
Drug Delivery Systems in the Development of Novel Strategies for Glioblastoma Treatment

 , and
, and
Abstract
:1. Introduction
2. GBM Traditional Treatments
2.1. Surgical Resection
2.2. Radiotherapy
2.3. Chemotherapy
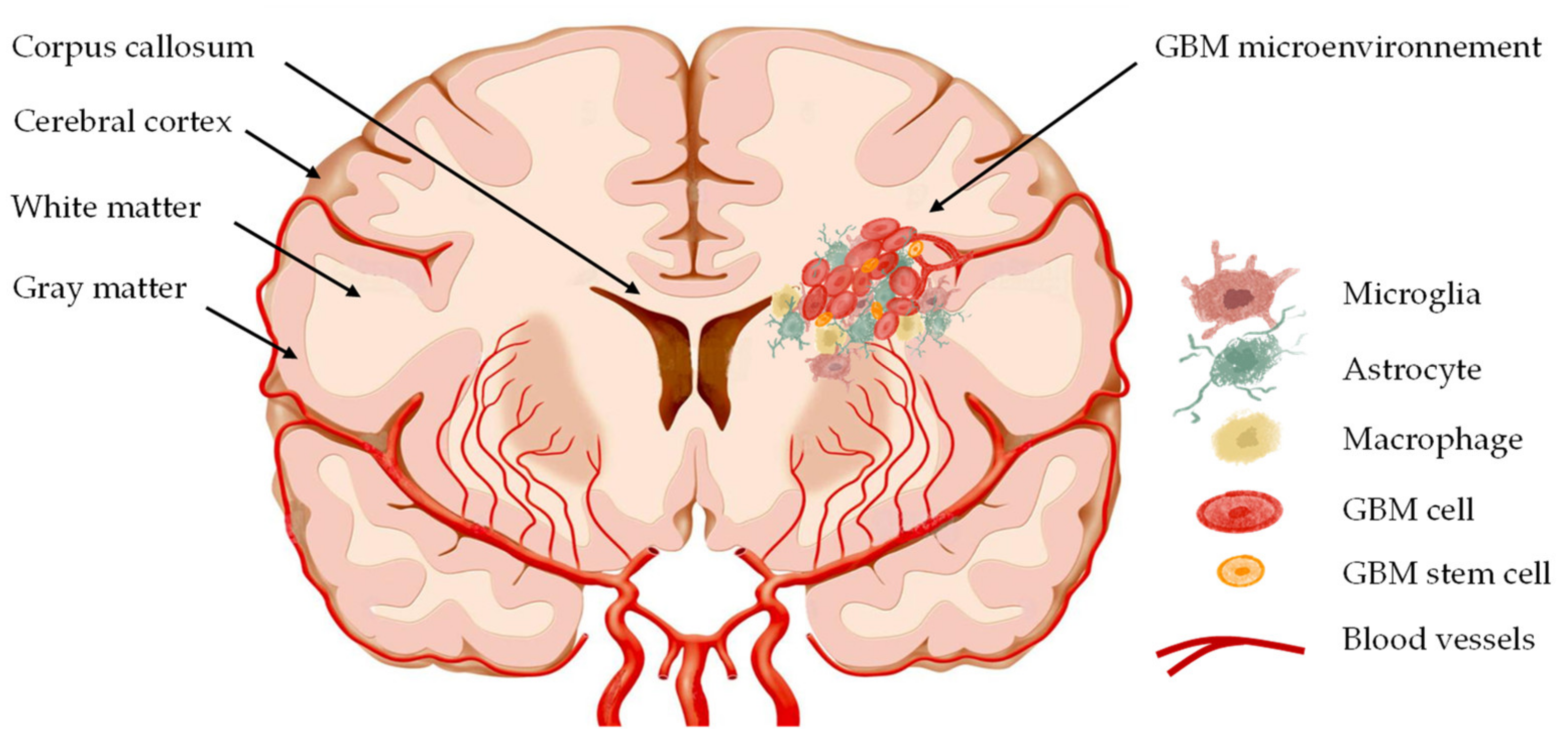
3. CXCL12–CXCR4 in Glioblastoma Invasion
3.1. The Pathways of GBM Cell Invasion

3.2. CXCL12–CXCR4 Axis in GBM Cell Migration
3.3. Chemokines Implicated in GBM Cell Invasion
- (a)
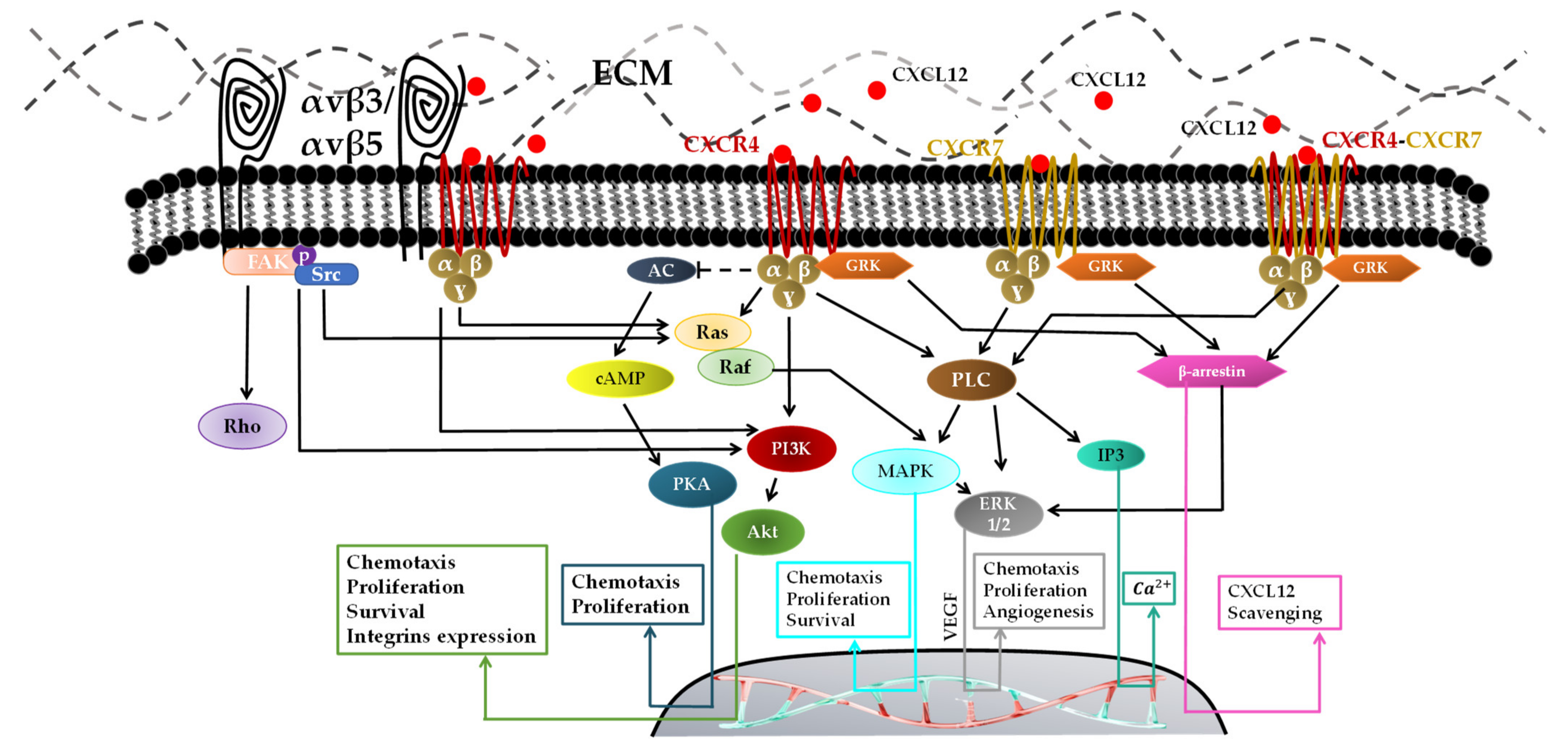
- Through CXCR7, which has been known for decades and has an inability to activate the G-protein complex [113]. Lately, it has been proved in vitro that CXCR7 activation by CXCL12 is mediated via G-protein and β-arrestin and increases the intracellular calcium concentration. β-arrestin has four isoforms and plays a key role in GPCR signal transduction [114]. It activates numerous intracellular signaling pathways such as MAPK-ERK1/2 pathways for cell proliferation and migration [115]. An in vivo study demonstrated that the inhibition of CXCR7 after irradiation prolonged survival and blocked tumor recurrence of intracranial U251 GBM in nude mice [116]. Yang Liu et al. showed that knocking down CXCR7 in GBM cells (U251MG and U373MG) using siRNA to block ERK1/2 in response to CXCL12 decreases cell proliferation, invasion, and migration [93].
- (b)
- CXCR7 can heterodimerize with CXCR4 in response to CXCL12, which modulates CXCR4-mediated G-protein and β-arrestin signaling pathways such as MAPK-ERK1/2 inducing cell migration [92].
- (c)
- One CXCL12 binds to its receptor CXCR4 [117,118], the tertiary structure of CXCR4 changes to activate the G-protein through its intracellular component. Multiple signals are activated via GRK, such as phospholipase C (PLC), PI3K, and MAPK/ERK, for vascular endothelial growth factor (VEGF) production, which is mainly responsible for recruiting new vessels for GBM neovascularizations [119]. The activation of the PI3K pathway followed by the activation of Akt contributes to the CXCL12/CXCR4-induced survival, invasion and proliferation. CXCL12–CXCR4-mediated migration is reported to be regulated by the PI3K and MAPK/ERK pathways [119].
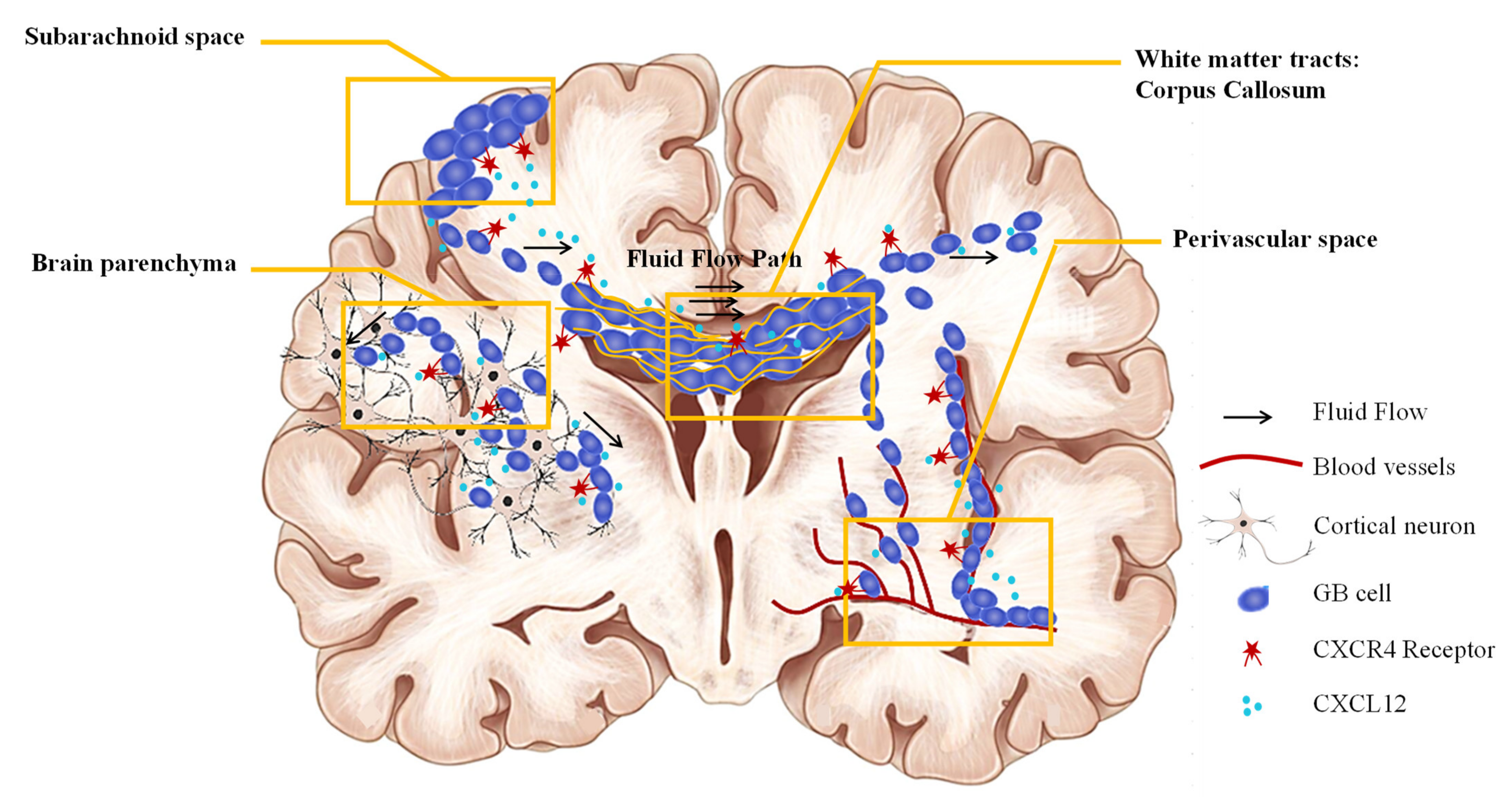
4. Interstitial Fluid Flow in Glioblastoma
4.1. Cerebral Fluids
4.2. Interstitial Fluid Flow and CXCL12 in the Migration of GBM
5. Models to Study Glioblastoma Cells Migration
5.1. Two-Dimensional Models for Glioblastoma Studies
5.2. Three-Dimensional Models for Glioblastoma Studies
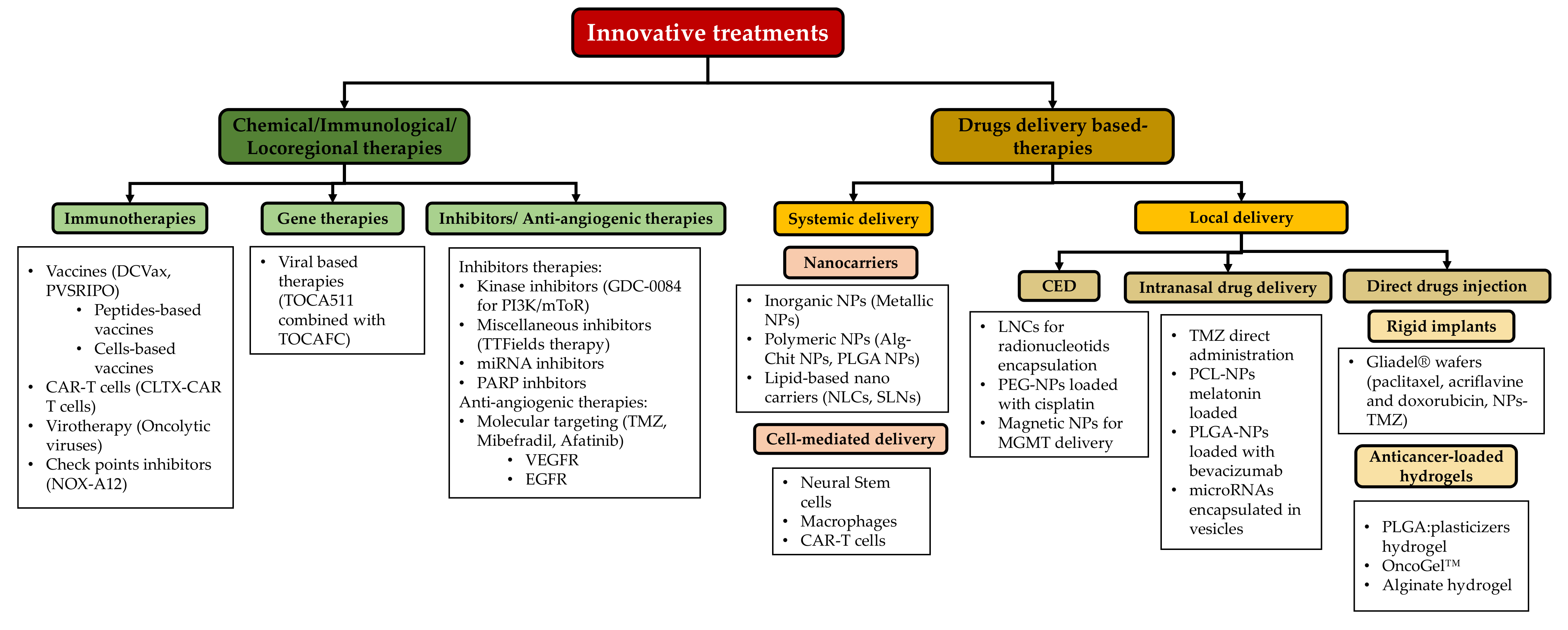
6. Innovative Treatments for Glioblastoma
6.1. Some Innovative Treatments in Glioblastoma
6.1.1. Gene Therapies
- mRNA and siRNA in GBM Gene Therapy:
6.1.2. Immunotherapies
6.2. Drugs Delivery Systems for Glioblastoma Treatments
6.2.1. Systemic Delivery
- Cell-Mediated Delivery:
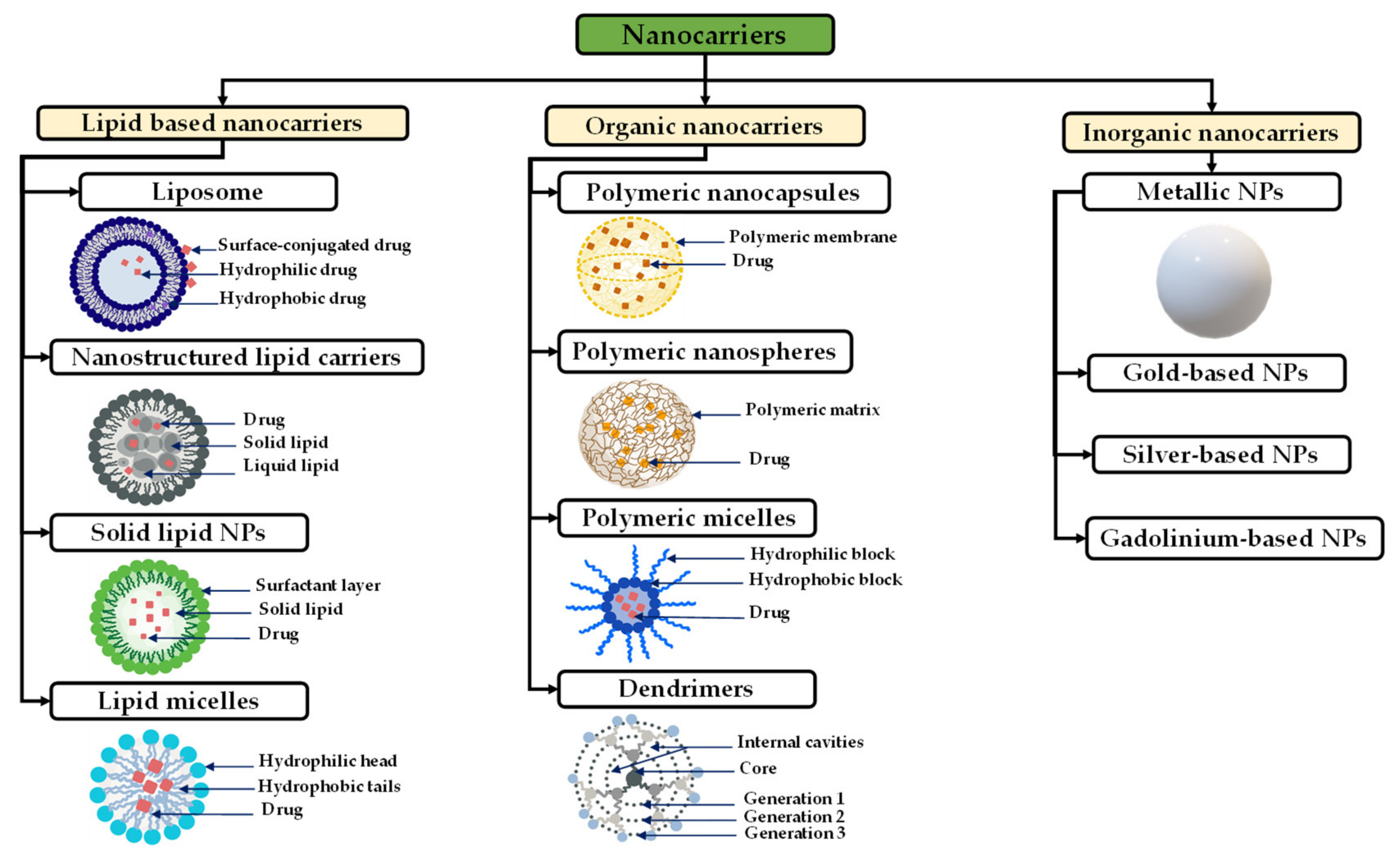
- Nanocarriers:
- Lipid-Based Nanocarriers for Glioblastoma Treatment
- Polymeric-Based Nanocarriers for Glioblastoma Treatment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer Type | Drug/Molecule Loaded | Particle Size (nm) | Ref |
|---|---|---|---|
| PLGA | DOX | ~120 | [245] |
| PLGA | TMZ | ~194 | [241] |
| PLGA-PEG | DOX | ~50 | [240] |
| PLGA-PEG-chitosan | Paclitaxel and R-flurbiprofen | 150–190 | [242] |
| PLGA | DTX and indocyanine green | ~220 | [246] |
| PLGA/PEG-PLGA | CXCL12 | 200–250 | [213] |
| Chitosan-Alginate | CXCL12 | ~250 | [243] |
| Chitosan-modified PLGA NPs | R-Flurbiprofen and Paclitaxel | 150–190 | [100] |
| Dextran | Paclitaxel | ~60 | [235] |
| Silk fibroin | Indocyanine green | ~209 | [247] |
| Synthetic protein | AMD3100 | 37–98 | [244] |
6.2.2. Local Delivery
- Intranasal Drugs Delivery:
- Convection Enhanced Delivery:
- Direct Drug Injection:
- Rigid Implants:
- New Innovative Drug-Delivery Approaches in Glioblastoma Treatments:
- Hydrogels:
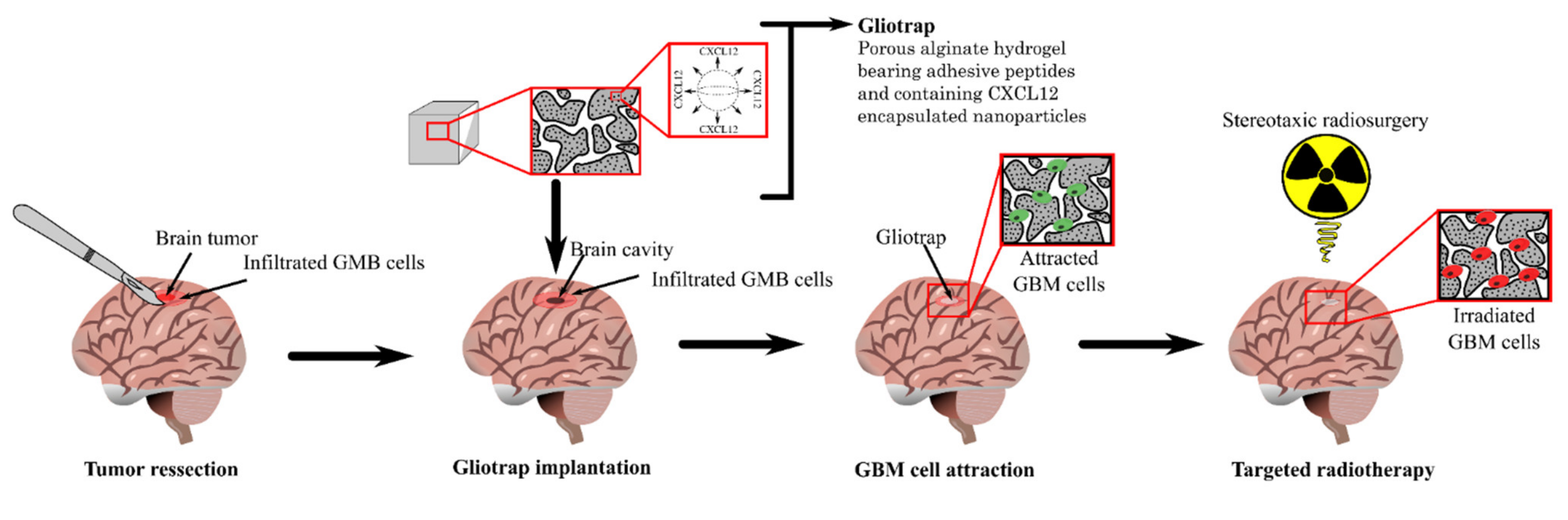
- Cancer Cells Trap:
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Schwartzbaum, J.A.; Fisher, J.L.; Aldape, K.D.; Wrensch, M. Epidemiology and molecular pathology of glioma. Nat. Clin. Pract. Neurol. 2006, 2, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Patel, A.P.; Fisher, J.L.; Nichols, E.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; Abraha, H.N.; Agius, D.; Alahdab, F.; Alam, T.; et al. Global, regional, and national burden of brain and other CNS cancer, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 376–393. [Google Scholar] [CrossRef] [Green Version]
- Dolecek, T.A.; Propp, J.M.; Stroup, N.E.; Kruchko, C. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2005–2009. Neuro-Oncology 2012, 14, v1–v49. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro-Oncology 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, I.; Cockburn, M.; Cozen, W.; Wang, Y.-P.; Preston-Martin, S. A population-based description of glioblastoma multiforme in Los Angeles County, 1974–1999. Cancer 2005, 104, 2798–2806. [Google Scholar] [CrossRef]
- Tamimi, A.F.; Juweid, M. Epidemiology and outcome of glioblastoma. Exon Publ. 2017, 143–153. [Google Scholar] [CrossRef]
- Lah, T.T.; Novak, M.; Breznik, B. Brain malignancies: Glioblastoma and brain metastases. Semin. Cancer Biol. 2020, 60, 262–273. [Google Scholar] [CrossRef]
- Al-Rikabi, A.C.; Al-Sohaibani, M.O.; Jamjoom, A.; Al-Rayess, M.M. Metastatic deposits of a high-grade malignant glioma in cervical lymph nodes diagnosed by fine needle aspiration (FNA) cytology-Case report and literature review. Cytopathology 1997, 8, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Widjaja, A.; Mix, H.; Gölkel, C.; Flemming, P.; Egensperger, R.; Holstein, A.; Rademaker, J.; Becker, H.; Hundt, M.; Wagner, S.; et al. Uncommon metastasis of a glioblastoma multiforme in liver and spleen. Digestion 2000, 61, 219–222. [Google Scholar] [CrossRef]
- Lin, C.; Liu, L.; Zeng, C.; Cui, Z.-K.; Chen, Y.; Lai, P.; Wang, H.; Shao, Y.; Zhang, H.; Zhang, R.; et al. Activation of mTORC1 in subchondral bone preosteoblasts promotes osteoarthritis by stimulating bone sclerosis and secretion of CXCL12. Bone Res. 2019, 7, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, S.; Bhardwaj, A.; Singh, S.; Srivastava, S.K.; McClellan, S.; Nirodi, C.S.; Piazza, G.A.; Grizzle, W.E.; Owen, L.B.; Singh, A.P. An undesired effect of chemotherapy: Gemcitabine promotes pancreatic cancer cell invasiveness through reactive oxygen species-dependent, nuclear factorκb- and hypoxia-inducible factor 1α-mediated up-regulation of CXCR4. J. Biol. Chem. 2013, 288, 21197–21207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, D.; Nandi, S.; Bhattacharjee, S. Combination therapy to checkmate Glioblastoma: Clinical challenges and advances. Clin. Transl. Med. 2018, 7, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherer, H.J. Structural Development in Gliomas. Am. J. Cancer 1938, 34, 333–351. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Barone, D.G.; Lawrie, T.A.; Hart, M.G. Image guided surgery for the resection of brain tumours. Cochrane Database Syst. Rev. 2014, 2014, CD009685. [Google Scholar] [CrossRef]
- Mrugala, M.M. Advances and challenges in the treatment of glioblastoma: A clinician’s perspective. Discov. Med. 2013, 15, 221–230. [Google Scholar]
- Azagury, D.E.; Dua, M.M.; Barrese, J.C.; Henderson, J.M.; Buchs, N.C.; Ris, F.; Cloyd, J.M.; Martinie, B.J.; Razzaque, S.; Nicolau, S.; et al. Image-guided surgery. Curr. Probl. Surg. 2015, 52, 476–520. [Google Scholar] [CrossRef] [Green Version]
- Klimberg, V.S.; Rivere, A. Ultrasound image-guided core biopsy of the breast. Chin. Clin. Oncol. 2016, 5, 33. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [Green Version]
- Desmarais, G.; Fortin, D.; Bujold, R.; Wagner, R.; Mathieu, D.; Paquette, B. Infiltration of glioma cells in brain parenchyma stimulated by radiation in the F98/Fischer rat model. Int. J. Radiat. Biol. 2012, 88, 565–574. [Google Scholar] [CrossRef]
- Mann, J.; Ramakrishna, R.; Magge, R.; Wernicke, A.G. Advances in radiotherapy for glioblastoma. Front. Neurol. 2018, 8, 748. [Google Scholar] [CrossRef] [Green Version]
- Fuller, C.D.D.; Choi, M.; Forthuber, B.; Wang, S.J.J.; Rajagiriyil, N.; Salter, B.J.J.; Fuss, M. Standard fractionation intensity modulated radiation therapy (IMRT) of primary and recurrent glioblastoma multiforme. Radiat. Oncol. 2007, 2, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; Curschmann, J.; Janzer, R.C. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar]
- Hatoum, A.; Mohammed, R.; Zakieh, O. The unique invasiveness of glioblastoma and possible drug targets on extracellular matrix. Cancer Manag. Res. 2019, 11, 1843–1855. [Google Scholar] [CrossRef] [Green Version]
- Solano, A.G.; Dupuy, J.; Therriault, H.; Liberelle, B.; Faucheux, N.; Lauzon, M.-A.; Virgilio, N.; Paquette, B. An alginate-based macroporous hydrogel matrix to trap cancer cells. Carbohydr. Polym. 2021, 266, 118115. [Google Scholar] [CrossRef]
- Desmarais, G.; Charest, G.; Therriault, H.; Shi, M.; Fortin, D.; Bujold, R.; Mathieu, D.; Paquette, B. Infiltration of F98 glioma cells in Fischer rat brain is temporary stimulated by radiation. Int. J. Radiat. Biol. 2016, 92, 444–450. [Google Scholar] [CrossRef]
- Combs, S.E.; Widmer, V.; Thilmann, C.; Hof, H.; Debus, J.; Schulz-Ertner, D. Stereotactic radiosurgery (SRS): Treatment option for recurrent glioblastoma multiforme (GBM). Cancer 2005, 104, 2168–2173. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nakai, K.; Matsumura, A. Boron neutron capture therapy for glioblastoma. Cancer Lett. 2008, 262, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Norden, A.D.; Wen, P.Y. Glioma therapy in adults. Neurologist 2006, 12, 279–292. [Google Scholar] [CrossRef]
