
Activation of Cellular Players in Adaptive Immunity via Exogenous Delivery of Tumor Cell Lysates


Abstract
:1. Introduction
2. Preparation of Tumor Cell Lysates
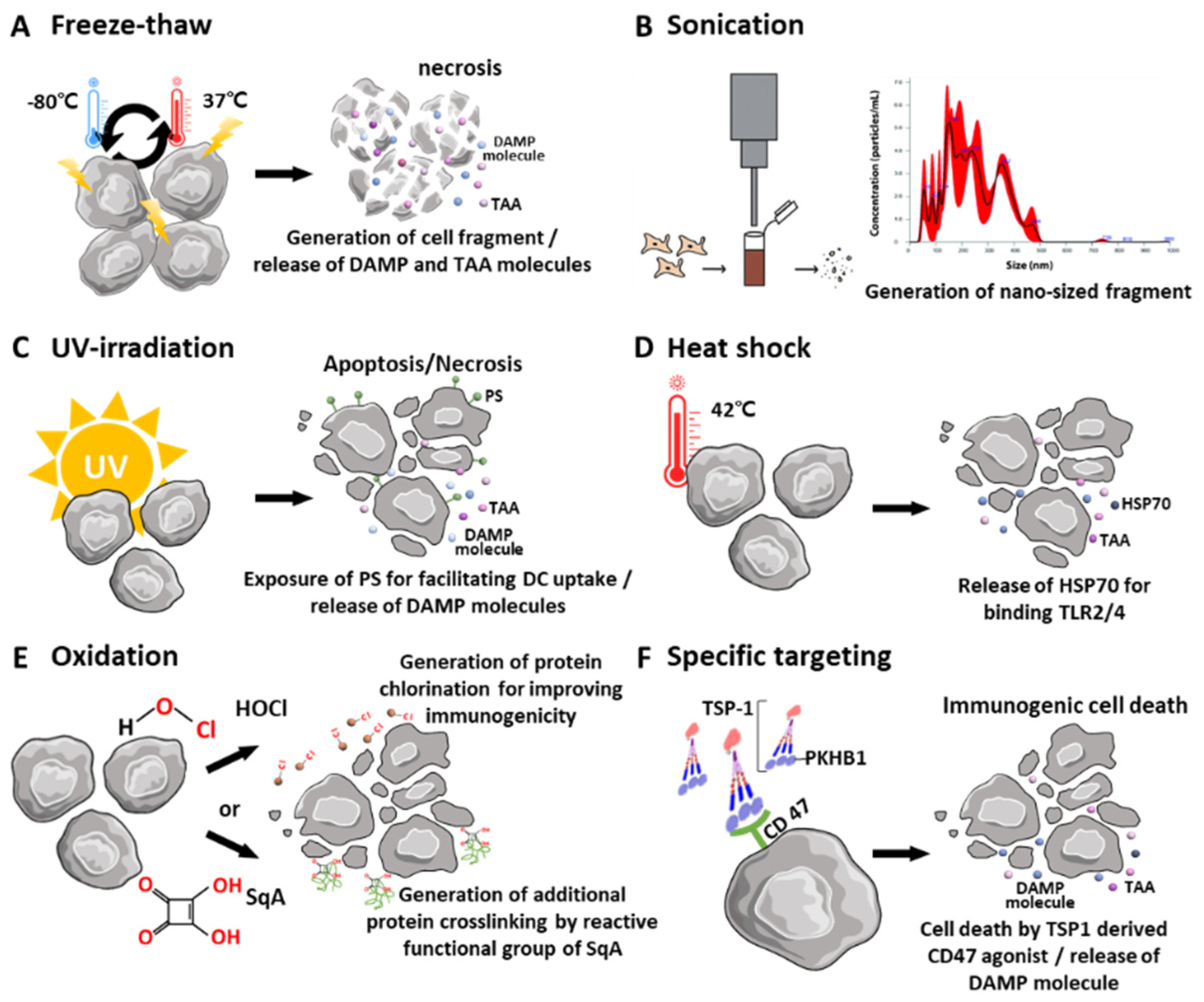
2.1. Physical Disruption and Stimulation of Tumor Cells to Obtain Whole Tumor Cells
2.2. Pretreatment of Source Tumor Cells
2.2.1. Heat Shock
2.2.2. Oxidation
2.2.3. Specific Targeting
2.2.4. Treatment with Natural Compounds
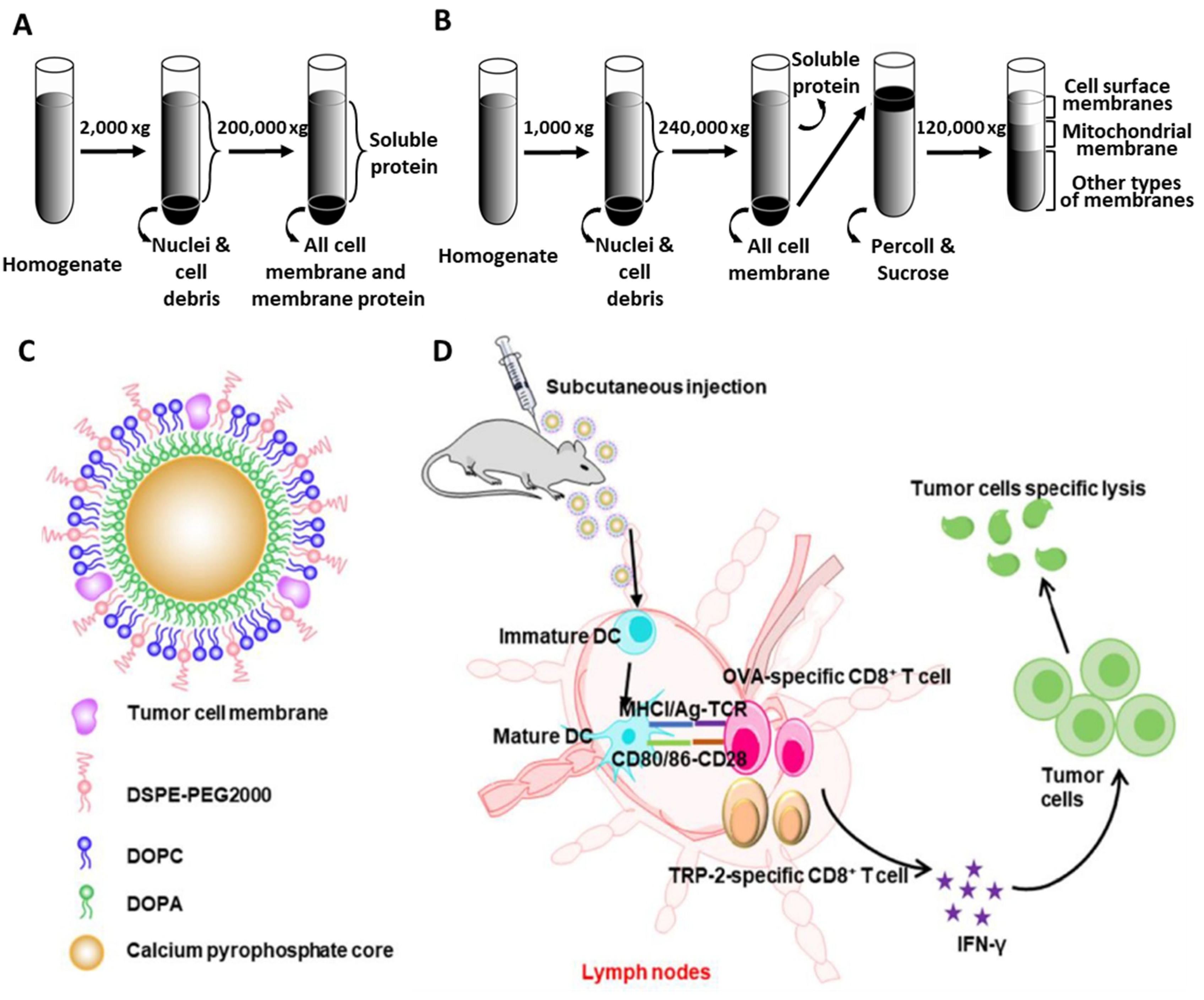
2.3. Preparation of Tumor Cell Membranes
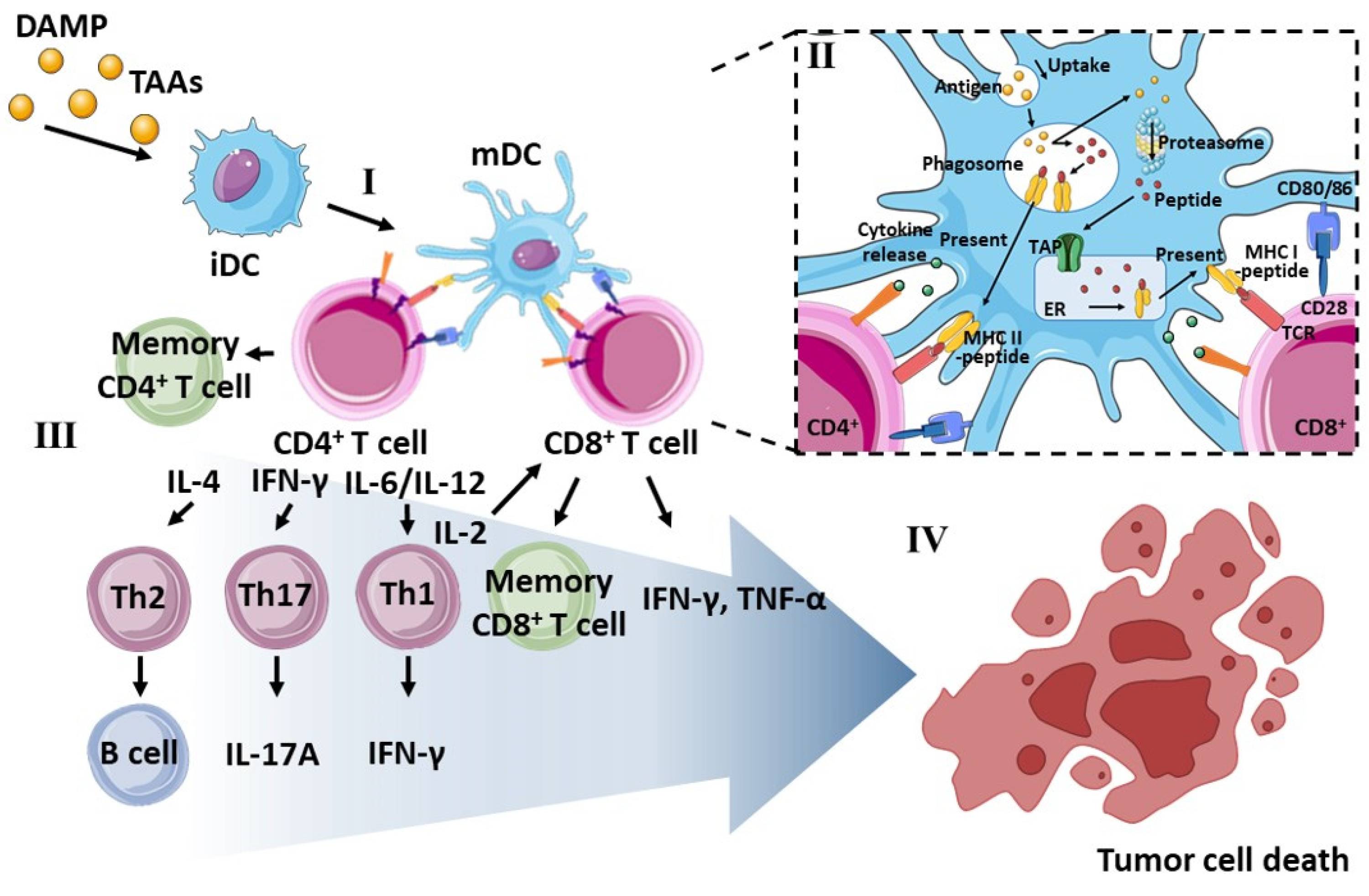
3. Role of DCs in Cancer Immunotherapy
3.1. Phenotype of Dendritic Cells
3.2. Antigen Presentation by MHC Molecules
3.3. Downstream T Cell Commitment by mDCs
3.4. Limitations of Ex Vivo Manipulation and the In Vivo Administration of DCs
4. Therapeutic Outcomes of Exogenous TCL Delivery Using Various Biomaterials
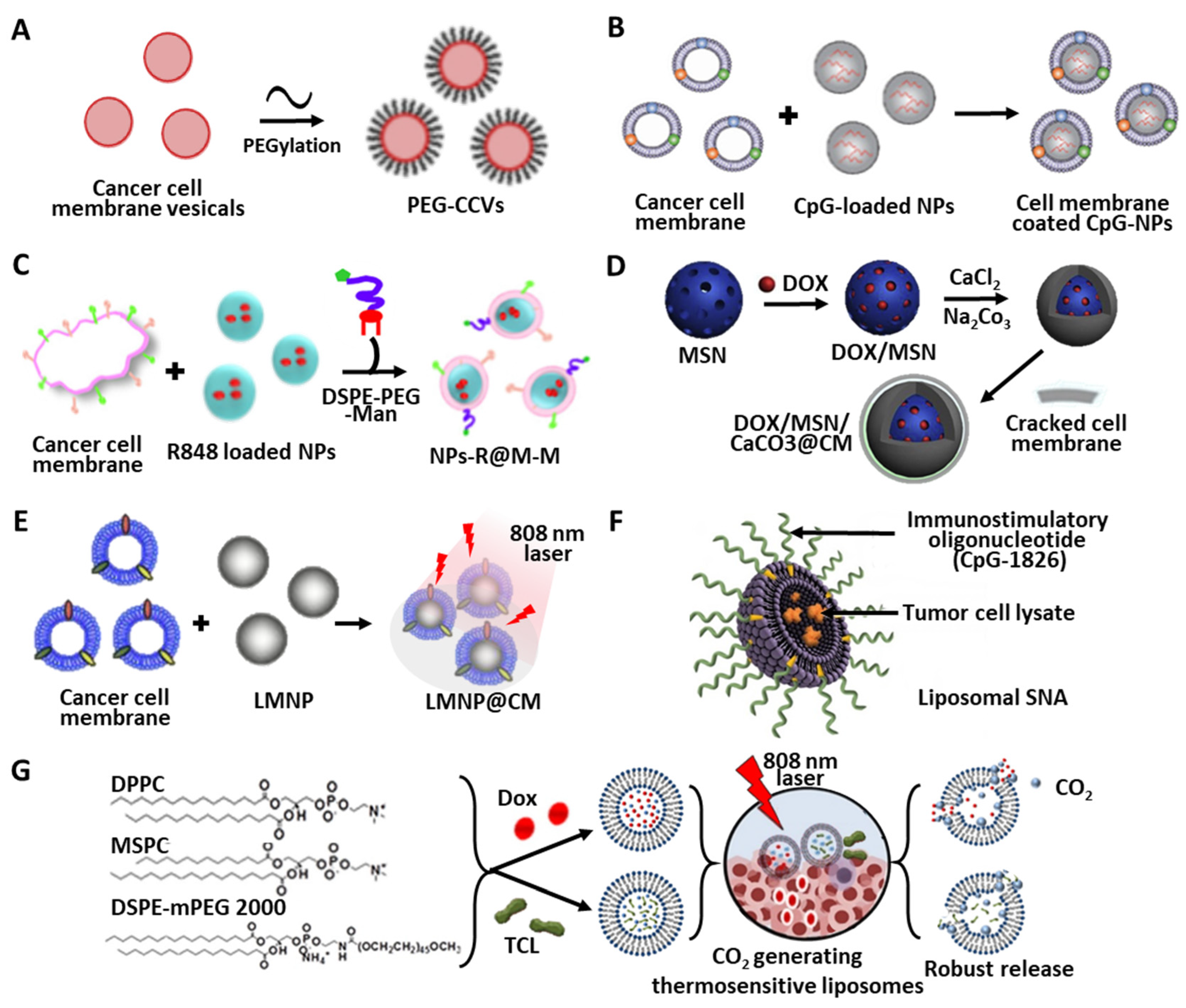
4.1. Nanoparticles
4.1.1. Design Parameters for TCL Carriers
4.1.2. Polymer-Based Materials
4.1.3. Camouflage Using Cancer Cell Membranes
4.1.4. Inorganic Templates for TCL Delivery
4.1.5. Adjuvant Activities of NPs
4.2. Liposome
4.3. 3D Polymeric Gel
4.4. Natural Components
4.5. Future Progress of Cancer Immunotherapy Using TCLs
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
| Th | T helper |
| APC | Antigen-presenting cell |
| CTL | Cytotoxic T lymphocyte |
| IFN | Interferon |
| IL | Interleukin |
| DC | Dendritic cell |
| MHC | Major histocompatibility complex |
| TCR | T cell receptor |
| iDC | Immature DC |
| mDC | Mature DC |
| TNF | Tumor necrosis factor |
| TAA | Tumor-associated antigens |
| TCL | Tumor cell lysate |
| DAMP | Damage-associated molecular patterns |
| PRR | Pattern recognition receptor |
| HSP | Heat shock protein |
| HMGB-1 | High-mobility group box-1 |
| TLR | Toll-like receptor |
| UV | Ultraviolet |
| ICD | Immunogenic cell death |
| HOCl | Hypochlorous acid |
| OVA | Ovalbumin |
| SqA | Squaric acid |
| TSP-1 | Thrombospondin-1 |
| ROS | Reactive oxygen species |
| LPS | Lipopolysaccharide |
| ER | Endoplasmic reticulum |
| TAP | Transporter associated with antigen processing |
| FDA | Food and Drug Administration |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| MAGE-1 | Melanoma-associated antigen-1 |
| NP | Nanoparticle |
| VEP | Virus envelope protein |
| PLGA | Poly(lactic-co-glycolic acid) |
| PEG-CCV | PEGlyated cancer cell membrane vesicle |
| FBS | Fatal bovine serum |
| CCNP | Cancer cell membrane nanoparticle |
| Man | Mannose |
| CaCO3 | Calcium carbonate |
| MSN | Mesoporous silica NP |
| HPMA | N -(hydroxypropyl) methacrylamide |
| APMA | N-(3-aminopropyl) methacrylamide |
| PAMP | Pathogen associated molecular pattern |
| DOX | Doxorubicin |
| LM | Liquid metal |
| AP | Aluminum phosphate |
| BMDC | Bone marrow dendritic cell |
| DA | Dopamine |
| PDA | Polydopamine |
| Lys-SNA | TCL-loaded liposomal spherical nucleic acid |
| CpG-1826 | Cholesteryl-modified immunostimulatory oligonucleotide adjuvants |
| BG-TSLs | CO2-generating thermosensitive liposomes |
| NIR | Near-infrared |
| PEV | Poly(L-valine) |
| PEA | Poly(L-alanine) |
| BGs | Bacterial ghosts |
| GPs | ß-glucan particles |
| MC38 | Murine colon adenocarcinoma cell |
References
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Skipper, H.E.; Heidelberger, C.; Welch, A.D. Some Biochemical Problems of Cancer Chemotherapy. Nature 1957, 179, 1159–1162. [Google Scholar] [CrossRef] [PubMed]
- Love, R.R.; Leventhal, H.; Ma, D.V.E.; Nerenz, D.R. Side effects and emotional distress during cancer chemotherapy. Cancer 1989, 63, 604–612. [Google Scholar] [CrossRef]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2019, 19, 23–38. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Kraehenbuehl, L.; Weng, C.-H.; Eghbali, S.; Wolchok, J.D.; Merghoub, T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 2021, 19, 37–50. [Google Scholar] [CrossRef]
- Harari, A.; Graciotti, M.; Bassani-Sternberg, M.; Kandalaft, L.E. Antitumour dendritic cell vaccination in a priming and boosting approach. Nat. Rev. Drug Discov. 2020, 19, 635–652. [Google Scholar] [CrossRef]
- Huang, H.; Hao, S.; Li, F.; Ye, Z.; Yang, J.; Xiang, J. CD4+Th1 cells promote CD8+Tc1 cell survival, memory response, tumor localization and therapy by targeted delivery of interleukin 2 via acquired pMHC I complexes. Immunology 2007, 120, 148–159. [Google Scholar] [CrossRef]
- Li, H.; Edin, M.L.; Gruzdev, A.; Cheng, J.; Bradbury, J.A.; Graves, J.P.; DeGraff, L.M.; Zeldin, D.C. Regulation of T helper cell subsets by cyclooxygenases and their metabolites. Prostaglandins Other Lipid Mediat. 2012, 104–105, 74–83. [Google Scholar] [CrossRef]
- Li, M.; Qin, M.; Song, G.; Deng, H.; Wang, D.; Wang, X.; Dai, W.; He, B.; Zhang, H.; Zhang, Q. A biomimetic antitumor nanovaccine based on biocompatible calcium pyrophosphate and tumor cell membrane antigens. Asian J. Pharm. Sci. 2020, 16, 97–109. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, M.J.; Nigro, A.; Mentucci, F.M.; Vittar, N.B.R.; Casolaro, V.; Col, J.D. Dendritic Cells and Immunogenic Cancer Cell Death: A Combination for Improving Antitumor Immunity. Pharmaceutics 2020, 12, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-G.; Cho, M.-Z.; Choi, J.-M. Bystander CD4+ T cells: Crossroads between innate and adaptive immunity. Exp. Mol. Med. 2020, 52, 1255–1263. [Google Scholar] [CrossRef]
