liu, R.; Li, F.; Xiao, J.; Yuan, L.; Zhou, Z.; Ma, J.; et al. Ilamycin E, a natural product of marine actinomycete, inhibits triple-negative breast cancer partially through ER stress-CHOP-Bcl-2. Int. J. Biol. Sci. 2019, 15, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Ju, X.; Wang, J.; Fan, Y.; Ren, M.; Zhang, H. Immunogenic cell death in anticancer chemotherapy and its impact on clinical studies. Cancer Lett. 2018, 438, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Coleman, L.G.; Maile, R.; Jones, S.W.; Cairns, B.A.; Crews, F.T. HMGB1/IL-1β complexes in plasma microvesicles modulate immune responses to burn injury. PLoS ONE 2018, 13, e0195335. [Google Scholar] [CrossRef] [Green Version]
- Mishchenko, T.; Mitroshina, E.; Balalaeva, I.; Krysko, O.; Vedunova, M.; Krysko, D.V. An emerging role for nanomaterials in increasing immunogenicity of cancer cell death. Biochim. Biophys. Acta 2019, 1871, 99–108. [Google Scholar] [CrossRef]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef]
- Spiro, R.G.; Zhu, Q.; Bhoyroo, V.; Söling, H.-D. Definition of the Lectin-like Properties of the Molecular Chaperone, Calreticulin, and Demonstration of Its Copurification with Endomannosidase from Rat Liver Golgi. J. Biol. Chem. 1996, 271, 11588–11594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.; Tait, S.W. Targeting immunogenic cell death in cancer. Mol. Oncol. 2020, 14, 2994–3006. [Google Scholar] [CrossRef] [PubMed]
- Schcolnik-Cabrera, A.; Oldak, B.; Juárez, M.; Cruz-Rivera, M.; Flisser, A.; Mendlovic, F. Calreticulin in phagocytosis and cancer: Opposite roles in immune response outcomes. Apoptosis 2019, 24, 245–255. [Google Scholar] [CrossRef]
- Calvet, C.Y.; Famin, D.; André, F.M.; Mir, L.M. Electrochemotherapy with bleomycin induces hallmarks of immunogenic cell death in murine colon cancer cells. OncoImmunology 2014, 3, e28131. [Google Scholar] [CrossRef] [Green Version]
- Pitt, J.M.; Kroemer, G.; Zitvogel, L. Immunogenic and Non-immunogenic Cell Death in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 65–79. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Khani, A.T.; Swaminathan, S. Type I interferons: One stone to concurrently kill two birds, viral infections and cancers. Curr. Res. Virol. Sci. 2021, 2, 100014. [Google Scholar] [CrossRef]
- Bruschi, M.; Petretto, A.; Vaglio, A.; Santucci, L.; Candiano, G.; Ghiggeri, G.M. Annexin A1 and Autoimmunity: From Basic Science to Clinical Applications. Int. J. Mol. Sci. 2018, 19, 1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavins, F.N.E.; Hickey, M.J. Annexin A1 and the regulation of innate and adaptive immunity. Front. Immunol. 2012, 3, 354. [Google Scholar] [CrossRef] [Green Version]
- Hernández, I.B.B.; Angelier, M.L.; D’Ondes, T.D.B.D.B.; Di Di Maggio, A.; Yu, Y.; Oliveira, S. The Potential of Nanobody-Targeted Photodynamic Therapy to Trigger Immune Responses. Cancers 2020, 12, 978. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, D.; Tanaka, K. A crosstalk between extracellular ATP and jasmonate signaling pathways for plant defense. Plant Signal. Behav. 2018, 13, e1432229. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Nuevo, A.; Zorzano, A. The sensing of mitochondrial DAMPs by non-immune cells. Cell Stress 2019, 3, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Wang, Y.; Michaud, M.; Ma, Y.; Sukkurwala, A.Q.; Shen, S.; Kepp, O.; Metivier, D.; Galluzzi, L.; Perfettini, J.-L.; et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2013, 21, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef]
- Deng, Z.; He, Z.; Maksaev, G.; Bitter, R.M.; Rau, M.; Fitzpatrick, J.A.J.; Yuan, P. Cryo-EM structures of the ATP release channel pannexin 1. Nat. Struct. Mol. Biol. 2020, 27, 373–381. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Borges Da Silva, H.; Beura, L.K.; Wang, H.; Hanse, E.A.; Gore, R.; Scott, M.C.; Walsh, D.A.; Block, K.E.; Fonseca, R.; Yan, Y.; et al. The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8+ T cells. Nature 2018, 559, 264–268. [Google Scholar] [CrossRef]
- Amores-Iniesta, J.; Barberà-Cremades, M.; Martínez, C.M.; Pons, J.A.; Revilla-Nuin, B.; Martínez-Alarcón, L.; Di Virgilio, F.; Parrilla, P.; Baroja-Mazo, A.; Pelegrín, P. Extracellular ATP Activates the NLRP3 Inflammasome and Is an Early Danger Signal of Skin Allograft Rejection. Cell Rep. 2017, 21, 3414–3426. [Google Scholar] [CrossRef] [Green Version]