- Williams, B.J.; Suki, D.; Fox, B.D.; Pelloski, C.E.; Maldaun, M.V.C.; Sawaya, R.E.; Lang, F.F.; Rao, G. Stereotactic radiosurgery for metastatic brain tumors: A comprehensive review of complications: Clinical article. J. Neurosurg. JNS 2009, 111, 439–448. [Google Scholar] [CrossRef]
- Alphandéry, E. Glioblastoma treatments: An account of recent industrial developments. Front. Pharmacol. 2018, 9, 879. [Google Scholar] [CrossRef] [Green Version]
- Neuwelt, E.A.; Rapoport, S.I. Modification of the blood-brain barrier in the chemotherapy of malignant brain tumors. Fed. Proc. 1984, 43, 214–219. [Google Scholar]
- Sayegh, E.T.; Kaur, G.; Bloch, O.; Parsa, A.T. Systematic review of protein biomarkers of invasive behavior in glioblastoma. Mol. Neurobiol. 2014, 49, 1212–1244. [Google Scholar] [CrossRef]
- Wu, W.; Klockow, J.L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H.E. Glioblastoma multiforme (GBM): An overview of current therapies and mechanisms of resistance. Pharmacol. Res. 2021, 171, 105780. [Google Scholar] [CrossRef]
- O’Reilly, S.M.; Newlands, E.S.; Brampton, M.; Glaser, M.G.; Rice-Edwards, J.M.; Illingworth, R.D.; Richards, P.G.; Kennard, C.; Colquhoun, I.R.; Lewis, P.; et al. Temozolomide: A new oral cytotoxic chemotherapeutic agent with promising activity against primary brain tumours. Eur. J. Cancer 1993, 29, 940–942. [Google Scholar] [CrossRef]
- Witthayanuwat, S.; Pesee, M.; Supaadirek, C.; Supakalin, N.; Thamronganantasakul, K.; Krusun, S. Survival analysis of Glioblastoma Multiforme. Asian Pac. J. Cancer Prev. 2018, 19, 2613–2617. [Google Scholar]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Gerber, D.E.; Grossman, S.A.; Zeltzman, M.; Parisi, M.A.; Kleinberg, L. The impact of thrombocytopenia from temozolomide and radiation in newly diagnosed adults with high-grade gliomas1. Neuro-Oncology 2007, 9, 47–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2022, in press. [Google Scholar] [CrossRef]
- Grossman, S.A.; Reinhard, C.; Colvin, O.M.; Chasin, M.; Brundrett, R.; Tamargo, R.J.; Brem, H. The intracerebral distribution of BCNU delivered by surgically implanted biodegradable polymers. J. Neurosurg. 1992, 76, 640–647. [Google Scholar] [CrossRef]
- Bartzatt, R. Lomustine Analogous Drug Structures for Intervention of Brain and Spinal Cord Tumors: The Benefit of In Silico Substructure Search and Analysis. Chemother. Res. Pract. 2013, 2013, 360624. [Google Scholar] [CrossRef] [Green Version]
- Binabaj, M.M.; Bahrami, A.; ShahidSales, S.; Joodi, M.; Joudi Mashhad, M.; Hassanian, S.M.; Anvari, K.; Avan, A. The prognostic value of MGMT promoter methylation in glioblastoma: A meta-analysis of clinical trials. J. Cell. Physiol. 2018, 233, 378–386. [Google Scholar] [CrossRef]
- Illic, R.; Somma, T.; Savic, D.; Frio, F.; Milicevic, M.; Solari, D.; Nikitovic, M.; Lavrnic, S.; Raicevic, S.; Milosevic, S.; et al. A Survival Analysis with Identification of Prognostic Factors in a Series of 110 Patients with Newly Diagnosed Glioblastoma Before and After Introduction of the Stupp Regimen: A Single-Center Observational Study. World Neurosurg. 2017, 104, 581–588. [Google Scholar] [CrossRef]
- Taphoorn, M.J.B.; Stupp, R.; Coens, C.; Osoba, D.; Kortmann, R.; van den Bent, M.J.; Mason, W.; Mirimanoff, R.O.; Baumert, B.G.; Eisenhauer, E. Health-related quality of life in patients with glioblastoma: A randomised controlled trial. Lancet Oncol. 2005, 6, 937–944. [Google Scholar] [CrossRef]
- Huijbers, I.J.; Iravani, M.; Popov, S.; Robertson, D.; Al-Sarraj, S.; Jones, C.; Isacke, C.M. A role for fibrillar collagen deposition and the collagen internalization receptor endo180 in glioma invasion. PLoS ONE 2010, 5, e9808. [Google Scholar] [CrossRef]
- Joo, K.M.; Jin, J.; Kim, E.; Kim, K.H.; Kim, Y.; Kang, B.G.; Kang, Y.J.; Lathia, J.D.; Cheong, K.H.; Song, P.H.; et al. MET signaling regulates glioblastoma stem cells. Cancer Res. 2012, 72, 3828–3838. [Google Scholar] [CrossRef] [Green Version]
- Veeravalli, K.K.; Rao, J.S. MMP-9 and uPAR regulated glioma cell migration. Cell Adhes. Migr. 2012, 6, 509–512. [Google Scholar] [CrossRef] [Green Version]
- Pencheva, N.; de Gooijer, M.C.; Vis, D.J.; Wessels, L.F.A.A.; Würdinger, T.; van Tellingen, O.; Bernards, R. Identification of a Druggable Pathway Controlling Glioblastoma Invasiveness. Cell Rep. 2017, 20, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Mohiuddin, E.; Wakimoto, H. Extracellular matrix in glioblastoma: Opportunities for emerging therapeutic approaches. Am. J. Cancer Res. 2021, 11, 3742–3754. [Google Scholar]
- Li, G.; Qin, Z.; Chen, Z.; Xie, L.; Wang, R.; Zhao, H. Tumor microenvironment in treatment of glioma. Open Med. 2017, 12, 247–251. [Google Scholar] [CrossRef]
- de Gooijer, M.C.; Guillén Navarro, M.; Bernards, R.; Wurdinger, T.; van Tellingen, O. An Experimenter’s Guide to Glioblastoma Invasion Pathways. Trends Mol. Med. 2018, 24, 763–780. [Google Scholar] [CrossRef]
- Tamkun, J.W.; DeSimone, D.W.; Fonda, D.; Patel, R.S.; Buck, C.; Horwitz, A.F.; Hynes, R.O. Structure of integrin, a glycoprotein involved in the transmembrane linkage between fibronectin and actin. Cell 1986, 46, 271–282. [Google Scholar] [CrossRef]
- Humphries, J.D.; Askari, J.A.; Zhang, X.-P.; Takada, Y.; Humphries, M.J.; Mould, A.P. Molecular Basis of Ligand Recognition by Integrin 5. J. Biol. Chem. 2000, 275, 20337–20345. [Google Scholar] [CrossRef] [Green Version]
- Bouvard, D.; Pouwels, J.; De Franceschi, N.; Ivaska, J. Integrin inactivators: Balancing cellular functions in vitro and in vivo. Nat. Rev. Mol. Cell Biol. 2013, 14, 430–442. [Google Scholar] [CrossRef]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [Green Version]
- Banères, J.L.; Roquet, F.; Green, M.; LeCalvez, H.; Parello, J. The cation-binding domain from the α subunit of integrin α5β1 is a minimal domain for fibronectin recognition. J. Biol. Chem. 1998, 273, 24744–24753. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Varner, J.A.; Cheresh, D.A. Integrins and cancer. Curr. Opin. Cell Biol. 1996, 8, 724–730. [Google Scholar] [CrossRef]
- Paolillo, M.; Serra, M.; Schinelli, S. Integrins in glioblastoma: Still an attractive target? Pharmacol. Res. 2016, 113, 55–61. [Google Scholar] [CrossRef]
- Muradashvili, N.; Lominadze, D. Role of fibrinogen in cerebrovascular dysfunction after traumatic brain injury. Brain Inj. 2013, 27, 1508–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gritsenko, P.G.; Ilina, O.; Friedl, P. Interstitial guidance of cancer invasion. J. Pathol. 2012, 226, 185–199. [Google Scholar] [CrossRef]
- Zimmermann, D.R.; Dours-Zimmermann, M.T. Extracellular matrix of the central nervous system: From neglect to challenge. Histochem. Cell Biol. 2008, 130, 635–653. [Google Scholar] [CrossRef] [Green Version]
- Maglott, A.; Bartik, P.; Cosgun, S.; Klotz, P.; Rondé, P.; Fuhrmann, G.; Takeda, K.; Martin, S.; Dontenwill, M. The Small α5β1 Integrin Antagonist, SJ749, Reduces Proliferation and Clonogenicity of Human Astrocytoma Cells. Cancer Res. 2006, 66, 6002–6007. [Google Scholar] [CrossRef] [Green Version]
- Mallawaaratchy, D.M.; Buckland, M.E.; McDonald, K.L.; Li, C.C.Y.; Ly, L.; Sykes, E.K.; Christopherson, R.I.; Kaufman, K.L. Membrane Proteome Analysis of Glioblastoma Cell Invasion. J. Neuropathol. Exp. Neurol. 2015, 74, 425–441. [Google Scholar] [CrossRef] [Green Version]
- Nakada, M.; Nambu, E.; Furuyama, N.; Yoshida, Y.; Takino, T.; Hayashi, Y.; Sato, H.; Sai, Y.; Tsuji, T.; Miyamoto, K.; et al. Integrin α3 is overexpressed in glioma stem-like cells and promotes invasion. Br. J. Cancer 2013, 108, 2516–2524. [Google Scholar] [CrossRef]
- Schnell, O.; Krebs, B.; Wagner, E.; Romagna, A.; Beer, A.J.; Grau, S.J.; Thon, N.; Goetz, C.; Kretzschmar, H.A.; Tonn, J.-C.; et al. Expression of Integrin αvβ3 in Gliomas Correlates with Tumor Grade and Is not Restricted to Tumor Vasculature. Brain Pathol. 2008, 18, 378–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blandin, A.-F.; Noulet, F.; Renner, G.; Mercier, M.-C.; Choulier, L.; Vauchelles, R.; Ronde, P.; Carreiras, F.; Etienne-Selloum, N.; Vereb, G.; et al. Glioma cell dispersion is driven by α5 integrin-mediated cell–matrix and cell–cell interactions. Cancer Lett. 2016, 376, 328–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, S.; Akutsu, N.; Matsunaga, Y.; Kadoya, K.; Nishiyama, T.; Champliaud, M.-F.; Burgeson, R.E.; Adachi, E. Importance of Balance between Extracellular Matrix Synthesis and Degradation in Basement Membrane Formation. Exp. Cell Res. 2001, 271, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Kubiatowski, T.; Jang, T.; Lachyankar, M.B.; Salmonsen, R.; Nabi, R.R.; Quesenberry, P.J.; Litofsky, N.S.; Ross, A.H.; Recht, L.D. Association of increased phosphatidylinositol 3-kinase signaling with increased invasiveness and gelatinase activity in malignant gliomas. J. Neurosurg. 2001, 95, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.Y.; Hu, D.X.; Chen, W.-Q.; Chen, R.Q.; Qian, S.R.; Li, C.Y.; Li, Y.J.; Xiong, X.X.; Liu, D.; Pan, F.; et al. PD-L1 confers glioblastoma multiforme malignancy via Ras binding and Ras/Erk/EMT activation. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 1754–1769. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Mikheev, A.M.; Mikheeva, S.A.; Trister, A.D.; Tokita, M.J.; Emerson, S.N.; Parada, C.A.; Born, D.E.; Carnemolla, B.; Frankel, S.; Kim, D.-H.; et al. Periostin is a novel therapeutic target that predicts and regulates glioma malignancy. Neuro-Oncology 2015, 17, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Serres, E.; Debarbieux, F.; Stanchi, F.; Maggiorella, L.; Grall, D.; Turchi, L.; Burel-Vandenbos, F.; Figarella-Branger, D.; Virolle, T.; Rougon, G.; et al. Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene 2014, 33, 3451–3462. [Google Scholar] [CrossRef]