- Dersh, D.; Hollý, J.; Yewdell, J.W. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat. Rev. Immunol. 2020, 21, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2017, 18, 168–182. [Google Scholar] [CrossRef]
- Vormehr, M.; Türeci, Ö.; Sahin, U. Harnessing Tumor Mutations for Truly Individualized Cancer Vaccines. Annu. Rev. Med. 2019, 70, 395–407. [Google Scholar] [CrossRef]
- Khodaei, T.; Sadri, B.; Nouraein, S.; Vahedi, N.; Mohammadi, J. Cancer vaccination: Various platforms and recent advances. J. Immun. Biol. 2020, 5, 151. [Google Scholar]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-gamma in tumor progression and regression: A review. Biomark Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Woodland, D.L.; Kohlmeier, J.E. Migration, maintenance and recall of memory T cells in peripheral tissues. Nat. Rev. Immunol. 2009, 9, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.-N.; Zhang, C.-N.; Xu, R.; Niu, J.-F.; Song, H.-J.; Zhang, X.-Y.; Wang, W.-W.; Wang, Y.-M.; Li, C.; Wei, X.-Q.; et al. Enhanced antitumor immunity by targeting dendritic cells with tumor cell lysate-loaded chitosan nanoparticles vaccine. Biomaterials 2017, 113, 191–202. [Google Scholar] [CrossRef]
- Lybaert, L.; Ryu, K.A.; Nuhn, L.; De Rycke, R.; De Wever, O.; Chon, A.C.; Esser-Kahn, A.P.; De Geest, B.G. Cancer Cell Lysate Entrapment in CaCO3 Engineered with Polymeric TLR-Agonists: Immune-Modulating Microparticles in View of Personalized Antitumor Vaccination. Chem. Mater. 2017, 29, 4209–4217. [Google Scholar] [CrossRef]
- Wang, X.; Wang, N.; Yang, Y.; Wang, X.; Liang, J.; Tian, X.; Zhang, H.; Leng, X. Polydopamine nanoparticles carrying tumor cell lysate as a potential vaccine for colorectal cancer immunotherapy. Biomater. Sci. 2019, 7, 3062–3075. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Yang, P.; Huang, P.; Zhang, C.; Kong, D.; Wang, W. Injectable polypeptide hydrogel-based co-delivery of vaccine and immune checkpoint inhibitors improves tumor immunotherapy. Theranostics 2019, 9, 2299–2314. [Google Scholar] [CrossRef]
- Ochyl, L.J.; Bazzill, J.D.; Park, C.; Xu, Y.; Kuai, R.; Moon, J.J. PEGylated tumor cell membrane vesicles as a new vaccine platform for cancer immunotherapy. Biomaterials 2018, 182, 157–166. [Google Scholar] [CrossRef]
- Dobrovolskienė, N.; Pašukonienė, V.; Darinskas, A.; Kraśko, J.; Žilionytė, K.; Mlynska, A.; Gudlevičienė, Ž.; Mišeikytė-Kaubrienė, E.; Schijns, V.; Lubitz, W.; et al. Tumor lysate-loaded Bacterial Ghosts as a tool for optimized production of therapeutic dendritic cell-based cancer vaccines. Vaccine 2018, 36, 4171–4180. [Google Scholar] [CrossRef]
- Hou, Y.; Liu, R.; Hong, X.; Zhang, Y.; Bai, S.; Luo, X.; Zhang, Y.; Gong, T.; Zhang, Z.; Sun, X. Engineering a sustained release vaccine with a pathogen-mimicking manner for robust and durable immune responses. J. Control. Release 2021, 333, 162–175. [Google Scholar] [CrossRef]
- Dombroski, J.A.; Jyotsana, N.; Crews, D.W.; Zhang, Z.; King, M.R. Fabrication and Characterization of Tumor Nano-Lysate as a Preventative Vaccine for Breast Cancer. Langmuir 2020, 36, 6531–6539. [Google Scholar] [CrossRef]
- Benencia, F.; Courrèges, M.C.; Coukos, G. Whole tumor antigen vaccination using dendritic cells: Comparison of RNA electroporation and pulsing with UV-irradiated tumor cells. J. Transl. Med. 2008, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, R.; Saffie, C.; Tittarelli, A.; González, F.E.; Ramírez, M.; Reyes, D.; Pereda, C.; Hevia, D.; García, T.; Salazar, L.; et al. Heat-Shock Induction of Tumor-Derived Danger Signals Mediates Rapid Monocyte Differentiation into Clinically Effective Dendritic Cells. Clin. Cancer Res. 2011, 17, 2474–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mookerjee, A.; Graciotti, M.; Kandalaft, L.E. A cancer vaccine with dendritic cells differentiated with GM-CSF and IFN alpha and pulsed with a squaric acid treated cell lysate improves T cell priming and tumor growth control in a mouse model. Bioimpacts 2018, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Torres, A.C.; Calvillo-Rodríguez, K.M.; Uscanga-Palomeque, A.C.; Gómez-Morales, L.; Mendoza-Reveles, R.; Caballero-Hernandez, D.; Karoyan, P.; Rodríguez-Padilla, C. PKHB1 Tumor Cell Lysate Induces Antitumor Immune System Stimulation and Tumor Regression in Syngeneic Mice with Tumoral T Lymphoblasts. J. Oncol. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, S.I.A.; Jantan, I.; Nafiah, M.A.; Seyed, M.A.; Chan, K.M. Dendritic cells pulsed with generated tumor cell lysate from Phyllanthus amarus Schum. & Thonn. induces anti-tumor immune response. BMC Complement. Altern. Med. 2018, 18, 232. [Google Scholar] [CrossRef]