- Andersson, U.; Yang, H.; Harris, H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. In Seminars in Immunology; Academic Press: New York, NY, USA, 2018. [Google Scholar]
- Tanaka, M.; Kataoka, H.; Yano, S.; Sawada, T.; Akashi, H.; Inoue, M.; Suzuki, S.; Inagaki, Y.; Hayashi, N.; Nishie, H.; et al. Immunogenic cell death due to a new photodynamic therapy (PDT) with glycoconjugated chlorin (G-chlorin). Oncotarget 2016, 7, 47242–47251. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Gene Funct. Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, L.; Loos, F.; Iribarren, K.; Lachkar, S.; Zhou, H.; da Silva, L.C.G.; Chen, G.; Bezu, L.; Boncompain, G.; et al. Identification of pharmacological agents that induce HMGB1 release. Sci. Rep. 2017, 7, 14915. [Google Scholar] [CrossRef]
- Rufo, N.; Garg, A.; Agostinis, P. The Unfolded Protein Response in Immunogenic Cell Death and Cancer Immunotherapy. Trends Cancer 2017, 3, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Guan, Z.; Wang, X.; Wang, Z.; Zeng, R.; Xu, L.; Cao, P. ALA-PDT promotes HPV-positive cervical cancer cells apoptosis and DCs maturation via miR-34a regulated HMGB1 exosomes secretion. Photodiagn. Photodyn. Ther. 2018, 24, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Antón, M.; Alén, F.; Gómez de Heras, R.; Serrano, A.; Pavón, F.J.; Leza, J.C.; García-Bueno, B.; Rodríguez de Fonseca, F.; Orio, L. Oleoylethanolamide prevents neuroimmune HMGB1/TLR4/NF-kB danger signaling in rat frontal cortex and depressive-like behavior induced by ethanol binge administration. Addict. Biol. 2017, 22, 724–741. [Google Scholar] [CrossRef]
- Radogna, F.; Dicato, M.; Diederich, M. Natural modulators of the hallmarks of immunogenic cell death. Biochem. Pharmacol. 2019, 162, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Nastasi, C.; Mannarino, L.; D’Incalci, M. DNA Damage Response and Immune Defense. Int. J. Mol. Sci. 2020, 21, 7504. [Google Scholar] [CrossRef]
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; De Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Gen. Physiol. 2012, 140, i6. [Google Scholar] [CrossRef] [Green Version]
- Preissner, K.T.; Fischer, S.; Deindl, E. Extracellular RNA as a Versatile DAMP and Alarm Signal That Influences Leukocyte Recruitment in Inflammation and Infection. Front. Cell Dev. Biol. 2020, 8, 619221. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, X.; Su, C.; Peng, B.; Du, J.; Jia, H.; Luo, M.; Fang, C.; Wei, Y. Uric acid enhances the antitumor immunity of dendritic cell-based vaccine. Sci. Rep. 2015, 5, 16427. [Google Scholar] [CrossRef]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; Van De Vijver, K.K.; De Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef]
- Bota, D.A.; Chung, J.; Dandekar, M.; Carrillo, J.A.; Kong, X.-T.; Fu, B.D.; Hsu, F.P.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C.; et al. Phase II study of ERC1671 plus bevacizumab versus bevacizumab plus placebo in recurrent glioblastoma: Interim results and correlations with CD4+ T-lymphocyte counts. CNS Oncol. 2018, 7, CNS22. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Jaffee, E.M. Regulatory T-cell Modulation Using Cyclophosphamide in Vaccine Approaches: A Current Perspective. Cancer Res. 2012, 72, 3439–3444. [Google Scholar] [CrossRef] [Green Version]
- Vaes, R.; Hendriks, L.; Vooijs, M.; De Ruysscher, D. Biomarkers of Radiotherapy-Induced Immunogenic Cell Death. Cells 2021, 10, 930. [Google Scholar] [CrossRef]
- Lämmer, F.; Delbridge, C.; Würstle, S.; Neff, F.; Meyer, B.; Schlegel, J.; Kessel, K.A.; Schmid, T.E.; Schilling, D.; Combs, S.E. Cytosolic Hsp70 as a biomarker to predict clinical outcome in patients with glioblastoma. PLoS ONE 2019, 14, e0221502. [Google Scholar] [CrossRef] [Green Version]
- Rothammer, A.; Sage, E.K.; Werner, C.; Combs, S.E.; Multhoff, G. Increased heat shock protein 70 (Hsp70) serum levels and low NK cell counts after radiotherapy—Potential markers for predicting breast cancer recurrence? Radiat. Oncol. 2019, 14, 78. [Google Scholar] [CrossRef]