- Xiong, J.; Balcioglu, H.E.; Danen, E.H.J. Integrin signaling in control of tumor growth and progression. Int. J. Biochem. Cell Biol. 2013, 45, 1012–1015. [Google Scholar] [CrossRef]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Kulbe, H.; Levinson, N.R.; Balkwill, F.; Wilson, J.L. The chemokine network in cancer-Much more than directing cell movement. Int. J. Dev. Biol. 2004, 48, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Balestrieri, M.L.; Balestrieri, A.; Mancini, F.P.; Napoli, C. Understanding the immunoangiostatic CXC chemokine network. Cardiovasc. Res. 2008, 78, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Bockaert, J.; Pin, J.P. Molecular tinkering of G protein-coupled receptors: An evolutionary success. EMBO J. 1999, 18, 1723–1729. [Google Scholar] [CrossRef] [Green Version]
- Bonavia, R.; Bajetto, A.; Barbero, S.; Pirani, P.; Florio, T.; Schettini, G. Chemokines and their receptors in the CNS: Expression of CXCL12/SDF-1 and CXCR4 and their role in astrocyte proliferation. Toxicol. Lett. 2003, 139, 181–189. [Google Scholar] [CrossRef]
- Johnson, G.L.; Dhanasekaran, N. The G-protein family and their interaction with receptors. Endocr. Rev. 1989, 10, 317–331. [Google Scholar] [CrossRef]
- L’Allemain, G.; Pouyssegur, J.; Weber, M.J. p42/mitogen-activated protein kinase as a converging target for different growth factor signaling pathways: Use of pertussis toxin as a discrimination factor. Cell Regul. 1991, 2, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Cheng, L.; Guryanova, O.A.; Wu, Q.; Bao, S. Cancer stem cells in glioblastoma—molecular signaling and therapeutic targeting. Protein Cell 2010, 1, 638–655. [Google Scholar] [CrossRef]
- Liu, R.; Tian, B.; Gearing, M.; Hunter, S.; Ye, K.; Mao, Z. Cdk5-mediated regulation of the PIKE-A-Akt pathway and glioblastoma cell invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 7570–7575. [Google Scholar] [CrossRef] [Green Version]
- Roland, J.; Murphy, B.J.; Ahr, B.; Robert-Hebmann, V.; Delauzun, V.; Nye, K.E.; Devaux, C.; Biard-Piechaczyk, M. Role of the intracellular domains of CXCR4 in SDF-1–mediated signaling. Blood 2003, 101, 399–406. [Google Scholar] [CrossRef]
- Würth, R.; Bajetto, A.; Harrison, J.K.; Barbieri, F.; Florio, T. CXCL12 modulation of CXCR4 and CXCR7 activity in human glioblastoma stem-like cells and regulation of the tumor microenvironment. Front. Cell. Neurosci. 2014, 8, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Décaillot, F.M.; Kazmi, M.A.; Lin, Y.; Ray-Saha, S.; Sakmar, T.P.; Sachdev, P. CXCR7/CXCR4 heterodimer constitutively recruits β-arrestin to enhance cell migration. J. Biol. Chem. 2011, 286, 32188–32197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Carson-Walter, E.; Walter, K.A. Targeting chemokine receptor CXCR7 inhibits glioma cell proliferation and mobility. Anticancer Res. 2015, 35, 53–64. [Google Scholar]
- Groblewska, M.; Litman-Zawadzka, A.; Mroczko, B. The role of selected chemokines and their receptors in the development of gliomas. Int. J. Mol. Sci. 2020, 21, 3704. [Google Scholar] [CrossRef] [PubMed]
- Lepore, F.; D’Alessandro, G.; Antonangeli, F.; Santoro, A.; Esposito, V.; Limatola, C.; Trettel, F. CXCL16/CXCR6 axis drives microglia/macrophages phenotype in physiological conditions and plays a crucial role in glioma. Front. Immunol. 2018, 9, 2750. [Google Scholar] [CrossRef] [Green Version]
- Novak, M.; Koprivnikar Krajnc, M.; Hrastar, B.; Breznik, B.; Majc, B.; Mlinar, M.; Rotter, A.; Porčnik, A.; Mlakar, J.; Stare, K.; et al. CCR5-Mediated Signaling is Involved in Invasion of Glioblastoma Cells in Its Microenvironment. Int. J. Mol. Sci. 2020, 21, 4199. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Latha, K.; Manyam, G.; Yang, Y.; Rao, A.; Rao, G. Role of CX3CR1 signaling in malignant transformation of gliomas. Neuro-Oncology 2020, 22, 1463–1473. [Google Scholar] [CrossRef]
- Rodero, M.; Marie, Y.; Coudert, M.; Blondet, E.; Mokhtari, K.; Rousseau, A.; Raoul, W.; Carpentier, C.; Sennlaub, F.; Deterre, P. Polymorphism in the microglial cell-mobilizing CX3CR1 gene is associated with survival in patients with glioblastoma. J. Clin. Oncol. 2008, 26, 5957–5964. [Google Scholar] [CrossRef] [Green Version]
- Munson, J.M.; Bellamkonda, R.V.; Swartz, M.A. Interstitial flow in a 3d microenvironment increases glioma invasion by a cxcr4-dependent mechanism. Cancer Res. 2013, 73, 1536–1546. [Google Scholar] [CrossRef] [Green Version]
- Calatozzolo, C.; Canazza, A.; Pollo, B.; Di Pierro, E.; Ciusani, E.; Maderna, E.; Salce, E.; Sponza, V.; Frigerio, S.; Di Meco, F.; et al. Expression of the new CXCL12 receptor, CXCR7, in gliomas. Cancer Biol. Ther. 2011, 11, 242–253. [Google Scholar] [CrossRef] [Green Version]
- Balabanian, K.; Lagane, B.; Infantino, S.; Chow, K.Y.C.; Harriague, J.; Moepps, B.; Arenzana-Seisdedos, F.; Thelen, M.; Bachelerie, F. The Chemokine SDF-1/CXCL12 Binds to and Signals through the Orphan Receptor RDC1 in T Lymphocytes. J. Biol. Chem. 2005, 280, 35760–35766. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Lu, Y.; Xu, Y.; Wang, J.; Zhang, C.; Du, Y.; Wang, L.; Li, L.; Wang, B.; Shen, J.; et al. Horizontal transfer of exosomal CXCR4 promotes murine hepatocarcinoma cell migration, invasion and lymphangiogenesis. Gene 2018, 676, 101–109. [Google Scholar] [CrossRef]
- Bai, R.; Jie, X.; Sun, J.; Liang, Z.; Yoon, Y.; Feng, A.; Oum, Y.; Yu, W.; Wu, R.; Sun, B.; et al. Development of CXCR4 modulators based on the lead compound RB-108. Eur. J. Med. Chem. 2019, 173, 32–43. [Google Scholar] [CrossRef]
- Brickute, D.; Braga, M.; Kaliszczak, M.A.; Barnes, C.; Lau, D.; Carroll, L.; Stevens, E.; Trousil, S.; Alam, I.S.; Nguyen, Q.-D.; et al. Development and Evaluation of an 18F-Radiolabeled Monocyclam Derivative for Imaging CXCR4 Expression. Mol. Pharm. 2019, 16, 2106–2117. [Google Scholar] [CrossRef] [Green Version]
- Truong, D.; Fiorelli, R.; Barrientos, E.S.; Melendez, E.L.; Sanai, N.; Mehta, S.; Nikkhah, M. A three-dimensional (3D) organotypic microfluidic model for glioma stem cells–Vascular interactions. Biomaterials 2019, 198, 63–77. [Google Scholar] [CrossRef]
- Cornelison, R.C.; Brennan, C.E.; Kingsmore, K.M.; Munson, J.M. Convective forces increase CXCR4-dependent glioblastoma cell invasion in GL261 murine model. Sci. Rep. 2018, 8, 17057. [Google Scholar] [CrossRef] [Green Version]
- Mortezaee, K. CXCL12/CXCR4 axis in the microenvironment of solid tumors: A critical mediator of metastasis. Life Sci. 2020, 249, 117534. [Google Scholar] [CrossRef]
- Domanska, U.M.; Kruizinga, R.C.; Nagengast, W.B.; Timmer-Bosscha, H.; Huls, G.; De Vries, E.G.E.; Walenkamp, A.M.E. A review on CXCR4/CXCL12 axis in oncology: No place to hide. Eur. J. Cancer 2013, 49, 219–230. [Google Scholar] [CrossRef]
- Duan, Y.; Zhang, S.; Wang, L.; Zhou, X.; He, Q.; Liu, S.; Yue, K.; Wang, X. Targeted silencing of CXCR4 inhibits epithelial-mesenchymal transition in oral squamous cell carcinoma. Oncol. Lett. 2016, 12, 2055–2061. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Zhang, P.; Luo, H.; Huang, H.; Wang, F. CXCL12 modulates the radiosensitivity of cervical cancer by regulating CD44. Mol. Med. Rep. 2018, 18, 5101–5108. [Google Scholar] [CrossRef] [Green Version]
- Duda, D.G.; Kozin, S.V.; Kirkpatrick, N.D.; Xu, L.; Fukumura, D.; Jain, R.K. CXCL12 (SDF1α)-CXCR4/CXCR7 Pathway Inhibition: An Emerging Sensitizer for Anticancer Therapies? Clin. Cancer Res. 2011, 17, 2074–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, Y.; Yao, X.; Jiang, J.; Zhao, L.; Yu, S.; Jiang, T.; Lin, M.C.M.; Chen, J.; Wang, B.; Zhang, R. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J. Pathol. 2011, 224, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.J. D6 and the atypical chemokine receptor family: Novel regulators of immune and inflammatory processes. Eur. J. Immunol. 2009, 39, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Lefkowitz, R.J. Multifaceted roles of β-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem. J. 2003, 375, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, M.; Li, Y.; Xu, D.; Wang, Y.; Song, A.; Zhu, B.; Huang, Y.; Zheng, J.C. CXCR7 Mediates Neural Progenitor Cells Migration to CXCL12 Independent of CXCR4. Stem Cells 2015, 33, 2574–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, M.J.; Ebsworth, K.; Berahovich, R.D.; Penfold, M.E.T.; Liu, S.-C.; Al Omran, R.; Kioi, M.; Chernikova, S.B.; Tseng, D.; Mulkearns-Hubert, E.E.; et al. Inhibition of CXCR7 extends survival following irradiation of brain tumours in mice and rats. Br. J. Cancer 2014, 110, 1179–1188. [Google Scholar] [CrossRef]
- Orsini, M.J.; Parent, J.-L.; Mundell, S.J.; Benovic, J.L. Trafficking of the HIV coreceptor CXCR4: Role of arrestins and identification of residues in the C-terminal tail that mediate receptor internalization. J. Biol. Chem. 1999, 274, 31076–31086. [Google Scholar] [CrossRef] [Green Version]
- Haribabu, B.; Richardson, R.M.; Fisher, I.; Sozzani, S.; Peiper, S.C.; Horuk, R.; Ali, H.; Snyderman, R. Regulation of human chemokine receptors CXCR4: Role of phosphorylation in desensitization and internalization. J. Biol. Chem. 1997, 272, 28726–28731. [Google Scholar] [CrossRef] [Green Version]