- Gan, J.; Du, G.; He, C.; Jiang, M.; Mou, X.; Xue, J.; Sun, X. Tumor cell membrane enveloped aluminum phosphate nanoparticles for enhanced cancer vaccination. J. Control. Release 2020, 326, 297–309. [Google Scholar] [CrossRef]
- Chiang, C.L.; Coukos, G.; Kandalaft, L.E. Whole Tumor Antigen Vaccines: Where Are We? Vaccines (Basel) 2015, 3, 344–372. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.H.; Hecht, M.H. Recombinant Proteins Can Be Isolated from E. coli Cells by Repeated Cycles of Freezing and Thawing. Nat. Biotechnol. 1994, 12, 1357–1360. [Google Scholar] [CrossRef]
- Melcher, A.; Gough, M.; Todryk, S.; Vile, R. Apoptosis or necrosis for tumor immunotherapy: What’s in a name? J. Mol. Med. (Berl.) 1999, 77, 824–833. [Google Scholar] [CrossRef]
- Land, W.G. The Role of Damage-Associated Molecular Patterns in Human Diseases: Part I—Promoting inflammation and immunity. Sultan Qaboos Univ. Med. J. 2015, 15, e9–e21. [Google Scholar]
- Pouwels, S.D.; Heijink, I.H.; Hacken, N.H.T.; Vandenabeele, P.; Krysko, D.V.; Nawijn, M.C.; van Oosterhout, A.J. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol. 2013, 7, 215–226. [Google Scholar] [CrossRef]
- Lin, F.-C.; Hsu, C.-H.; Lin, Y.-Y. Nano-therapeutic cancer immunotherapy using hyperthermia-induced heat shock proteins: Insights from mathematical modeling. Int. J. Nanomed. 2018, 13, 3529–3539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettaieb, A.; Averill-Bates, D.A. Thermotolerance induced at a mild temperature of 40 degrees C alleviates heat shock-induced ER stress and apoptosis in HeLa cells. Biochim. Biophys Acta 2015, 1853, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salimu, J.C.; Spary, L.; Al-Taei, S.; Clayton, A.; Mason, M.D.; Staffurth, J.; Tabi, Z. Cross-presentation of the oncofoetal tumor antigen 5T4 from irradiated prostate cancer cells—A key role for Hsp70. J. Immunother. Cancer 2014, 2, P161. [Google Scholar] [CrossRef] [Green Version]
- Grant, M.L.; Shields, N.; Neumann, S.; Kramer, K.; Bonato, A.; Jackson, C.; A Baird, M.; Young, S.L. Combining dendritic cells and B cells for presentation of oxidised tumour antigens to CD8+ T cells. Clin. Transl. Immunol. 2017, 6, e149. [Google Scholar] [CrossRef]
- Marcinkiewicz, J.; Chain, B.; Olszowska, E.; Olszowski, S.; Zgliczyński, J. Enhancement of immunogenic properties of ovalbumin as a result of its chlorination. Int. J. Biochem. 1991, 23, 1393–1395. [Google Scholar] [CrossRef]
- Allison, M.; Fearon, D.T. Enhanced immunogenicity of aldehyde-bearing antigens: A possible link between innate and adaptive immunity. Eur. J. Immunol. 2000, 30, 2881–2887. [Google Scholar] [CrossRef]
- Chiang, C.L.-L.; Kandalaft, L.E.; Tanyi, J.; Hagemann, A.R.; Motz, G.T.; Svoronos, N.; Montone, K.; Mantia-Smaldone, G.M.; Smith, L.; Nisenbaum, H.L.; et al. A Dendritic Cell Vaccine Pulsed with Autologous Hypochlorous Acid-Oxidized Ovarian Cancer Lysate Primes Effective Broad Antitumor Immunity: From Bench to Bedside. Clin. Cancer Res. 2013, 19, 4801–4815. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Torres, A.C.; Quiney, C.; Attout, T.; Boullet, H.; Herbi, L.; Vela, L.; Barbier, S.; Chateau, D.; Chapiro, E.; Nguyen-Khac, F.; et al. CD47 agonist peptides induce programmed cell death in refractory chronic lymphocytic leukemia B cells via PLCgamma1 activation: Evidence from mice and humans. PLoS Med. 2015, 12, e1001796. [Google Scholar] [CrossRef]
- Denèfle, T.; Boullet, H.; Herbi, L.; Newton, C.; Martinez-Torres, A.C.; Guez, A.; Pramil, E.; Quiney, C.; Pourcelot, M.; Levasseur, M.; et al. Thrombospondin-1 Mimetic Agonist Peptides Induce Selective Death in Tumor Cells: Design, Synthesis, and Structure–Activity Relationship Studies. J. Med. Chem. 2016, 59, 8412–8421. [Google Scholar] [CrossRef]
- Leth-Larsen, R.; Lund, R.; Hansen, H.V.; Laenkholm, A.-V.; Tarin, D.; Jensen, O.N.; Ditzel, H.J. Metastasis-related Plasma Membrane Proteins of Human Breast Cancer Cells Identified by Comparative Quantitative Mass Spectrometry. Mol. Cell. Proteom. 2009, 8, 1436–1449. [Google Scholar] [CrossRef] [Green Version]
- Lund, R.; Leth-Larsen, R.; Jensen, O.N.; Ditzel, H.J. Efficient Isolation and Quantitative Proteomic Analysis of Cancer Cell Plasma Membrane Proteins for Identification of Metastasis-Associated Cell Surface Markers. J. Proteome Res. 2009, 8, 3078–3090. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Aoshi, T.; Kogai, Y.; Nomi, D.; Haseda, Y.; Kuroda, E.; Kobiyama, K.; Ishii, K.J. Optimization of physiological properties of hydroxyapatite as a vaccine adjuvant. Vaccine 2016, 34, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. Dendritic cells: Versatile controllers of the immune system. Nat. Med. 2007, 13, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Dalod, M.; Chelbi, R.; Malissen, B.; Lawrence, T. Dendritic cell maturation: Functional specialization through signaling specificity and transcriptional programming. EMBO J. 2014, 33, 1104–1116. [Google Scholar] [CrossRef]