- Hongo, K.; Kazama, S.; Tsuno, N.H.; Ishihara, S.; Sunami, E.; Kitayama, J.; Watanabe, T. Immunohistochemical detection of high-mobility group box 1 correlates with resistance of preoperative chemoradiotherapy for lower rectal cancer: A retrospective study. World J. Surg. Oncol. 2015, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-Y.; Chiang, S.-F.; Ke, T.-W.; Chen, T.-W.; Lan, Y.-C.; You, Y.-S.; Shiau, A.-C.; Chen, W.T.-L.; Chao, K.S.C. Cytosolic high-mobility group box protein 1 (HMGB1) and/or PD-1+ TILs in the tumor microenvironment may be contributing prognostic biomarkers for patients with locally advanced rectal cancer who have undergone neoadjuvant chemoradiotherapy. Cancer Immunol. Immunother. 2018, 67, 551–562. [Google Scholar] [CrossRef]
- Muehlmann, L.; Ma, B.; Longo, J.P.; Santos, M.; Azevedo, R. Aluminum–phthalocyanine chloride associated to poly(methyl vinyl ether-co-maleic anhydride) nanoparticles as a new third-generation photosensitizer for anticancer photodynamic therapy. Int. J. Nanomed. 2014, 9, 1199–1213. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Agostinis, P. ER stress, autophagy and immunogenic cell death in photodynamic therapy-induced anti-cancer immune responses. Photochem. Photobiol. Sci. 2014, 13, 474–487. [Google Scholar] [CrossRef]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Feng, X.; Wan, C.; Lovell, J.F.; Jin, H.; Ding, J. Role of nanoparticle-mediated immunogenic cell death in cancer immunotherapy. Asian J. Pharm. Sci. 2020, 16, 129–132. [Google Scholar] [CrossRef]
- Longo, J.P.F.; Muehlmann, L.A. Nanomedicine beyond tumor passive targeting: What next? Nanomedicine 2020, 15, 1819–1822. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DAMP | Abbreviation | Effect on Immune Cells | References |
|---|---|---|---|
| Annexin A1 | ANXA1 | Expressed in different cells (neutrophils, eosinophils, and monocytes), ANXA1 has a role in the regulation and resolution of inflammation. It can act as a negative regulator of innate immunity, with neutrophils being its main target; it activates the migration of APCs towards the dying cells, facilitating their engulfment and processing. | [34,74] |
| Adenosine triphosphate | ATP | ATP acts as a strong chemoattractant and promotes not only the recruitment of immune cells but also their maturation. | [73,75] |
| Calreticulin | CRT | Calreticulin acts as phagocytosis inducer. Its exposure and the release of ANXA1, ATP and HMGB1 result in the attraction and maturation of DCs in the tumor microenvironment. | [36,39] |
| Deoxyribonucleic acid | DNA | With its accumulation in the cytoplasm, DNA can stimulate innate immune responses. | [93] |
| High mobility group box 1 protein | HMGB1 | Acts as an essential DAMP in the DCs activation, stimulating the production of pro-inflammatory factors, strongly contributing to the immunogenicity of ICD. | [29,94] |
| Heat-shock protein | HSP 70 HSP 90 | HSP act as eat-me signals for phagocytes. They can induce DC maturation and promote target engulfment by APC cells. | [29] |
| Type I interferon | IFN-I | IFN-Is acts as potent immunostimulatory proteins and have a crucial role in ICD. It can modulate the maturation, differentiation, and migration of DC cells, increase primary antibody responses, and activate B and T cells directly or indirectly. | [29,71] |
| Ribonucleic acid | RNA | It recruits leukocyte and M1-type macrophages. | [95] |
| Uric acid | UA | Crystalline UA can produce inflammatory mediators through macrophage activation and the enhancement of T cells. | [96] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, M.C.; Morais, J.A.V.; Ganassin, R.; Oliveira, G.R.T.; Costa, F.C.; Morais, A.A.C.; Silveira, A.P.; Silva, V.C.M.; Longo, J.P.F.; Muehlmann, L.A. An Overview on Immunogenic Cell Death in Cancer Biology and Therapy. Pharmaceutics 2022, 14, 1564. https://doi.org/10.3390/pharmaceutics14081564
Rodrigues MC, Morais JAV, Ganassin R, Oliveira GRT, Costa FC, Morais AAC, Silveira AP, Silva VCM, Longo JPF, Muehlmann LA. An Overview on Immunogenic Cell Death in Cancer Biology and Therapy. Pharmaceutics. 2022; 14(8):1564. https://doi.org/10.3390/pharmaceutics14081564
Chicago/Turabian StyleRodrigues, Mosar Corrêa, José Athayde Vasconcelos Morais, Rayane Ganassin, Giulia Rosa Tavares Oliveira, Fabiana Chagas Costa, Amanda Alencar Cabral Morais, Ariane Pandolfo Silveira, Victor Carlos Mello Silva, João Paulo Figueiró Longo, and Luis Alexandre Muehlmann. 2022. "An Overview on Immunogenic Cell Death in Cancer Biology and Therapy" Pharmaceutics 14, no. 8: 1564. https://doi.org/10.3390/pharmaceutics14081564