- Mousavi, A. CXCL12/CXCR4 signal transduction in diseases and its molecular approaches in targeted-therapy. Immunol. Lett. 2020, 217, 91–115. [Google Scholar] [CrossRef]
- Urbantat, R.M.; Vajkoczy, P.; Brandenburg, S. Advances in Chemokine Signaling Pathways as Therapeutic Targets in Glioblastoma. Cancers 2021, 13, 2983. [Google Scholar] [CrossRef]
- Fricker, S.P. Physiology and pharmacology of plerixafor. Transfus. Med. Hemother. 2013, 40, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, T.N.; Burger, J.A.; Glodek, A.; Fujii, N.; Burger, M. CXCR4 chemokine receptor and integrin signaling co-operate in mediating adhesion and chemoresistance in small cell lung cancer (SCLC) cells. Oncogene 2005, 24, 4462–4471. [Google Scholar] [CrossRef] [Green Version]
- Engl, T.; Relja, B.; Marian, D.; Blumenberg, C.; Müller, I.; Beecken, W.-D.; Jones, J.; Ringel, E.M.; Bereiter-Hahn, J.; Jonas, D.; et al. CXCR4 Chemokine Receptor Mediates Prostate Tumor Cell Adhesion through α5 and β3 Integrins. Neoplasia 2006, 8, 290–301. [Google Scholar] [CrossRef] [Green Version]
- Maurer, G.D.; Tritschler, I.; Adams, B.; Tabatabai, G.; Wick, W.; Stupp, R.; Weller, M. Cilengitide modulates attachment and viability of human glioma cells, but not sensitivity to irradiation or temozolomide in vitro. Neuro-Oncology 2009, 11, 747–756. [Google Scholar] [CrossRef] [Green Version]
- Kingsmore, K.M.; Logsdon, D.K.; Floyd, D.H.; Peirce, S.M.; Purow, B.W.; Munson, J.M. Interstitial flow differentially increases patient-derived glioblastoma stem cell invasion: Via CXCR4, CXCL12, and CD44-mediated mechanisms. Integr. Biol. 2016, 8, 1246–1260. [Google Scholar] [CrossRef]
- Lei, Y.; Han, H.; Yuan, F.; Javeed, A.; Zhao, Y. The brain interstitial system: Anatomy, modeling, in vivo measurement, and applications. Prog. Neurobiol. 2017, 157, 230–246. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J. Evidence for bulk flow of brain interstitial fluid: Significance for physiology and pathology. Neurochem. Int. 2004, 45, 545–552. [Google Scholar] [CrossRef]
- Munson, J.M. Interstitial fluid flow under the microscope: Is it a future drug target for high grade brain tumours such as glioblastoma? Expert Opin. Ther. Targets 2019, 23, 725–728. [Google Scholar] [CrossRef]
- Boucher, Y.; Salehi, H.; Witwer, B.; Harsh, G.R.; Jain, R.K. Interstitial fluid pressure in intracranial tumours in patients and in rodents. Br. J. Cancer 1997, 75, 829–836. [Google Scholar] [CrossRef]
- Zagzag, D.; Lukyanov, Y.; Lan, L.; Ali, M.A.; Esencay, M.; Mendez, O.; Yee, H.; Voura, E.B.; Newcomb, E.W. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: Implications for angiogenesis and glioma cell invasion. Lab. Investig. 2006, 86, 1221–1232. [Google Scholar] [CrossRef] [Green Version]
- Burridge, K.; Monaghan-Benson, E.; Graham, D.M. Mechanotransduction: From the cell surface to the nucleus via RhoA. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180229. [Google Scholar] [CrossRef] [PubMed]
- Qazi, H.; Shi, Z.-D.D.; Tarbell, J.M. Fluid shear stress regulates the invasive potential of glioma cells via modulation of migratory activity and matrix metalloproteinase expression. PLoS ONE 2011, 6, e20348. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, G.M.; Zhong, J.; Paul, A.; Kellie, S.J. Mesenchymal migration as a therapeutic target in glioblastoma. J. Oncol. 2010, 2010, 430142. [Google Scholar]
- Vollmann-Zwerenz, A.; Leidgens, V.; Feliciello, G.; Klein, C.A.; Hau, P. Tumor Cell Invasion in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 1932. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Griffiths, S.; Veljanoski, D.; Vaughn-Beaucaire, P.; Speirs, V.; Brüning-Richardson, A. Preclinical models of glioblastoma: Limitations of current models and the promise of new developments. Expert Rev. Mol. Med. 2021, 23, e20. [Google Scholar] [CrossRef]
- Paolillo, M.; Comincini, S.; Schinelli, S. In vitro glioblastoma models: A journey into the third dimension. Cancers 2021, 13, 2449. [Google Scholar] [CrossRef]
- Heffernan, J.M.; Overstreet, D.J.; Le, L.D.; Vernon, B.L.; Sirianni, R.W. Bioengineered scaffolds for 3D analysis of glioblastoma proliferation and invasion. Ann. Biomed. Eng. 2015, 43, 1965–1977. [Google Scholar] [CrossRef]
- Cai, X.; Briggs, R.G.; Homburg, H.B.; Young, I.M.; Davis, E.J.; Lin, Y.-H.; Battiste, J.D.; Sughrue, M.E. Application of microfluidic devices for glioblastoma study: Current status and future directions. Biomed. Microdevices 2020, 22, 60. [Google Scholar] [CrossRef]
- Hutter-Schmid, B.; Kniewallner, K.; Humpel, C. Organotypic brain slice cultures as a model to study angiogenesis of brain vessels. Front. Cell Dev. Biol. 2015, 3, 52. [Google Scholar] [CrossRef] [Green Version]
- Haddad, A.F.; Young, J.S.; Amara, D.; Berger, M.S.; Raleigh, D.R.; Aghi, M.K.; Butowski, N.A. Mouse models of glioblastoma for the evaluation of novel therapeutic strategies. Neuro-oncology Adv. 2021, 3, vdab100. [Google Scholar] [CrossRef]
- Rao, S.S.; Lannutti, J.J.; Viapiano, M.S.; Sarkar, A.; Winter, J.O. Toward 3D Biomimetic Models to Understand the Behavior of Glioblastoma Multiforme Cells. Tissue Eng. Part B Rev. 2014, 20, 314–327. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, J.T.S.; Chutna, O.; Chu, V.; Conde, J.P.; Outeiro, T.F. A Novel Microfluidic Cell Co-culture Platform for the Study of the Molecular Mechanisms of Parkinson’s Disease and Other Synucleinopathies. Front. Neurosci. 2016, 10, 511. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Holtkamp, N.; Afanasieva, A.; Elstner, A.; van Landeghem, F.K.H.; Könneker, M.; Kuhn, S.A.; Kettenmann, H.; Deimling, A. von Brain slice invasion model reveals genes differentially regulated in glioma invasion. Biochem. Biophys. Res. Commun. 2005, 336, 1227–1233. [Google Scholar] [CrossRef]
- Kwiatkowska, A.; Nandhu, M.S.; Behera, P.; Chiocca, E.A.; Viapiano, M.S. Strategies in gene therapy for glioblastoma. Cancers 2013, 5, 1271–1305. [Google Scholar] [CrossRef]
- Zanders, E.D.; Svensson, F.; Bailey, D.S. Therapy for glioblastoma: Is it working? Drug Discov. Today 2019, 24, 1193–1201. [Google Scholar] [CrossRef]
- Yamamoto, M.; Curiel, D.T.T.; Ph, D.; South, S.; South, S. Cancer gene therapy. Technol. Cancer Res. Treat. 2005, 4, 315–330. [Google Scholar] [CrossRef] [Green Version]
- Oka, N.; Soeda, A.; Inagaki, A.; Onodera, M.; Maruyama, H.; Hara, A.; Kunisada, T.; Mori, H.; Iwama, T. VEGF promotes tumorigenesis and angiogenesis of human glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2007, 360, 553–559. [Google Scholar] [CrossRef]
- Triozzi, P.L.; Borden, E.C. VB-111 for cancer. Expert Opin. Biol. Ther. 2011, 11, 1669–1676. [Google Scholar] [CrossRef]
- Arend, R.C.; Beer, H.M.; Cohen, Y.C.; Berlin, S.; Birrer, M.J.; Campos, S.M.; Rachmilewitz Minei, T.; Harats, D.; Wall, J.A.; Foxall, M.E.; et al. Ofranergene obadenovec (VB-111) in platinum-resistant ovarian cancer; favorable response rates in a phase I/II study are associated with an immunotherapeutic effect. Gynecol. Oncol. 2020, 157, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Brenner, A.; De Groot, J.F.; Butowski, N.A.; Zach, L.; Campian, J.L.; Ellingson, B.M.; Freedman, L.S.; Cohen, Y.C.; Lowenton-Spier, N. A randomized controlled phase III study of VB-111 combined with bevacizumab vs bevacizumab monotherapy in patients with recurrent glioblastoma (GLOBE). Neuro-Oncology 2020, 22, 705–717. [Google Scholar] [CrossRef]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.A.; Paleologos, N.; Nicholas, M.K.; Jensen, R. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, G.; Wei, Z.-P.; Pei, D.-S.; Xin, Y.; Liu, Y.-Q.; Zheng, J.-N. A novel approach to overcome temozolomide resistance in glioma and melanoma: Inactivation of MGMT by gene therapy. Biochem. Biophys. Res. Commun. 2011, 406, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Przystal, J.M.; Waramit, S.; Pranjol, M.Z.I.; Yan, W.; Chu, G.; Chongchai, A.; Samarth, G.; Olaciregui, N.G.; Tabatabai, G.; Carcaboso, A.M. Efficacy of systemic temozolomide-activated phage-targeted gene therapy in human glioblastoma. EMBO Mol. Med. 2019, 11, e8492. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Woo, J.S.; Jeong, C.H.; Ryu, C.H.; Jang, J.-D.; Jeun, S.-S. Potential application of temozolomide in mesenchymal stem cell-based TRAIL gene therapy against malignant glioma. Stem Cells Transl. Med. 2014, 3, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Peng, H.; Xu, P.; Zhang, L.; Fu, R.; Tu, H.; Guo, X.; Huang, K.; Lu, J.; Chen, H. Synthetic mRNA-based gene therapy for glioblastoma: TRAIL-mRNA synergistically enhances PTEN-mRNA-based therapy. Mol. Ther. 2022, 24, 707–718. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, S.; Fu, R.; Zhang, L.; Huang, K.; Peng, H.; Dai, L.; Chen, Q. Therapeutic prospects of mRNA-based gene therapy for glioblastoma. Front. Oncol. 2019, 9, 1208. [Google Scholar] [CrossRef]
- Long, Y.; Tao, H.; Karachi, A.; Grippin, A.J.; Jin, L.; Chang, Y.E.; Zhang, W.; Dyson, K.A.; Hou, A.Y.; Na, M. Dysregulation of glutamate transport enhances treg function that promotes VEGF blockade resistance in glioblastoma. Cancer Res. 2020, 80, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Tian, R.-F.; Li, X.-F.; Xu, C.; Wu, H.; Liu, L.; Wang, L.-H.; He, D.; Cao, K.; Cao, P.-G.; Ma, J.K. SiRNA targeting PFK1 inhibits proliferation and migration and enhances radiosensitivity by suppressing glycolysis in colorectal cancer. Am. J. Transl. Res. 2020, 12, 4923. [Google Scholar]
- Yang, B.; Hao, A.; Chen, L. Mirror siRNAs loading for dual delivery of doxorubicin and autophagy regulation siRNA for multidrug reversing chemotherapy. Biomed. Pharmacother. 2020, 130, 110490. [Google Scholar] [CrossRef]