- Kim, M.K.; Kim, J. Properties of immature and mature dendritic cells: Phenotype, morphology, phagocytosis, and migration. RSC Adv. 2019, 9, 11230–11238. [Google Scholar] [CrossRef] [Green Version]
- Lanzavecchia, A.; Sallusto, F. Regulation of T Cell Immunity by Dendritic Cells. Cell 2001, 106, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Fang, P.; Li, X.; Dai, J.; Cole, L.; Camacho, J.A.; Zhang, Y.; Ji, Y.; Wang, J.; Yang, X.-F.; Wang, H. Immune cell subset differentiation and tissue inflammation. J. Hematol. Oncol. 2018, 11, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Roche, P.A.; Cresswell, P. Antigen Processing and Presentation Mechanisms in Myeloid Cells. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Albert, M.L.; Sauter, B.; Bhardwaj, N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 1998, 392, 86–89. [Google Scholar] [CrossRef]
- Embgenbroich, M.; Burgdorf, S. Current Concepts of Antigen Cross-Presentation. Front. Immunol. 2018, 9, 1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Hiltbold, E.M.; A Roche, P. Trafficking of MHC class II molecules in the late secretory pathway. Curr. Opin. Immunol. 2002, 14, 30–35. [Google Scholar] [CrossRef]
- Turley, S.J.; Inaba, K.; Garrett, W.S.; Ebersold, M.; Unternaehrer, J.; Steinman, R.M.; Mellman, I. Transport of Peptide-MHC Class II Complexes in Developing Dendritic Cells. Science 2000, 288, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Decker, W.K.; Xing, D.; Shpall, E.J. Dendritic Cell Immunotherapy for the Treatment of Neoplastic Disease. Biol. Blood Marrow Transplant. 2006, 12, 113–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.-J.; Ishido, S.; Eisenlohr, L.C.; Roche, P.A. Activation of Dendritic Cells Alters the Mechanism of MHC Class II Antigen Presentation to CD4 T Cells. J. Immunol. 2020, 204, 1621–1629. [Google Scholar] [CrossRef]
- Landsverk, O.J.B.; Bakke, O.; Gregers, T.F. MHC II and the Endocytic Pathway: Regulation by Invariant Chain. Scand. J. Immunol. 2009, 70, 184–193. [Google Scholar] [CrossRef]
- Kloetzel, P.-M.; Ossendorp, F. Proteasome and peptidase function in MHC-class-I-mediated antigen presentation. Curr. Opin. Immunol. 2003, 16, 76–81. [Google Scholar] [CrossRef]
- Shen, L.; Sigal, L.J.; Boes, M.; Rock, K.L. Important Role of Cathepsin S in Generating Peptides for TAP-Independent MHC Class I Crosspresentation In Vivo. Immunity 2004, 21, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Guermonprez, P.; Saveanu, L.; Kleijmeer, M.J.; Davoust, J.; van Endert, P.; Amigorena, S. ER–phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 2003, 425, 397–402. [Google Scholar] [CrossRef]
- Tang-Huau, T.-L.; Gueguen, P.; Goudot, C.; Durand, M.; Bohec, M.; Baulande, S.; Pasquier, B.; Amigorena, S.; Segura, E. Human in vivo-generated monocyte-derived dendritic cells and macrophages cross-present antigens through a vacuolar pathway. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cruz, F.M.; Colbert, J.D.; Merino, E.; Kriegsman, B.A.; Rock, K.L. The Biology and Underlying Mechanisms of Cross-Presentation of Exogenous Antigens on MHC-I Molecules. Annu. Rev. Immunol. 2017, 35, 149–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, Y.; Wang, Q.; Korner, H.; Zhang, L.; Wei, W. Molecular Mechanisms of T Cells Activation by Dendritic Cells in Autoimmune Diseases. Front. Pharmacol. 2018, 9, 642. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A. The instructive role of dendritic cells on T-cell responses. Arthritis Res. Ther. 2002, 4, S127–S132. [Google Scholar] [CrossRef]
- Basu, A.; Ramamoorthi, G.; Albert, G.; Gallen, C.; Beyer, A.; Snyder, C.; Koski, G.; Disis, M.L.; Czerniecki, B.J.; Kodumudi, K. Differentiation and Regulation of TH Cells: A Balancing Act for Cancer Immunotherapy. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Owen, D.; Mahmud, S.; Vang, K.B.; Kelly, R.M.; Blazar, B.R.; Smith, K.A.; Farrar, M.A. Identification of Cellular Sources of IL-2 Needed for Regulatory T Cell Development and Homeostasis. J. Immunol. 2018, 200, 3926–3933. [Google Scholar] [CrossRef]
- Cheng, L.E.; Greenberg, P.D. Selective Delivery of Augmented IL-2 Receptor Signals to Responding CD8+T Cells Increases the Size of the Acute Antiviral Response and of the Resulting Memory T Cell Pool. J. Immunol. 2002, 169, 4990–4997. [Google Scholar] [CrossRef] [Green Version]
- Lanzavecchia, A.; Sallusto, F. Dynamics of T Lymphocyte Responses: Intermediates, Effectors, and Memory Cells. Science 2000, 290, 92–97. [Google Scholar] [CrossRef]
- Lanzavecchia, A.; Sallusto, F. Antigen decoding by T lymphocytes: From synapses to fate determination. Nat. Immunol. 2001, 2, 487–492. [Google Scholar] [CrossRef]
- Miller, M.J.; Safrina, O.; Parker, I.; Cahalan, M.D. Imaging the Single Cell Dynamics of CD4+ T Cell Activation by Dendritic Cells in Lymph Nodes. J. Exp. Med. 2004, 200, 847–856. [Google Scholar] [CrossRef]