- Önay Uçar, E.; Şengelen, A. Resveratrol and siRNA in combination reduces Hsp27 expression and induces caspase-3 activity in human glioblastoma cells. Cell Stress Chaperones 2019, 24, 763–775. [Google Scholar] [CrossRef]
- Zhang, C.; Yuan, W.; Wu, Y.; Wan, X.; Gong, Y. Co-delivery of EGFR and BRD4 siRNA by cell-penetrating peptides-modified redox-responsive complex in triple negative breast cancer cells. Life Sci. 2021, 266, 118886. [Google Scholar] [CrossRef]
- Mirzaei, S.; Mahabady, M.K.; Zabolian, A.; Abbaspour, A.; Fallahzadeh, P.; Noori, M.; Hashemi, F.; Hushmandi, K.; Daneshi, S.; Kumar, A.P.; et al. Small interfering RNA (siRNA) to target genes and molecular pathways in glioblastoma therapy: Current status with an emphasis on delivery systems. Life Sci. 2021, 275, 119368. [Google Scholar] [CrossRef]
- Huang, W.; Liang, Y.; Sang, C.; Mei, C.; Li, X.; Chen, T. Therapeutic nanosystems co-deliver anticancer drugs and oncogene SiRNA to achieve synergetic precise cancer chemo-gene therapy. J. Mater. Chem. B 2018, 6, 3013–3022. [Google Scholar] [CrossRef]
- Danhier, F.; Messaoudi, K.; Lemaire, L.; Benoit, J.-P.; Lagarce, F. Combined anti-Galectin-1 and anti-EGFR siRNA-loaded chitosan-lipid nanocapsules decrease temozolomide resistance in glioblastoma: In vivo evaluation. Int. J. Pharm. 2015, 481, 154–161. [Google Scholar] [CrossRef]
- Egorova, A.; Shubina, A.; Sokolov, D.; Selkov, S.; Baranov, V.; Kiselev, A. CXCR4-targeted modular peptide carriers for efficient anti-VEGF siRNA delivery. Int. J. Pharm. 2016, 515, 431–440. [Google Scholar] [CrossRef]
- Liu, E.K.; Sulman, E.P.; Wen, P.Y.; Kurz, S.C. Novel Therapies for Glioblastoma. Curr. Neurol. Neurosci. Rep. 2020, 20, 19. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Maxwell, R.; Jackson, C.M.; Lim, M. Clinical Trials Investigating Immune Checkpoint Blockade in Glioblastoma. Curr. Treat. Options Oncol. 2017, 18, 51. [Google Scholar] [CrossRef]
- Romani, M.; Pistillo, M.P.; Carosio, R.; Morabito, A.; Banelli, B. Immune Checkpoints and Innovative Therapies in Glioblastoma. Front. Oncol. 2018, 8, 464. [Google Scholar] [CrossRef] [PubMed]
- Lukas, R.V.; Rodon, J.; Becker, K.; Wong, E.T.; Shih, K.; Touat, M.; Fassò, M.; Osborne, S.; Molinero, L.; O’Hear, C. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J. Neuro-Oncol. 2018, 140, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Gardell, J.L.; Matsumoto, L.R.; Chinn, H.; DeGolier, K.R.; Kreuser, S.A.; Prieskorn, B.; Balcaitis, S.; Davis, A.; Ellenbogen, R.G.; Crane, C.A. Human macrophages engineered to secrete a bispecific T cell engager support antigen-dependent T cell responses to glioblastoma. J. Immunother. Cancer 2020, 8, e001202. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T cell therapy for solid tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Polyzoidis, S.; Ashkan, K. DCVax®-L—Developed by northwest biotherapeutics. Hum. Vaccin. Immunother. 2014, 10, 3139–3145. [Google Scholar] [CrossRef] [Green Version]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar]
- Jain, K.K. A critical overview of targeted therapies for glioblastoma. Front. Oncol. 2018, 8, 419. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Herndon, J.E., 2nd; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Gopalan, D.; Pandey, A.; Udupa, N.; Mutalik, S. Receptor specific, stimuli responsive and subcellular targeted approaches for effective therapy of Alzheimer: Role of surface engineered nanocarriers. J. Control Release 2020, 319, 183–200. [Google Scholar] [CrossRef]
- Angeli, E.; Nguyen, T.T.; Janin, A.; Bousquet, G. How to make anticancer drugs cross the blood–brain barrier to treat brain metastases. Int. J. Mol. Sci. 2019, 21, 22. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Li, K.; Li, T.; Chen, Z.; Wen, Y.; Liu, X.; Jia, X.; Zhang, Y.; Xu, Y.; Han, M.; et al. Monocyte-mediated chemotherapy drug delivery in glioblastoma. Nanomedicine 2017, 13, 157–178. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Cell-mediated delivery of nanoparticles: Taking advantage of circulatory cells to target nanoparticles. J. Control. Release 2014, 190, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Xie, Z.; Kim, G.B.; Dong, C.; Yang, J. Design strategies and applications of circulating cell-mediated drug delivery systems. ACS Biomater. Sci. Eng. 2015, 1, 201–217. [Google Scholar] [CrossRef] [Green Version]
- Ehtesham, M.; Kabos, P.; Gutierrez, M.A.R.; Chung, N.H.C.; Griffith, T.S.; Black, K.L.; Yu, J.S. Induction of Glioblastoma Apoptosis Using Neural Stem Cell-mediated Delivery of Tumor Necrosis Factor-related Apoptosis-inducing Ligand1. Cancer Res. 2002, 62, 7170–7174. [Google Scholar]
- Pang, L.; Qin, J.; Han, L.; Zhao, W.; Liang, J.; Xie, Z.; Yang, P.; Wang, J. Exploiting macrophages as targeted carrier to guide nanoparticles into glioma. Oncotarget 2016, 7, 37081–37091. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.; Fan, S.; Zheng, Y.; Liao, S.; Xiong, Y.; Li, Y.; Liu, J. Recent advances on glioblastoma multiforme and nano-drug carriers: A review. Curr. Med. Chem. 2019, 26, 5862–5874. [Google Scholar] [CrossRef]
- Reis, C.P.; Neufeld, R.J.; Ribeiro, A.J.; Veiga, F. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2006, 2, 8–21. [Google Scholar] [CrossRef] [Green Version]
- Frellsen, A.F.; Hansen, A.E.; Jølck, R.I.; Kempen, P.J.; Severin, G.W.; Rasmussen, P.H.; Kjær, A.; Jensen, A.T.I.; Andresen, T.L. Mouse Positron Emission Tomography Study of the Biodistribution of Gold Nanoparticles with Different Surface Coatings Using Embedded Copper-64. ACS Nano 2016, 10, 9887–9898. [Google Scholar] [CrossRef]
- Malikmammadov, E.; Tanir, T.E.; Kiziltay, A.; Hasirci, V.; Hasirci, N. PCL and PCL-based materials in biomedical applications. J. Biomater. Sci. Polym. Ed. 2018, 29, 863–893. [Google Scholar] [CrossRef]
- Bregoli, L.; Movia, D.; Gavigan-Imedio, J.D.; Lysaght, J.; Reynolds, J.; Prina-Mello, A. Nanomedicine applied to translational oncology: A future perspective on cancer treatment. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 81–103. [Google Scholar] [CrossRef] [Green Version]
- Rejman, J.; Oberle, V.; Zuhorn, I.S.; Hoekstra, D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem. J. 2004, 377, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.Z.; Zhu, K.J. A novel approach to prepare tripolyphosphate/chitosan complex beads for controlled release drug delivery. Int. J. Pharm. 2000, 201, 51–58. [Google Scholar] [CrossRef]
- Jo, D.H.; Kim, J.H.J.H.; Lee, T.G.; Kim, J.H.J.H. Size, surface charge, and shape determine therapeutic effects of nanoparticles on brain and retinal diseases. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Truong, N.P.; Whittaker, M.R.; Mak, C.W.; Davis, T.P. The importance of nanoparticle shape in cancer drug delivery. Expert Opin. Drug Deliv. 2015, 12, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.-F.; Chu, S.-M.; Liao, C.-C.; Wang, C.-J.; Wang, Y.-S.; Lai, M.-Y.; Wang, H.-C.; Huang, H.-R.; Tsai, M.-H. Nanotechnology and nanocarrier-based drug delivery as the potential therapeutic strategy for glioblastoma multiforme: An update. Cancers 2021, 13, 195. [Google Scholar] [CrossRef] [PubMed]
- Paroha, S.; Chandel, A.K.S.; Dubey, R.D. Nanosystems for drug delivery of coenzyme Q10. Environ. Chem. Lett. 2018, 16, 71–77. [Google Scholar] [CrossRef]
- Paroha, S.; Chandel, A.K.S.; Dubey, R.D. Nanotechnology delivery systems of coenzyme Q10: Pharmacokinetic and clinical implications. In Nanoscience in Food and Agriculture 4; Springer: Cham, Switzerland, 2017; pp. 213–228. [Google Scholar]
- Glaser, T.; Han, I.; Wu, L.; Zeng, X. Targeted nanotechnology in glioblastoma multiforme. Front. Pharmacol. 2017, 8, 166. [Google Scholar] [CrossRef] [Green Version]
- Hosseini, M.; Haji-Fatahaliha, M.; Jadidi-Niaragh, F.; Majidi, J.; Yousefi, M. The use of nanoparticles as a promising therapeutic approach in cancer immunotherapy. Artif. Cells Nanomed. Biotechnol. 2016, 44, 1051–1061. [Google Scholar] [CrossRef]
- Gonçalves, C.; Ramalho, M.J.; Silva, R.; Silva, V.; Marques-Oliveira, R.; Silva, A.C.; Pereira, M.C.; Loureiro, J.A. Lipid Nanoparticles Containing Mixtures of Antioxidants to Improve Skin Care and Cancer Prevention. Pharmaceutics 2021, 13, 2042. [Google Scholar] [CrossRef]
- Patidar, A.; Thakur, D.S.; Kumar, P.; Verma, J. A review on novel lipid based nanocarriers. Int. J. Pharm. Pharm. Sci. 2010, 2, 30–35. [Google Scholar]
- Jnaidi, R.; Almeida, A.J.; Gonçalves, L.M. Solid Lipid Nanoparticles and Nanostructured Lipid Carriers as Smart Drug Delivery Systems in the Treatment of Glioblastoma Multiforme. Pharmaceutics 2020, 12, 860. [Google Scholar] [CrossRef]
- Shevtsov, M.A.; Nikolaev, B.P.; Yakovleva, L.Y.; Marchenko, Y.Y.; Dobrodumov, A.V.; Mikhrina, A.L.; Martynova, M.G.; Bystrova, O.A.; Yakovenko, I.V.; Ischenko, A.M. Superparamagnetic iron oxide nanoparticles conjugated with epidermal growth factor (SPION-EGF) for targeting brain tumors. Int. J. Nanomedicine 2014, 9, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y. Self-Assembled Gold Nanoplexes for Cancer-Targeted SiRNA Delivery. Master’s Thesis, University of Southern Mississippi, Hattiesburg, MS, USA, 2014. Available online: https://aquila.usm.edu/cgi/viewcontent.cgi?article=1052&context=masters_theses (accessed on 30 April 2022).
- Guglielmelli, A.; Rosa, P.; Contardi, M.; Prato, M.; Mangino, G.; Miglietta, S.; Petrozza, V.; Pani, R.; Calogero, A.; Athanassiou, A. Biomimetic keratin gold nanoparticle-mediated in vitro photothermal therapy on glioblastoma multiforme. Nanomedicine 2020, 16, 121–138. [Google Scholar] [CrossRef]
- Liu, D.; Cheng, Y.; Cai, R.; Wang, W.; Cui, H.; Liu, M.; Mei, Q.; Zhou, S. The enhancement of siPLK1 penetration across BBB and its anti glioblastoma activity in vivo by magnet and transferrin co-modified nanoparticle. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhou, H.; Liu, Y.; Wen, Y.; Wei, C.; Yu, Q.; Liu, J. Transferrin/aptamer conjugated mesoporous ruthenium nanosystem for redox-controlled and targeted chemo-photodynamic therapy of glioma. Acta Biomater. 2018, 82, 143–157. [Google Scholar] [CrossRef]
- Taghipour-Sabzevar, V.; Sharifi, T.; Moghaddam, M.M. Polymeric nanoparticles as carrier for targeted and controlled delivery of anticancer agents. Ther. Deliv. 2019, 10, 527–550. [Google Scholar] [CrossRef]
- Shi, J.; Xiao, Z.; Kamaly, N.; Farokhzad, O.C. Self-assembled targeted nanoparticles: Evolution of technologies and bench to bedside translation. Acc. Chem. Res. 2011, 44, 1123–1134. [Google Scholar] [CrossRef]
- Chenthamara, D.; Subramaniam, S.; Ramakrishnan, S.G.; Krishnaswamy, S.; Essa, M.M.; Lin, F.-H.; Qoronfleh, M.W. Therapeutic efficacy of nanoparticles and routes of administration. Biomater. Res. 2019, 23, 1–29. [Google Scholar] [CrossRef]
- Owens, D.E., III; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef]
- Haji Mansor, M.; Najberg, M.; Contini, A.; Alvarez-Lorenzo, C.; Garcion, E.; Jérôme, C.; Boury, F. Development of a non-toxic and non-denaturing formulation process for encapsulation of SDF-1α into PLGA/PEG-PLGA nanoparticles to achieve sustained release. Eur. J. Pharm. Biopharm. 2018, 125, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Pourgholi, F.; Hajivalili, M.; Farhad, J.-N.; Kafil, H.S.; Yousefi, M. Nanoparticles: Novel vehicles in treatment of Glioblastoma. Biomed. Pharmacother. 2016, 77, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Couvreur, P.; Vauthier, C. Nanotechnology: Intelligent design to treat complex disease. Pharm. Res. 2006, 23, 1417–1450. [Google Scholar] [CrossRef]
- Rescignano, N.; Fortunati, E.; Armentano, I.; Hernandez, R.; Mijangos, C.; Pasquino, R.; Kenny, J.M. Use of alginate, chitosan and cellulose nanocrystals as emulsion stabilizers in the synthesis of biodegradable polymeric nanoparticles. J. Colloid Interface Sci. 2015, 445, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Allouss, D.; Makhado, E.; Zahouily, M. Recent Progress in Polysaccharide-Based Hydrogel Beads as Adsorbent for Water Pollution Remediation. Funct. Polym. Nanocomposites Wastewater Treat. 2022, 323, 55–88. [Google Scholar]
- He, W.; Hosseinkhani, H.; Mohammadinejad, R.; Roveimiab, Z.; Hueng, D.; Ou, K.; Domb, A.J. Polymeric nanoparticles for therapy and imaging. Polym. Adv. Technol. 2014, 25, 1216–1225. [Google Scholar] [CrossRef]
- Elzoghby, A.O.; Abd-Elwakil, M.M.; Abd-Elsalam, K.; Elsayed, M.T.; Hashem, Y.; Mohamed, O. Natural polymeric nanoparticles for brain-targeting: Implications on drug and gene delivery. Curr. Pharm. Des. 2016, 22, 3305–3323. [Google Scholar] [CrossRef]
- Morshed, R.; Cheng, Y.; Auffinger, B.; Wegscheid, M.; Lesniak, M.S. The potential of polymeric micelles in the context of glioblastoma therapy. Front. Pharmacol. 2013, 4, 157. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, N.; Matsumura, Y.; Kataoka, K. Development of polymeric micelles for targeting intractable cancers. Cancer Sci. 2016, 107, 867–874. [Google Scholar] [CrossRef]
- Saxena, V.; Hussain, M.D. Formulation and in vitro evaluation of 17-allyamino-17-demethoxygeldanamycin (17-AAG) loaded polymeric mixed micelles for glioblastoma multiforme. Colloids Surf. B Biointerfaces 2013, 112, 350–355. [Google Scholar] [CrossRef]
- Fu, Z.; Xiang, J. Aptamer-functionalized nanoparticles in targeted delivery and cancer therapy. Int. J. Mol. Sci. 2020, 21, 9123. [Google Scholar] [CrossRef] [PubMed]
- Stenström, P.; Manzanares, D.; Zhang, Y.; Ceña, V.; Malkoch, M. Evaluation of amino-functional polyester dendrimers based on Bis-MPA as nonviral vectors for siRNA delivery. Molecules 2018, 23, 2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torchilin, V.P. Nanocarriers. Pharm. Res. 2007, 24, 2333–2334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qu, H.; Xue, X. Blood–brain barrier penetrating liposomes with synergistic chemotherapy for glioblastoma treatment. Biomater. Sci. 2022, 10, 423–434. [Google Scholar] [CrossRef]
- Papachristodoulou, A.; Signorell, R.D.; Werner, B.; Brambilla, D.; Luciani, P.; Cavusoglu, M.; Grandjean, J.; Silginer, M.; Rudin, M.; Martin, E.; et al. Chemotherapy sensitization of glioblastoma by focused ultrasound-mediated delivery of therapeutic liposomes. J. Control. Release 2019, 295, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Jhaveri, A.; Deshpande, P.; Pattni, B.; Torchilin, V. Transferrin-targeted, resveratrol-loaded liposomes for the treatment of glioblastoma. J. Control. Release 2018, 277, 89–101. [Google Scholar] [CrossRef]
- Shirazi, A.S.; Varshochian, R.; Rezaei, M.; Ardakani, Y.H.; Dinarvand, R. SN38 loaded nanostructured lipid carriers (NLCs); preparation and in vitro evaluations against glioblastoma. J. Mater. Sci. Mater. Med. 2021, 32, 78. [Google Scholar] [CrossRef]
- Paraiso, W.K.D.; Garcia-Chica, J.; Ariza, X.; Zagmutt, S.; Fukushima, S.; Garcia, J.; Mochida, Y.; Serra, D.; Herrero, L.; Kinoh, H. Poly-ion complex micelles effectively deliver CoA-conjugated CPT1A inhibitors to modulate lipid metabolism in brain cells. Biomater. Sci. 2021, 9, 7076–7091. [Google Scholar] [CrossRef]
- Song, S.; Mao, G.; Du, J.; Zhu, X. Novel RGD containing, temozolomide-loading nanostructured lipid carriers for glioblastoma multiforme chemotherapy. Drug Deliv. 2016, 23, 1404–1408. [Google Scholar] [CrossRef] [Green Version]
- Zwain, T.; Alder, J.E.; Sabagh, B.; Shaw, A.; Burrow, A.J.; Singh, K.K. Tailoring functional nanostructured lipid carriers for glioblastoma treatment with enhanced permeability through in-vitro 3D BBB/BBTB models. Mater. Sci. Eng. C 2021, 121, 111774. [Google Scholar] [CrossRef]
- Kuo, Y.-C.; Liang, C.-T. Inhibition of human brain malignant glioblastoma cells using carmustine-loaded catanionic solid lipid nanoparticles with surface anti-epithelial growth factor receptor. Biomaterials 2011, 32, 3340–3350. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, Y.; Liu, Y.; Zou, Y.; Zheng, M.; Shi, B. Recent advances in polymeric nanomedicines for glioblastoma therapy. Sci. Sin. Vitae 2021, 51, 819–835. [Google Scholar] [CrossRef]
- Han, H.; Zhang, Y.; Jin, S.; Chen, P.; Liu, S.; Xie, Z.; Jing, X.; Wang, Z. Paclitaxel-loaded dextran nanoparticles decorated with RVG29 peptide for targeted chemotherapy of glioma: An in vivo study. New J. Chem. 2020, 44, 5692–5701. [Google Scholar] [CrossRef]
- Esfandiarpour-Boroujeni, S.; Bagheri-Khoulenjani, S.; Mirzadeh, H.; Amanpour, S. Fabrication and study of curcumin loaded nanoparticles based on folate-chitosan for breast cancer therapy application. Carbohydr. Polym. 2017, 168, 14–21. [Google Scholar] [CrossRef]
- Sekar, V.; Rajendran, K.; Vallinayagam, S.; Deepak, V.; Mahadevan, S. Synthesis and characterization of chitosan ascorbate nanoparticles for therapeutic inhibition for cervical cancer and their in silico modeling. J. Ind. Eng. Chem. 2018, 62, 239–249. [Google Scholar] [CrossRef]
- Yin, T.; Bader, A.R.; Hou, T.K.; Maron, B.A.; Kao, D.D.; Qian, R.; Kohane, D.S.; Handy, D.E.; Loscalzo, J.; Zhang, Y.-Y. SDF-1α in glycan nanoparticles exhibits full activity and reduces pulmonary hypertension in rats. Biomacromolecules 2013, 14, 4009–4020. [Google Scholar] [CrossRef] [Green Version]
- Saulnier, P.; Benoit, J. Active targeting of brain tumors using nanocarriers. Biomaterials 2007, 28, 4947–4967. [Google Scholar]
- Geldenhuys, W.; Wehrung, D.; Groshev, A.; Hirani, A.; Sutariya, V. Brain-targeted delivery of doxorubicin using glutathione-coated nanoparticles for brain cancers. Pharm. Dev. Technol. 2015, 20, 497–506. [Google Scholar] [CrossRef]
- Ramalho, M.J.; Sevin, E.; Gosselet, F.; Lima, J.; Coelho, M.A.N.; Loureiro, J.A.; Pereira, M.C. Receptor-mediated PLGA nanoparticles for glioblastoma multiforme treatment. Int. J. Pharm. 2018, 545, 84–92. [Google Scholar] [CrossRef]
- Caban-Toktas, S.; Sahin, A.; Lule, S.; Esendagli, G.; Vural, I.; Oguz, K.K.; Soylemezoglu, F.; Mut, M.; Dalkara, T.; Khan, M. Combination of Paclitaxel and R-flurbiprofen loaded PLGA nanoparticles suppresses glioblastoma growth on systemic administration. Int. J. Pharm. 2020, 578, 119076. [Google Scholar] [CrossRef]
- Gascon, S.; Solano, A.G.; El Kheir, W.; Therriault, H.; Berthelin, P.; Cattier, B.; Marcos, B.; Virgilio, N.; Paquette, B.; Faucheux, N.; et al. Characterization and mathematical modeling of alginate/chitosan-based nanoparticles releasing the chemokine cxcl12 to attract glioblastoma cells. Pharmaceutics 2020, 12, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alghamri, M.S.; Banerjee, K.; Mujeeb, A.A.; Mauser, A.; Taher, A.; Thalla, R.; McClellan, B.L.; Varela, M.L.; Stamatovic, S.M.; Martinez-Revollar, G.; et al. Systemic Delivery of an Adjuvant CXCR4-CXCL12 Signaling Inhibitor Encapsulated in Synthetic Protein Nanoparticles for Glioma Immunotherapy. ACS Nano 2022. [Google Scholar] [CrossRef] [PubMed]
- Malinovskaya, Y.; Melnikov, P.; Baklaushev, V.; Gabashvili, A.; Osipova, N.; Mantrov, S.; Ermolenko, Y.; Maksimenko, O.; Gorshkova, M.; Balabanyan, V. Delivery of doxorubicin-loaded PLGA nanoparticles into U87 human glioblastoma cells. Int. J. Pharm. 2017, 524, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Wang, L.; Zhao, Y.; Meng, D.; Li, D.; Li, H.; Zhang, B.; Shi, J.; Zhang, H.; Zhang, Z. Targeted imaging and chemo-phototherapy of brain cancer by a multifunctional drug delivery system. Macromol. Biosci. 2015, 15, 1571–1585. [Google Scholar] [CrossRef]