- Mempel, T.R.; Henrickson, S.E.; Von Adrian, U.H. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 2004, 427, 154–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaech, S.M.; Ahmed, R. Memory CD8+ T cell differentiation: Initial antigen encounter triggers a developmental program in naïve cells. Nat. Immunol. 2001, 2, 415–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainone, V.; Martelli, C.; Ottobrini, L.; Biasin, M.; Borelli, M.; Lucignani, G.; Trabattoni, D.; Clerici, M. Immunological characterization of whole tumour lysate-loaded dendritic cells for cancer immunotherapy. PLoS ONE 2016, 11, e0146622. [Google Scholar] [CrossRef] [PubMed]
- Weigel, B.J.; Nath, N.; Taylor, P.A.; Panoskaltsis-Mortari, A.; Chen, W.; Krieg, A.M.; Brasel, K.; Blazar, B.R. Comparative analysis of murine marrow–derived dendritic cells generated by Flt3L or GM-CSF/IL-4 and matured with immune stimulatory agents on the in vivo induction of antileukemia responses. Blood 2002, 100, 4169–4176. [Google Scholar] [CrossRef]
- Nestle, F.O.; Alijagic, S.; Gilliet, M.; Sun, Y.; Grabbe, S.; Dummer, R.; Burg, G.; Schadendorf, D. Vaccination of melanoma patients with peptide- or tumorlysate-pulsed dendritic cells. Nat. Med. 1998, 4, 328–332. [Google Scholar] [CrossRef]
- Aarntzen, E.H.; Srinivas, M.; Bonetto, F.; Cruz, L.J.; Verdijk, P.; Schreibelt, G.; van de Rakt, M.; Lesterhuis, W.J.; van Riel, M.; Punt, C.J.; et al. Targeting of 111In-Labeled Dendritic Cell Human Vaccines Improved by Reducing Number of Cells. Clin. Cancer Res. 2013, 19, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- Celli, S.; Day, M.; Müller, A.J.; Molina-Paris, C.; Lythe, G.; Bousso, P. How many dendritic cells are required to initiate a T-cell response? Blood. J. Am. Soc. Hematol. 2012, 120, 3945–3948. [Google Scholar]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Anguille, S.; Smits, E.L.; Bryant, C.; Van Acker, H.H.; Goossens, H.; Lion, E.; Fromm, P.D.; Hart, D.N.; Van Tendeloo, V.F.; Berneman, Z.N. Dendritic Cells as Pharmacological Tools for Cancer Immunotherapy. Pharmacol. Rev. 2015, 67, 731–753. [Google Scholar] [CrossRef]
- Ali, O.A.; Huebsch, N.; Cao, L.; Dranoff, G.; Mooney, D.J. Infection-mimicking materials to program dendritic cells in situ. Nat. Mater. 2009, 8, 151–158. [Google Scholar] [CrossRef]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef]
- Gu, Y.-Z.; Zhao, X.; Song, X.-R. Ex vivo pulsed dendritic cell vaccination against cancer. Acta Pharmacol. Sin. 2020, 41, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Su, Y.; Xin, H.; Zhang, L.; Ding, J.; Chen, X. Immunologically Effective Biomaterials. ACS Appl. Mater. Interfaces 2021, 13, 56719–56724. [Google Scholar] [CrossRef] [PubMed]
- Vandenberk, L.; Belmans, J.; Van Woensel, M.; Riva, M.; Van Gool, S.W. Exploiting the Immunogenic Potential of Cancer Cells for Improved Dendritic Cell Vaccines. Front. Immunol. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Fang, R.H.; Kroll, A.V.; Gao, W.W.; Zhang, L.F. Cell Membrane Coating Nanotechnology. Adv. Mater. 2018, 30, e1706759. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef]
- Kwon, Y.J.; James, E.; Shastri, N.; Fréchet, J.M. In vivo targeting of dendritic cells for activation of cellular immunity using vaccine carriers based on pH-responsive microparticles. Proc. Natl. Acad. Sci. USA 2005, 102, 18264–18268. [Google Scholar] [CrossRef] [Green Version]
- Hardy, C.L.; LeMasurier, J.S.; Mohamud, R.; Yao, J.; Xiang, S.D.; Rolland, J.M.; O’Hehir, R.E.; Plebanski, M. Differential Uptake of Nanoparticles and Microparticles by Pulmonary APC Subsets Induces Discrete Immunological Imprints. J. Immunol. 2013, 191, 5278–5290. [Google Scholar] [CrossRef] [Green Version]
- Fifis, T.; Gamvrellis, A.; Crimeen-Irwin, B.; Pietersz, G.A.; Li, J.; Mottram, P.L.; McKenzie, I.F.C.; Plebanski, M. Size-Dependent Immunogenicity: Therapeutic and Protective Properties of Nano-Vaccines against Tumors. J. Immunol. 2004, 173, 3148–3154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooley, M.; Sarode, A.; Hoore, M.; Fedosov, D.A.; Mitragotri, S.; Gupta, A.S. Influence of particle size and shape on their margination and wall-adhesion: Implications in drug delivery vehicle design across nano-to-micro scale. Nanoscale 2018, 10, 15350–15364. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhang, Y.; Yuan, H.; Gao, H.; Zhang, S. Role of Nanoparticle Geometry in Endocytosis: Laying Down to Stand Up. Nano Lett. 2013, 13, 4546–4550. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kröger, M.; Liu, W.K. Shape effect in cellular uptake of PEGylated nanoparticles: Comparison between sphere, rod, cube and disk. Nanoscale 2015, 7, 16631–16646. [Google Scholar] [CrossRef]
- Niikura, K.; Matsunaga, T.; Suzuki, T.; Kobayashi, S.; Yamaguchi, H.; Orba, Y.; Kawaguchi, A.; Hasegawa, H.; Kajino, K.; Ninomiya, T.; et al. Gold Nanoparticles as a Vaccine Platform: Influence of Size and Shape on Immunological Responses in Vitro and in Vivo. ACS Nano 2013, 7, 3926–3938. [Google Scholar] [CrossRef]
- Neek, M.; Kim, T.I.; Wang, S.-W. Protein-based nanoparticles in cancer vaccine development. Nanomed. Nanotechnol. Biol. Med. 2018, 15, 164–174. [Google Scholar] [CrossRef]