- Xu, H.-L.; ZhuGe, D.-L.; Chen, P.-P.; Tong, M.-Q.; Lin, M.-T.; Jiang, X.; Zheng, Y.-W.; Chen, B.; Li, X.-K.; Zhao, Y.-Z. Silk fibroin nanoparticles dyeing indocyanine green for imaging-guided photo-thermal therapy of glioblastoma. Drug Deliv. 2018, 25, 364–375. [Google Scholar] [CrossRef]
- Bastiancich, C.; Bozzato, E.; Henley, I.; Newland, B. Does local drug delivery still hold therapeutic promise for brain cancer? A systematic review. J. Control. Release 2021, 337, 296–305. [Google Scholar] [CrossRef]
- Bregy, A.; Shah, A.H.; Diaz, M.V.; Pierce, H.E.; Ames, P.L.; Diaz, D.; Komotar, R.J. The role of Gliadel wafers in the treatment of high-grade gliomas. Expert Rev. Anticancer Ther. 2013, 13, 1453–1461. [Google Scholar] [CrossRef]
- Alghamdi, M.; Gumbleton, M.; Newland, B. Local delivery to malignant brain tumors: Potential biomaterial-based therapeutic/adjuvant strategies. Biomater. Sci. 2021, 9, 6037–6051. [Google Scholar] [CrossRef]
- Bruinsmann, F.A.; Richter Vaz, G.; de Cristo Soares Alves, A.; Aguirre, T.; Raffin Pohlmann, A.; Stanisçuaski Guterres, S.; Sonvico, F. Nasal Drug Delivery of Anticancer Drugs for the Treatment of Glioblastoma: Preclinical and Clinical Trials. Molecules 2019, 24, 4312. [Google Scholar] [CrossRef] [Green Version]
- Blacher, E.; Ben Baruch, B.; Levy, A.; Geva, N.; Green, K.D.; Garneau-Tsodikova, S.; Fridman, M.; Stein, R. Inhibition of glioma progression by a newly discovered CD38 inhibitor. Int. J. Cancer 2015, 136, 1422–1433. [Google Scholar] [CrossRef]
- Levy, A.; Blacher, E.; Vaknine, H.; Lund, F.E.; Stein, R.; Mayo, L. CD38 deficiency in the tumor microenvironment attenuates glioma progression and modulates features of tumor-associated microglia/macrophages. Neuro-Oncology 2012, 14, 1037–1049. [Google Scholar] [CrossRef]
- Mayo, L.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Moutin, M.-J.; Lund, F.E.; Stein, R. Dual role of CD38 in microglial activation and activation-induced cell death. J. Immunol. 2008, 181, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Gao, Y.; Liu, G.; Zhou, X.; Wang, Y.; Ma, L. Intranasal administration of temozolomide for brain-targeting delivery: Therapeutic effect on glioma in rats. J. South. Med. Univ. 2014, 34, 631–635. [Google Scholar]
- Ferreira, N.N.; Granja, S.; Boni, F.I.; Prezotti, F.G.; Ferreira, L.M.B.; Cury, B.S.F.; Reis, R.M.; Baltazar, F.; Gremião, M.P.D. Modulating chitosan-PLGA nanoparticle properties to design a co-delivery platform for glioblastoma therapy intended for nose-to-brain route. Drug Deliv. Transl. Res. 2020, 10, 1729–1747. [Google Scholar] [CrossRef]
- Alex, A.T.; Joseph, A.; Shavi, G.; Rao, J.V.; Udupa, N. Development and evaluation of carboplatin-loaded PCL nanoparticles for intranasal delivery. Drug Deliv. 2016, 23, 2144–2153. [Google Scholar] [CrossRef]
- de Oliveira Junior, E.R.; Nascimento, T.L.; Salomão, M.A.; da Silva, A.C.G.; Valadares, M.C.; Lima, E.M. Increased nose-to-brain delivery of melatonin mediated by polycaprolactone nanoparticles for the treatment of glioblastoma. Pharm. Res. 2019, 36, 131. [Google Scholar] [CrossRef]
- Madane, R.G.; Mahajan, H.S. Curcumin-loaded nanostructured lipid carriers (NLCs) for nasal administration: Design, characterization, and in vivo study. Drug Deliv. 2016, 23, 1326–1334. [Google Scholar] [CrossRef]
- Ferreira, N.N.; de Oliveira Junior, E.; Granja, S.; Boni, F.I.; Ferreira, L.M.B.; Cury, B.S.F.; Santos, L.C.R.; Reis, R.M.; Lima, E.M.; Baltazar, F.; et al. Nose-to-brain co-delivery of drugs for glioblastoma treatment using nanostructured system. Int. J. Pharm. 2021, 603, 120714. [Google Scholar] [CrossRef]
- Sousa, F.; Dhaliwal, H.K.; Gattacceca, F.; Sarmento, B.; Amiji, M.M. Enhanced anti-angiogenic effects of bevacizumab in glioblastoma treatment upon intranasal administration in polymeric nanoparticles. J. Control. Release 2019, 309, 37–47. [Google Scholar] [CrossRef]
- D’Amico, R.S.; Aghi, M.K.; Vogelbaum, M.A.; Bruce, J.N. Convection-enhanced drug delivery for glioblastoma: A review. J. Neuro-Oncol. 2021, 151, 415–427. [Google Scholar] [CrossRef]
- Ung, T.H.; Malone, H.; Canoll, P.; Bruce, J.N. Convection-enhanced delivery for glioblastoma: Targeted delivery of antitumor therapeutics. CNS Oncol. 2015, 4, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Chakroun, R.W.; Zhang, P.; Lin, R.; Schiapparelli, P.; Quinones-Hinojosa, A.; Cui, H. Nanotherapeutic systems for local treatment of brain tumors. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 10, e1479. [Google Scholar] [CrossRef] [PubMed]
- Debinski, W.; Tatter, S.B. Convection-enhanced delivery for the treatment of brain tumors. Expert Rev. Neurother. 2009, 9, 1519–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobo, R.H.; Laske, D.W.; Akbasak, A.; Morrison, P.F.; Dedrick, R.L.; Oldfield, E.H. Convection-enhanced delivery of macromolecules in the brain. Proc. Natl. Acad. Sci. USA 1994, 91, 2076–2080. [Google Scholar] [CrossRef] [Green Version]
- Lidar, Z.; Mardor, Y.; Jonas, T.; Pfeffer, R.; Faibel, M.; Nass, D.; Hadani, M.; Ram, Z. Convection-enhanced delivery of paclitaxel for the treatment of recurrent malignant glioma: A Phase I/II clinical study. J. Neurosurg. 2004, 100, 472–479. [Google Scholar] [CrossRef]
- Séhédic, D.; Chourpa, I.; Tétaud, C.; Griveau, A.; Loussouarn, C.; Avril, S.; Legendre, C.; Lepareur, N.; Wion, D.; Hindré, F.; et al. Locoregional Confinement and Major Clinical Benefit of (188)Re-Loaded CXCR4-Targeted Nanocarriers in an Orthotopic Human to Mouse Model of Glioblastoma. Theranostics 2017, 7, 4517–4536. [Google Scholar] [CrossRef]
- Zhang, C.; Nance, E.A.; Mastorakos, P.; Chisholm, J.; Berry, S.; Eberhart, C.; Tyler, B.; Brem, H.; Suk, J.S.; Hanes, J. Convection enhanced delivery of cisplatin-loaded brain penetrating nanoparticles cures malignant glioma in rats. J. Control. Release 2017, 263, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Stephen, Z.R.; Kievit, F.M.; Veiseh, O.; Chiarelli, P.A.; Fang, C.; Wang, K.; Hatzinger, S.J.; Ellenbogen, R.G.; Silber, J.R.; Zhang, M. Redox-responsive magnetic nanoparticle for targeted convection-enhanced delivery of O 6-benzylguanine to brain tumors. ACS Nano 2014, 8, 10383–10395. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-Y.; Ozawa, T.; Drummond, D.C.; Kalra, A.; Fitzgerald, J.B.; Kirpotin, D.B.; Wei, K.-C.; Butowski, N.; Prados, M.D.; Berger, M.S.; et al. Comparing routes of delivery for nanoliposomal irinotecan shows superior anti-tumor activity of local administration in treating intracranial glioblastoma xenografts. Neuro-Oncology 2013, 15, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Cruickshank, G.; Fayeye, O.; Ngoga, D.; Connor, J.; Detta, A. ATNT-05: Intraoperative Intraparenchymal Injection of Irinotecan Drug Loaded Beads in Patients with Recurrent Glioblastoma (Gbm): A Safe New Depot Approach for Loco-Regional Therapy (NCT02433392). Neuro-Oncology 2015, 17, v11. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.G.; Kim, K.H.; Seo, Y.J.; Yang, H.; Marcusson, E.G.; Son, E.; Lee, K.; Sa, J.K.; Lee, H.W.; Nam, D.-H. Anti-miR delivery strategies to bypass the blood-brain barrier in glioblastoma therapy. Oncotarget 2016, 7, 29400. [Google Scholar] [CrossRef]
- Wait, S.D.; Prabhu, R.S.; Burri, S.H.; Atkins, T.G.; Asher, A.L. Polymeric drug delivery for the treatment of glioblastoma. Neuro-Oncology 2015, 17, ii9–ii23. [Google Scholar] [CrossRef] [Green Version]
- Valtonen, S.; Timonen, U.I.; Toivanen, P.; Kalimo, H.; Kivipelto, L.; Heiskanen, O.; Unsgaard, G.; Kuurne, T. Interstitial chemotherapy with carmustine-loaded polymers for high-grade gliomas: A randomized double-blind study. Neurosurgery 1997, 41, 44–49. [Google Scholar] [CrossRef]
- Walter, K.A.; Cahan, M.A.; Gur, A.; Tyler, B.; Hilton, J.; Colvin, O.M.; Burger, P.C.; Domb, A.; Brem, H. Interstitial taxol delivered from a biodegradable polymer implant against experimental malignant glioma. Cancer Res. 1994, 54, 2207–2212. [Google Scholar]
- Mangraviti, A.; Raghavan, T.; Volpin, F.; Skuli, N.; Gullotti, D.; Zhou, J.; Asnaghi, L.; Sankey, E.; Liu, A.; Wang, Y. HIF-1α-targeting acriflavine provides long term survival and radiological tumor response in brain cancer therapy. Sci. Rep. 2017, 7, 14978. [Google Scholar]
- Lesniak, M.S.; Upadhyay, U.; Goodwin, R.; Tyler, B.; Brem, H. Local delivery of doxorubicin for the treatment of malignant brain tumors in rats. Anticancer Res. 2005, 25, 3825–3831. [Google Scholar]
- Ranganath, S.H.; Fu, Y.; Arifin, D.Y.; Kee, I.; Zheng, L.; Lee, H.-S.; Chow, P.K.-H.; Wang, C.-H. The use of submicron/nanoscale PLGA implants to deliver paclitaxel with enhanced pharmacokinetics and therapeutic efficacy in intracranial glioblastoma in mice. Biomaterials 2010, 31, 5199–5207. [Google Scholar] [CrossRef]
- Ranganath, S.H.; Wang, C.-H. Biodegradable microfiber implants delivering paclitaxel for post-surgical chemotherapy against malignant glioma. Biomaterials 2008, 29, 2996–3003. [Google Scholar] [CrossRef]
- Shapira-Furman, T.; Serra, R.; Gorelick, N.; Doglioli, M.; Tagliaferri, V.; Cecia, A.; Peters, M.; Kumar, A.; Rottenberg, Y.; Langer, R. Biodegradable wafers releasing Temozolomide and Carmustine for the treatment of brain cancer. J. Control. Release 2019, 295, 93–101. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Khademhosseini, A. Advances in engineering hydrogels. Science 2017, 356, eaaf3627. [Google Scholar] [CrossRef]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2012, 64, 18–23. [Google Scholar] [CrossRef]