- Verma, A.; Stellacci, F. Effect of surface properties on nanoparticle–cell interactions. Small 2010, 6, 12–21. [Google Scholar] [CrossRef]
- Hühn, D.; Kantner, K.; Geidel, C.; Brandholt, S.; De Cock, I.; Soenen, S.J.H.; Rivera_Gil, P.; Montenegro, J.-M.; Braeckmans, K.; Müllen, K.; et al. Polymer-Coated Nanoparticles Interacting with Proteins and Cells: Focusing on the Sign of the Net Charge. ACS Nano 2013, 7, 3253–3263. [Google Scholar] [CrossRef]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Ambrogio, M.W.; Toro-González, M.; Keever, T.J.; McKnight, T.E.; Davern, S.M. Poly(lactic-co-glycolic acid) Nanoparticles as Delivery Systems for the Improved Administration of Radiotherapeutic Anticancer Agents. ACS Appl. Nano Mater. 2020, 3, 10565–10570. [Google Scholar] [CrossRef]
- Chung, Y.-I.; Kim, J.C.; Kim, Y.H.; Tae, G.; Lee, S.-Y.; Kim, K.; Kwon, I.C. The effect of surface functionalization of PLGA nanoparticles by heparin- or chitosan-conjugated Pluronic on tumor targeting. J. Control. Release 2010, 143, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Pavot, V.; Berthet, M.; Rességuier, J.; Legaz, S.; Handké, N.; Gilbert, S.C.; Paul, S.; Verrier, B. Poly(lactic acid) and poly(lactic-co-glycolic acid) particles as versatile carrier platforms for vaccine delivery. Nanomedicine 2014, 9, 2703–2718. [Google Scholar] [CrossRef] [PubMed]
- Kohnepoushi, C.; Nejati, V.; Delirezh, N.; Biparva, P. Poly Lactic-co-Glycolic Acid Nanoparticles Containing Human Gastric Tumor Lysates as Antigen Delivery Vehicles for Dendritic Cell-Based Antitumor Immunotherapy. Immunol. Investig. 2019, 48, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Kroll, A.V.; Fang, R.H.; Jiang, Y.; Zhou, J.; Wei, X.; Yu, C.L.; Gao, J.; Luk, B.T.; Dehaini, D.; Gao, W.; et al. Nanoparticulate Delivery of Cancer Cell Membrane Elicits Multiantigenic Antitumor Immunity. Adv. Mater. 2017, 29. [Google Scholar] [CrossRef]
- Yang, R.; Xu, J.; Xu, L.; Sun, X.; Chen, Q.; Zhao, Y.; Peng, R.; Liu, Z. Cancer Cell Membrane-Coated Adjuvant Nanoparticles with Mannose Modification for Effective Anticancer Vaccination. ACS Nano 2018, 12, 5121–5129. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, M.D.; Li, C.X.; Li, B.; Zhang, X.Z. Tumor Cell Membrane-Coated Liquid Metal Nanovaccine for Tumor Prevention. Chin. J. Chem 2020, 38, 595–600. [Google Scholar] [CrossRef]
- Mishra, P.; Nayak, B.; Dey, R. PEGylation in anti-cancer therapy: An overview. Asian J. Pharm. Sci. 2016, 11, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Cruz, L.J.; Tacken, P.J.; Fokkink, R.; Figdor, C.G. The influence of PEG chain length and targeting moiety on antibody-mediated delivery of nanoparticle vaccines to human dendritic cells. Biomaterials 2011, 32, 6791–6803. [Google Scholar] [CrossRef]
- Liu, C.M.; Chen, G.B.; Chen, H.H.; Zhang, J.B.; Li, H.Z.; Sheng, M.X.; Weng, W.B.; Guo, S.M. Cancer cell membrane-cloaked mesoporous silica nanoparticles with a pH-sensitive gatekeeper for cancer treatment. Colloid Surf. B 2019, 175, 477–486. [Google Scholar] [CrossRef]
- Callmann, C.E.; Cole, L.E.; Kusmierz, C.D.; Huang, Z.; Horiuchi, D.; Mirkin, C.A. Tumor cell lysate-loaded immunostimulatory spherical nucleic acids as therapeutics for triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 17543–17550. [Google Scholar] [CrossRef]
- Won, J.E.; Byeon, Y.; Wi, T.I.; Lee, J.M.; Kang, T.H.; Lee, J.W.; Shin, B.C.; Han, H.D.; Park, Y.-M. Enhanced Antitumor Immunity Using a Tumor Cell Lysate-Encapsulated CO2-Generating Liposomal Carrier System and Photothermal Irradiation. ACS Appl. Bio Mater. 2019, 2, 2481–2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, R.H.; Hu, C.-M.J.; Luk, B.T.; Gao, W.; Copp, J.A.; Tai, Y.; O’Connor, D.E.; Zhang, L. Cancer Cell Membrane-Coated Nanoparticles for Anticancer Vaccination and Drug Delivery. Nano Lett. 2014, 14, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Kastl, L.; Sasse, D.; Wulf, V.; Hartmann, R.; Mircheski, J.; Ranke, C.; Carregal-Romero, S.; Martínez-López, J.A.; Fernández-Chacón, R.; Parak, W.J.; et al. Multiple Internalization Pathways of Polyelectrolyte Multilayer Capsules into Mammalian Cells. ACS Nano 2013, 7, 6605–6618. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, E.J.; Parker, A.E.; O’Neill, L.A.J. Targeting Toll-like receptors: Emerging therapeutics? Nat. Rev. Drug Discov. 2010, 9, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Antonios, D.; Ade, N.; Kerdine-Romer, S.; Assaf-Vandecasteele, H.; Larange, A.; Azouri, H.; Pallardy, M. Metallic haptens induce differential phenotype of human dendritic cells through activation of mitogen-activated protein kinase and NF-kappa B pathways. Toxicol Vitr. 2009, 23, 227–234. [Google Scholar] [CrossRef]
- Méndez, I.Z.R.; Shi, Y.; HogenEsch, H.; Hem, S.L. Potentiation of the immune response to non-adsorbed antigens by aluminum-containing adjuvants. Vaccine 2007, 25, 825–833. [Google Scholar] [CrossRef]
- Hem, S.L.; HogenEsch, H. Relationship between physical and chemical properties of aluminum-containing adjuvants and immunopotentiation. Expert Rev. Vaccines 2007, 6, 685–698. [Google Scholar] [CrossRef]
- Carroll, E.C.; Jin, L.; Mori, A.; Munoz-Wolf, N.; Oleszycka, E.; Moran, H.B.T.; Mansouri, S.; McEntee, C.P.; Lambe, E.; Agger, E.M.; et al. The Vaccine Adjuvant Chitosan Promotes Cellular Immunity via DNA Sensor cGAS-STING-Dependent Induction of Type I Interferons. Immunity 2016, 44, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Engering, A.J.; Cella, M.; Fluitsma, D.; Brockhaus, M.; Hoefsmit, E.C.M.; Lanzavecchia, A.; Pieters, J. The mannose receptor functions as a high capacity and broad specificity antigen receptor in human dendritic cells. Eur. J. Immunol. 1997, 27, 2417–2425. [Google Scholar] [CrossRef]
- Nolan, R.A.; Reeb, K.L.; Rong, Y.; Matt, S.M.; Johnson, H.S.; Runner, K.; Gaskill, P.J. Dopamine activates NF-kappaB and primes the NLRP3 inflammasome in primary human macrophages. Brain Behav. Immun Health 2020, 2, 100030. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H. Thin-film hydration followed by extrusion method for liposome preparation. In Liposomes: Methods and Protocols; Springer: New York, NY, USA, 2017; Volume 1522, pp. 17–22. ISBN 9781461491644. [Google Scholar]
- Song, H.; Huang, P.; Niu, J.; Shi, G.; Zhang, C.; Kong, D.; Wang, W. Injectable polypeptide hydrogel for dual-delivery of antigen and TLR3 agonist to modulate dendritic cells in vivo and enhance potent cytotoxic T-lymphocyte response against melanoma. Biomaterials 2018, 159, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.K.; Das, B. Introduction to polymeric gels. In Polymeric Gels; Woodhead Publishing: Philadelphia, PA, USA, 2018; pp. 3–27. [Google Scholar]
- Bencherif, S.A.; Sands, R.W.; Ali, O.A.; Li, A.; Lewin, S.A.; Braschler, T.M.; Shih, T.-Y.; Verbeke, C.S.; Bhatta, D.; Dranoff, G.; et al. Injectable cryogel-based whole-cell cancer vaccines. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zariri, A.; van der Ley, P. Biosynthetically engineered lipopolysaccharide as vaccine adjuvant. Expert Rev Vaccines 2015, 14, 861–876. [Google Scholar] [CrossRef]
- Han, J.E.; Wui, S.R.; Kim, K.S.; Cho, Y.J.; Cho, W.J.; Lee, N.G. Characterization of the Structure and Immunostimulatory Activity of a Vaccine Adjuvant, De-O-Acylated Lipooligosaccharide. PLoS ONE 2014, 9, e85838. [Google Scholar] [CrossRef] [Green Version]
- Chou, Y.-J.; Lin, C.-C.; Dzhagalov, I.; Chen, N.-J.; Lin, C.-H.; Lin, C.-C.; Chen, S.-T.; Chen, K.-H.; Fu, S.-L. Vaccine adjuvant activity of a TLR4-activating synthetic glycolipid by promoting autophagy. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Jeong, S.; Choi, Y.; Kim, K. Engineering Therapeutic Strategies in Cancer Immunotherapy via Exogenous Delivery of Toll-like Receptor Agonists. Pharmaceutics 2021, 13, 1374. [Google Scholar] [CrossRef]
- Xie, J.; Qian, J.; Wang, S.; Freeman, M.E.; Epstein, J.; Yi, Q. Novel and Detrimental Effects of Lipopolysaccharide on In Vitro Generation of Immature Dendritic Cells: Involvement of Mitogen-Activated Protein Kinase p38. J. Immunol. 2003, 171, 4792–4800. [Google Scholar] [CrossRef]
- Herve, C.; Laupeze, B.; Del Giudice, G.; Didierlaurent, A.M.; Da Silva, F.T. The how’s and what’s of vaccine reactogenicity. NPJ Vaccines 2019, 4, 39. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 1–46. [Google Scholar] [CrossRef]
- Penna, G.; Roncari, A.; Amuchastegui, S.; Daniel, K.C.; Berti, E.; Colonna, M.; Adorini, L. Expression of the inhibitory receptor ILT3 on dendritic cells is dispensable for induction of CD4+Foxp3+ regulatory T cells by 1,25-dihydroxyvitamin D. Blood 2005, 106, 3490–3497. [Google Scholar] [CrossRef] [PubMed]
- Mirza, Z.; Soto, E.R.; Dikengil, F.; Levitz, S.M.; Ostroff, G.R. Beta-Glucan Particles as Vaccine Adjuvant Carriers. Bacteriophages 2017, 1625, 143–157. [Google Scholar] [CrossRef]
- Soto, E.R.; Caras, A.C.; Kut, L.C.; Castle, M.K.; Ostroff, G.R. Glucan Particles for Macrophage Targeted Delivery of Nanoparticles. J. Drug Deliv. 2011, 2012, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattner, F.; Fleitmann, J.-K.; Lingnau, K.; Schmidt, W.; Egyed, A.; Fritz, J.; Zauner, W.; Wittmann, B.; Gorny, I.; Berger, M.; et al. Vaccination with poly-L-arginine as immunostimulant for peptide vaccines: Induction of potent and long-lasting T-cell responses against cancer antigens. Cancer Res. 2002, 62. [Google Scholar]
- Schmidt, W.; Buschle, M.; Zauner, W.; Kirlappos, H.; Mechtler, K.; Trska, B.; Birnstiel, M.L. Cell-free tumor antigen peptide-based cancer vaccines. Proc. Natl. Acad. Sci. USA 1997, 94, 3262–3267. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Castejon, G.; Brough, D. Understanding the mechanism of IL-1beta secretion. Cytokine Growth Factor Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef]
- Kawahara, M.; Takaku, H. A tumor lysate is an effective vaccine antigen for the stimulation of CD4+T-cell function and subsequent induction of antitumor immunity mediated by CD8+T cells. Cancer Biol. Ther. 2015, 16, 1616–1625. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.; Zhang, Y.; Zhou, X.; Ji, J.; Liu, Z. Nanovaccines with cell-derived components for cancer immunotherapy. Adv. Drug Deliv. Rev. 2022, 182, 114107. [Google Scholar] [CrossRef]