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv. Mater. 2006, 18, 1345–1360. [Google Scholar] [CrossRef]
- Peppas, N.A.; Bures, P.; Leobandung, W.S.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Wang, C.; Tong, X.; Yang, F. Bioengineered 3D brain tumor model to elucidate the effects of matrix stiffness on glioblastoma cell behavior using PEG-based hydrogels. Mol. Pharm. 2014, 11, 2115–2125. [Google Scholar] [CrossRef]
- Hosseinzadeh, R.; Mirani, B.; Pagan, E.; Mirzaaghaei, S.; Nasimian, A.; Kawalec, P.; da Silva Rosa, S.C.; Hamdi, D.; Fernandez, N.P.; Toyota, B.D. A drug-eluting 3D-printed mesh (GlioMesh) for management of glioblastoma. Adv. Ther. 2019, 2, 1900113. [Google Scholar] [CrossRef]
- Schiapparelli, P.; Zhang, P.; Lara-Velazquez, M.; Guerrero-Cazares, H.; Lin, R.; Su, H.; Chakroun, R.W.; Tusa, M.; Quiñones-Hinojosa, A.; Cui, H. Self-assembling and self-formulating prodrug hydrogelator extends survival in a glioblastoma resection and recurrence model. J. Control. Release 2020, 319, 311–321. [Google Scholar] [CrossRef]
- Turabee, M.H.; Jeong, T.H.; Ramalingam, P.; Kang, J.H.; Ko, Y.T. N, N, N-trimethyl chitosan embedded in situ Pluronic F127 hydrogel for the treatment of brain tumor. Carbohydr. Polym. 2019, 203, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Bastiancich, C.; Danhier, P.; Préat, V.; Danhier, F. Anticancer drug-loaded hydrogels as drug delivery systems for the local treatment of glioblastoma. J. Control. Release 2016, 243, 29–42. [Google Scholar] [CrossRef]
- Akbar, U.; Jones, T.; Winestone, J.; Michael, M.; Shukla, A.; Sun, Y.; Duntsch, C. Delivery of temozolomide to the tumor bed via biodegradable gel matrices in a novel model of intracranial glioma with resection. J. Neuro-Oncol. 2009, 94, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Bouché, M.; Dong, Y.C.; Sheikh, S.; Taing, K.; Saxena, D.; Hsu, J.C.; Chen, M.H.; Salinas, R.D.; Song, H.; Burdick, J.A.; et al. Novel Treatment for Glioblastoma Delivered by a Radiation Responsive and Radiopaque Hydrogel. ACS Biomater. Sci. Eng. 2021, 7, 3209–3220. [Google Scholar] [CrossRef] [PubMed]
- Vellimana, A.K.; Recinos, V.R.; Hwang, L.; Fowers, K.D.; Li, K.W.; Zhang, Y.; Okonma, S.; Eberhart, C.G.; Brem, H.; Tyler, B.M. Combination of paclitaxel thermal gel depot with temozolomide and radiotherapy significantly prolongs survival in an experimental rodent glioma model. J. Neuro-Oncol. 2013, 111, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Zentner, G.M.; Rathi, R.; Shih, C.; McRea, J.C.; Seo, M.-H.; Oh, H.; Rhee, B.G.; Mestecky, J.; Moldoveanu, Z.; Morgan, M. Biodegradable block copolymers for delivery of proteins and water-insoluble drugs. J. Control. Release 2001, 72, 203–215. [Google Scholar] [CrossRef]
- Déry, L.; Charest, G.; Guérin, B.; Akbari, M.; Fortin, D. Chemoattraction of Neoplastic Glial Cells with CXCL10, CCL2 and CCL11 as a Paradigm for a Promising Therapeutic Approach for Primary Brain Tumors. Int. J. Mol. Sci. 2021, 22, 12150. [Google Scholar] [CrossRef]
- Ye, E.; Loh, X.J. Polymeric hydrogels and nanoparticles: A merging and emerging field. Aust. J. Chem. 2013, 66, 997–1007. [Google Scholar] [CrossRef]
- Najberg, M.; Mansor, M.H.; Boury, F.; Alvarez-Lorenzo, C.; Garcion, E. Reversing the Tumor Target: Establishment of a Tumor Trap. Front. Pharmacol. 2019, 10, 887. [Google Scholar] [CrossRef] [Green Version]
- Brachi, G.; Ruiz-Ramírez, J.; Dogra, P.; Wang, Z.; Cristini, V.; Ciardelli, G.; Rostomily, R.C.; Ferrari, M.; Mikheev, A.M.; Blanco, E. Intratumoral injection of hydrogel-embedded nanoparticles enhances retention in glioblastoma. Nanoscale 2020, 12, 23838–23850. [Google Scholar] [CrossRef]
- Zhao, M.; Danhier, F.; Bastiancich, C.; Joudiou, N.; Ganipineni, L.P.; Tsakiris, N.; Gallez, B.; des Rieux, A.; Jankovski, A.; Bianco, J.; et al. Post-resection treatment of glioblastoma with an injectable nanomedicine-loaded photopolymerizable hydrogel induces long-term survival. Int. J. Pharm. 2018, 548, 522–529. [Google Scholar] [CrossRef]
- Nutan, B.; Chandel, A.K.S.; Jewrajka, S.K. Liquid prepolymer-based in situ formation of degradable poly (ethylene glycol)-linked-poly (caprolactone)-linked-poly (2-dimethylaminoethyl) methacrylate amphiphilic conetwork gels showing polarity driven gelation and bioadhesion. ACS Appl. Bio Mater. 2018, 1, 1606–1619. [Google Scholar] [CrossRef]
- Chandel, A.K.S.; Kannan, D.; Nutan, B.; Singh, S.; Jewrajka, S.K. Dually crosslinked injectable hydrogels of poly (ethylene glycol) and poly [(2-dimethylamino) ethyl methacrylate]-b-poly (N-isopropyl acrylamide) as a wound healing promoter. J. Mater. Chem. B 2017, 5, 4955–4965. [Google Scholar] [CrossRef]
- Chandel, A.K.S.; Nutan, B.; Raval, I.H.; Jewrajka, S.K. Self-Assembly of Partially Alkylated Dextran- graft -poly[(2-dimethylamino)ethyl methacrylate] Copolymer Facilitating Hydrophobic/Hydrophilic Drug Delivery and Improving Conetwork Hydrogel Properties. Biomacromolecules 2018, 19, 1142–1153. [Google Scholar] [CrossRef]
- Giarra, S.; Ierano, C.; Biondi, M.; Napolitano, M.; Campani, V.; Pacelli, R.; Scala, S.; De Rosa, G.; Mayol, L. Engineering of thermoresponsive gels as a fake metastatic niche. Carbohydr. Polym. 2018, 191, 112–118. [Google Scholar] [CrossRef]
- Molina-Peña, R.; Haji Mansor, M.; Najberg, M.; Thomassin, J.M.; Gueza, B.; Alvarez-Lorenzo, C.; Garcion, E.; Jérôme, C.; Boury, F. Nanoparticle-containing electrospun nanofibrous scaffolds for sustained release of SDF-1α. Int. J. Pharm. 2021, 610, 121205. [Google Scholar] [CrossRef]





| Axis | Actions in GBM | |||
|---|---|---|---|---|
| Migration and Invasiveness | Proliferation | Growth, Survival, and Apoptosis | Ref | |
| CXCL12–CXCR4 | Chemotaxis | Activation of Ras, Raf kinase | Ca2 + mobilization via inhibition of cAMP (survival) | [91] |
| ERK1/2 phosphorylation | ||||
| CXCL12–CXCR7 | Activation of β-arrestin by heterodimerization with CXCR4 | Activation of ERK1/2 via GRK | [92,93] | |
| Activation of ERK1/2 | ||||
| CXCL8–CXCR1-2 | Overexpression of MMP-9 and MMP-2 | High density of macrophage promotes a high degree of microvascular proliferation | Activation of IL-6 | [94] |
| Activation of JAK pathway | ||||
| EMT transition | Increase of anti-apoptotic protein secretion | |||
| CXCL16–CXCR6 | Overexpression of anti-inflammatory genes and modulating microglia polarization | Establishing a pro-tumoral microenvironment in the brain | [95] | |
| Increase of MMP-9 and MMP-2 expression | ||||
| CCL5–CCR5 | Activation of Akt kinase | Stimulation of AKT pathway | [94,96] | |
| CX3CL1–CX3CR1 | Modulation of the activation of TGF-beta1 | Not clear | CX3CR1 polymorphism through isoleucine V249I (survival) | [97,98] |
| GBM Study Models | Advantages | Disadvantages | Applications | Ref | ||
|---|---|---|---|---|---|---|
| 2D | In vitro | Scratch assays | • Easily implemented • Low cost • Real-time cell tracking | • Low physiological relevance • Migration and invasion are not distinguished | Migration Matrix remodeling Drug screening | [137] |
| Transwell assays | • Technically easy • Low cost • A matrix can be added to study ECM | • Lacks tumor complexity • Lack of 3D environment without matrix • Difficult to distinguish between migration and invasion | ||||
| 3D | 3D bioscaffolds (Spheroids) | • Simple • Control of spheroids size • Control of growth parameters • High throughput • Allow ECM interactions study • Allow pathways signaling study | • Long-term culture difficulties • Lack tumors complexity • Lack vascularization | Migration/Invasion Cell biology (proliferation, apoptosis, etc.) Drug Screening | [138] | |
| Microfluidic co-culture | • Real-time cell tracking • Allow ECM interactions study • Allow pathways signaling study • Cell–Cell interactions study | • High cost • Lack of native microenvironment • Lack of tumor complexity | Migration/Invasion Cell adhesion | [139] | ||
| Ex vivo | Organotypic brain slices cultures | • Native ECM composition • Real-time cells tracking • Maintain tumor heterogeneity • Allow ECM interactions study • Allow cell signaling study | • Ethical issues associated with animal studies • Lacks blood • Flow • Lack of tumor complexity | Migration/Invasion Tumor growth Study of drugs delivery | [140] | |
| In vivo | Orthotopic xenograft | • Native microenvironment • Studies under the effect of the flow • Cell–Cell interactions study • Allow pathway’s signaling study | • Ethical issues associated with animal studies • High cost • Less experimental control • Time consuming | Migration/Invasion Signaling Study of drugs delivery Tumor growth | [141] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Kheir, W.; Marcos, B.; Virgilio, N.; Paquette, B.; Faucheux, N.; Lauzon, M.-A. Drug Delivery Systems in the Development of Novel Strategies for Glioblastoma Treatment. Pharmaceutics 2022, 14, 1189. https://doi.org/10.3390/pharmaceutics14061189
El Kheir W, Marcos B, Virgilio N, Paquette B, Faucheux N, Lauzon M-A. Drug Delivery Systems in the Development of Novel Strategies for Glioblastoma Treatment. Pharmaceutics. 2022; 14(6):1189. https://doi.org/10.3390/pharmaceutics14061189
Chicago/Turabian StyleEl Kheir, Wiam, Bernard Marcos, Nick Virgilio, Benoit Paquette, Nathalie Faucheux, and Marc-Antoine Lauzon. 2022. "Drug Delivery Systems in the Development of Novel Strategies for Glioblastoma Treatment" Pharmaceutics 14, no. 6: 1189. https://doi.org/10.3390/pharmaceutics14061189