- Matsuo, K.; Yoshie, O.; Kitahata, K.; Kamei, M.; Hara, Y.; Nakayama, T. Recent Progress in Dendritic Cell-Based Cancer Immunotherapy. Cancers 2021, 13, 2495. [Google Scholar] [CrossRef]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 1–26. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification of Process | Condition | Ref. | |
|---|---|---|---|
| Physical disruption | Freeze–thaw cycle | Freeze at −80 °C and thaw at 37 °C (repeat) | [22,23,24,25,26,27,28] |
| Sonication | Sonicate 3 times for 10 s | [29] | |
| UV irradiation | Irradiate with 1500 μW/cm2 UVB | [30] | |
| Pretreatment of source tumor cells | Heat shock | 1. Treat at 42 °C for 1 h and 37 °C for 2 h 2. Additional physical disruption | [31,32] |
| CD47 agonist | Treat 150 or 300 μM of PKHB1 for 2 h | [33] | |
| Phyllanthus amarus | 1. Treat 1000 μg/mL Phyllanthus amarus 2. Additional physical disruption | [34] | |
| Cell membrane isolation | Sucrose-dependent | 1. Mix 0.0759 M sucrose and 0.225 M D-mannitol-containing buffer 2. Centrifuge at 10,000× g for 25 min 3. Centrifuge the supernatant at 150,000× g for 35 min | [10] |
| Sucrose-independent | 1. Centrifuge at 10,000× g for 25 min 2. Centrifuge the supernatant at 150,000× g for 40 min | [35] | |
| Material | TCL Type | Specificity | Material Platform | Target Cancer | Outcome | Ref. |
|---|---|---|---|---|---|---|
| PLGA | Whole TCLs | Human | TCL-loaded PLGA NPs | Gastric cancer | Increased IL-12 and IFN-γ in DCs Th1 immune system pathway activation | [113] |
| CM | Mouse | Cell membrane coated-CpG-PLGA NPs | Melanoma | Stability and longer circulation High recognition of specific tumor antigens 86% survival in vaccination group | [114] | |
| CM | Mouse | Cell membrane coated-R848-PLGA NP–mannose moiety conjugate | Melanoma | Specific binding by mannose Homotypic targeting on cancer cell surface antigens | [115] | |
| PEG | CM | Mouse | Co-delivery of PEGylated cell membrane and CpG | Melanoma | Enhanced serum stability Efficient trafficking to LNs 63% tumor regression | [26] |
| PEGylated LM | CM | Mouse | Cell membrane coated-PEG-LM NPs | Breast | Immune adjuvant effect and photothermal conversion efficacy with irradiation Metal-induced NF-kB immune pathway activation | [116] |
| CTS | Whole TCLs | Mouse | Mannose-coated TCLs-CTS NPs | Melanoma | Mitochondrial stress, ROS generation, and cGAS-STING pathway activation Improvement in NP uptake efficacy | [22] |
| PDA | Whole TCLs | Mouse | TCL-loaded PDA NPs | Colorectal cancer | Reacted with dopamine receptor Increased the subpopulation of T cells | [24] |
| Platform | Material | Specificity | Material Platform | Target Cancer | Outcome | Ref. |
|---|---|---|---|---|---|---|
| Liposome | Liposomal spherical nucleic acids | Mouse | CpG-1826-coated and TCL-loaded liposome | Triple-negative breast cancer cell | Increased population of CTLs Decreased population of myeloid derived suppressor cells | [120] |
| CO2-generating thermosensitive liposomes | Mouse | Co-delivery of DOX-loaded liposome and TCL-loaded liposome | Melanoma | High expression of pro-inflammatory cytokines and suppressed tumor growth by external NIR irradiation and generated CO2 bubbles | [121] | |
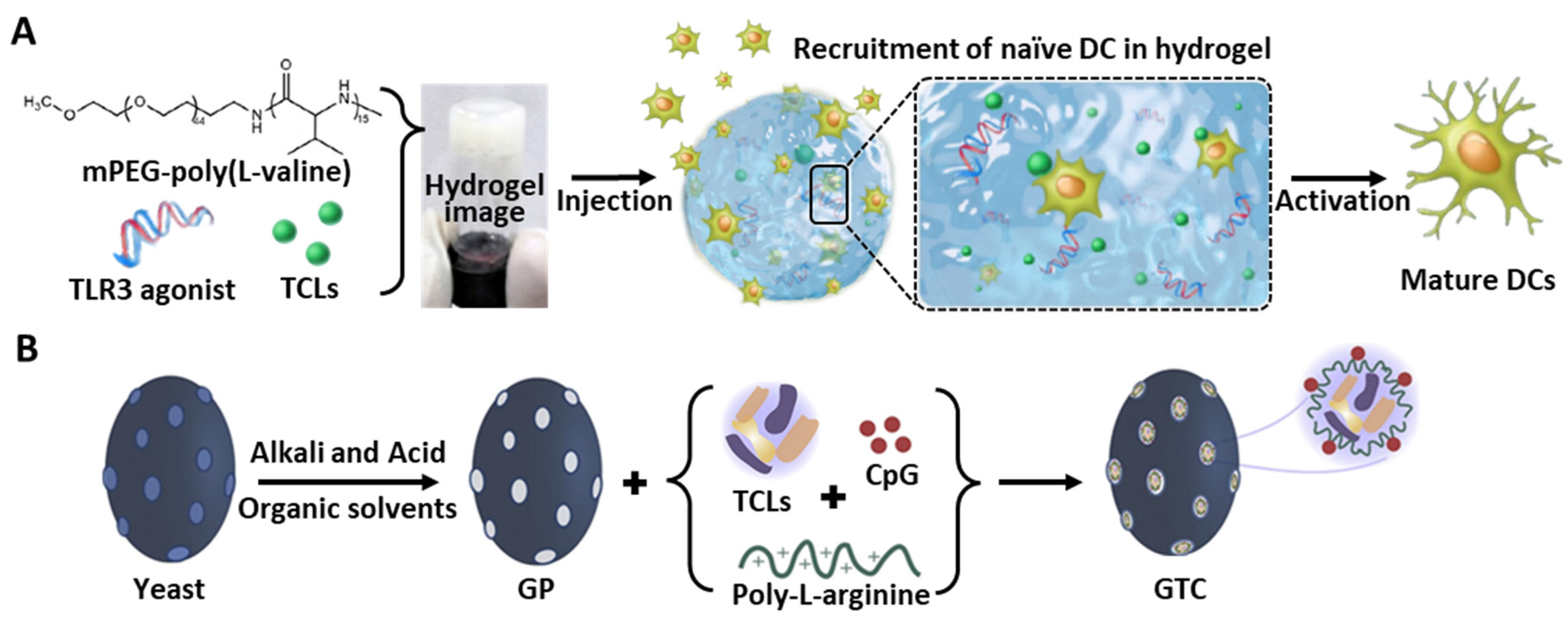
| 3D polymeric gel | PEV-based hydrogel | Mouse | TCL- and TLR3-loaded PEV hydrogel | Melanoma | Localization of injectable hydrogel and induction of sustained release Highest percentage of CTLs in LN | [133] |
| PEA-based hydrogel | Mouse | TCL, GM-CSF, and anti-CTLA4/PD-1 Ab-loaded PEA hydrogel | Melanoma | Persistent and synergistic DCs activation Augmented expansion of effector CD8+ T cells | [25] | |
| Cryogel | Mouse | CpG ODN, GM-CSF, and RGD-loaded cryogel-containing TCLs | Melanoma | Enhanced DC activation Leukocyte recruitment Greater survival rates | [135] | |
| Natural component | Empty envelope of bacterial ghost | Human | Combination of TCL-loaded bacterial ghost and IFN-γ | Melanoma, renal cell carcinoma, glioblastoma | Decreased expression of ILT3 and inhibitory receptor | [27] |
| Yeast derived ß-glucan particle | Mouse | TCL, CpG, and poly-L-arginine-loaded ß-glucan | Colorectal cancer | High internalization in DC NLRP3 inflammasome-mediated DC activation | [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seong, J.; Kim, K. Activation of Cellular Players in Adaptive Immunity via Exogenous Delivery of Tumor Cell Lysates. Pharmaceutics 2022, 14, 1358. https://doi.org/10.3390/pharmaceutics14071358
Seong J, Kim K. Activation of Cellular Players in Adaptive Immunity via Exogenous Delivery of Tumor Cell Lysates. Pharmaceutics. 2022; 14(7):1358. https://doi.org/10.3390/pharmaceutics14071358
Chicago/Turabian StyleSeong, Jihyun, and Kyobum Kim. 2022. "Activation of Cellular Players in Adaptive Immunity via Exogenous Delivery of Tumor Cell Lysates" Pharmaceutics 14, no. 7: 1358. https://doi.org/10.3390/pharmaceutics14071358