
Pharmacogenetics and Pain Treatment with a Focus on Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) and Antidepressants: A Systematic Review

 , , , ,
, , , ,
Abstract
:1. Introduction
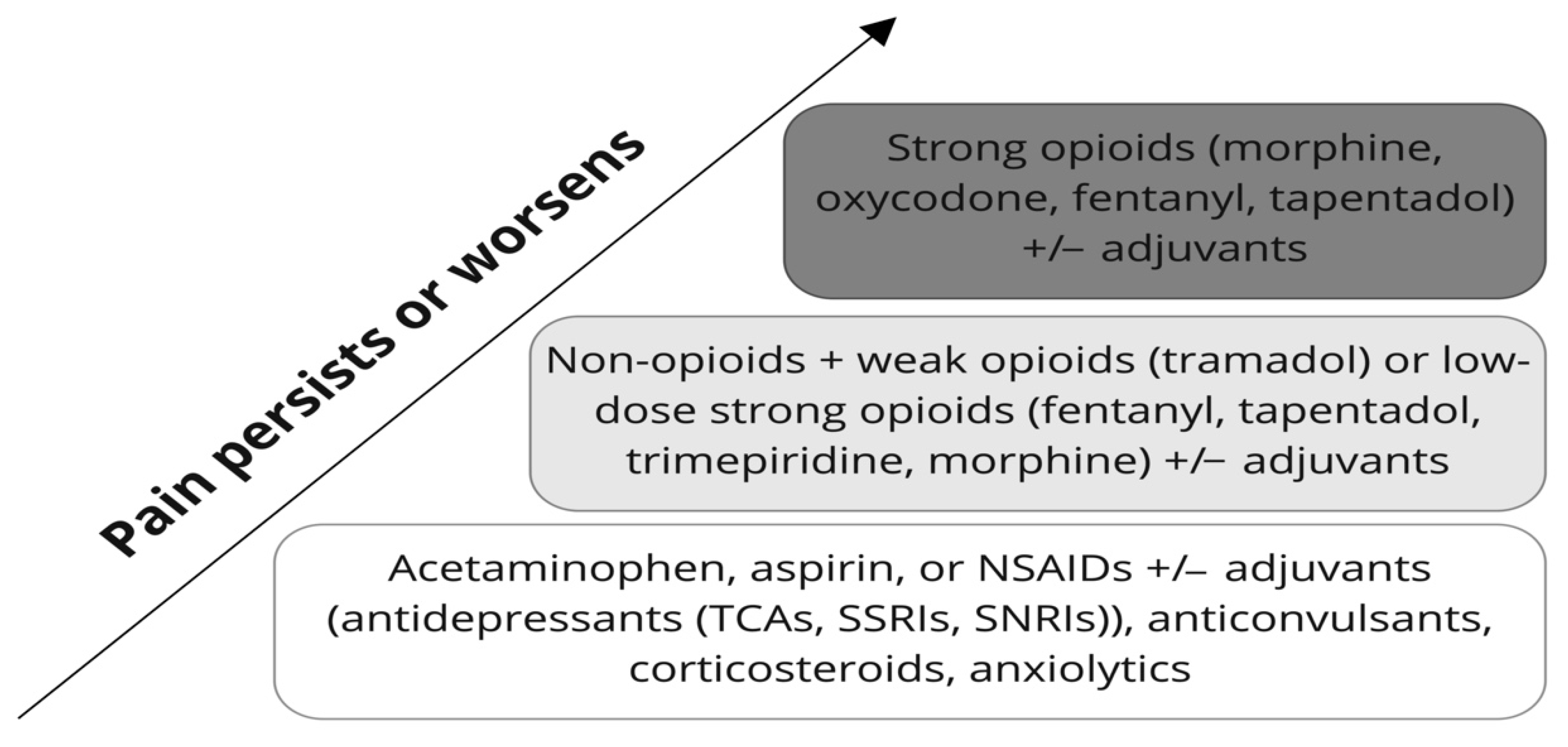
2. Pain Management
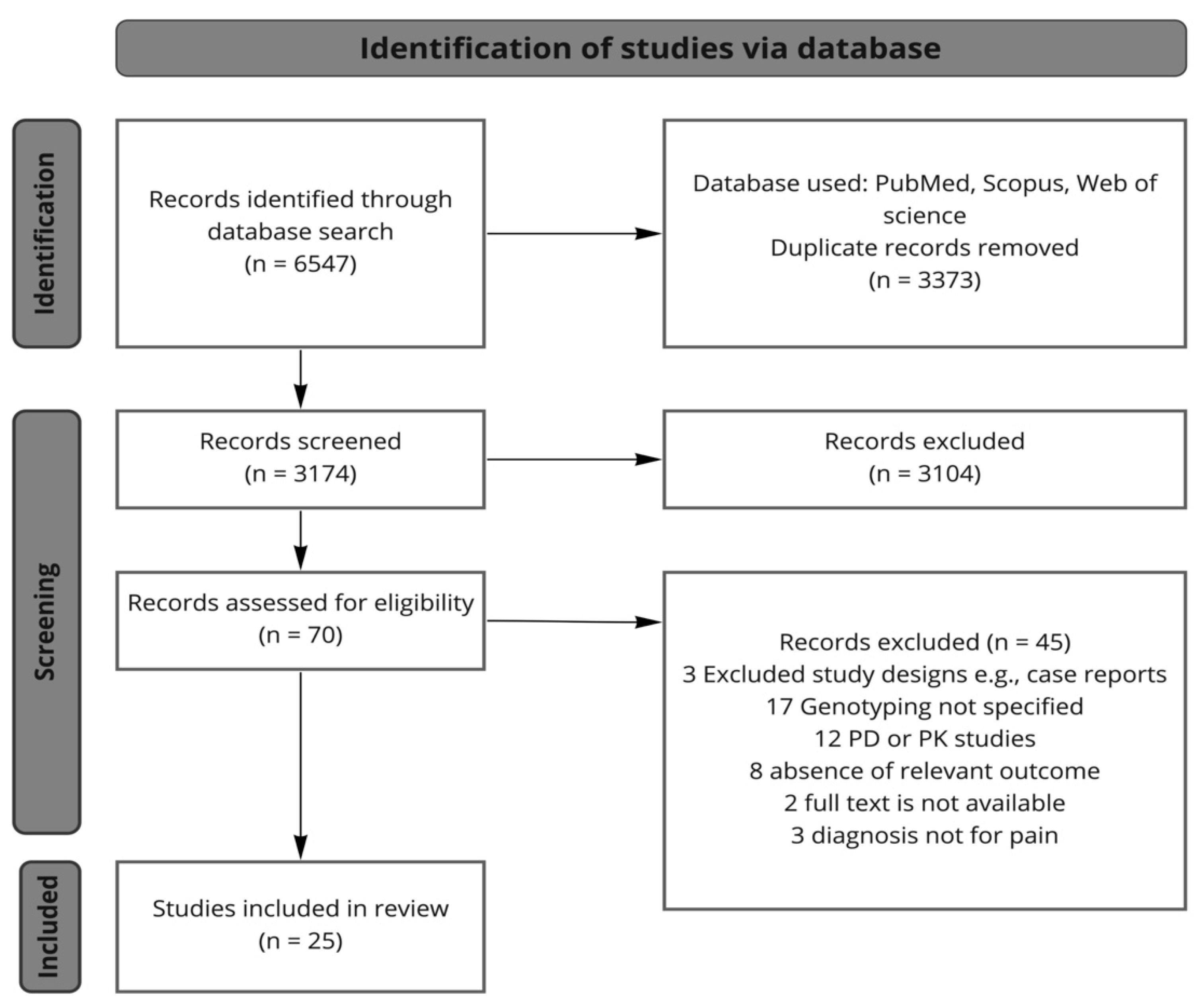
3. Methods
3.1. Search Strategy
3.2. Study Selection
3.3. Data Extraction and Synthesis
4. Drug Groups
4.1. Nonsteroidal Anti-Inflammatory Drugs and Aspirin
4.1.1. Ibuprofen
4.1.2. Celecoxib
4.1.3. Piroxicam
4.1.4. Dexketoprofen
4.1.5. Diclofenac
4.1.6. Meloxicam
4.1.7. Aspirin

{kind=link}
{kind=link}
| Study | Drug | Ethnicity | Study Design | Outcome | Gene Assessed | Variants | Findings (Effect or Safety) |
|---|---|---|---|---|---|---|---|
| Martinez et al. (2004) [40] | Celecoxib, diclofenac, ibuprofen, piroxicam | N/A | Patients with GI bleeding (n = 94) and healthy individuals (n = 124) | Adverse effects of different NSAIDs | CYP2C9 | CYP2C9*1/*1, *1/*2, *1/*3, *2/*2, *2/*3, *3/*3 | CYP2C9*2allele frequency increased in patients with acute bleeding |
| Saiz-Rodriguez et al. (2021) [37] | Ibuprofen | White | 43 patients with moderate to severe pain after dental surgery | Ibuprofen response | CYP2B6 CYP2C8 CYP2C9 CYP2C19 CYP2D6 CYP3A4 PTGS2 | CYP2B6 G/G, G/T, T/T CYP2C8 PMs, IMs, and NMs CYP2C9 PMs and IMs CYP2C19 IMs, NMs, and UMs CYP2D6 PMs, IMs, NMs, and UMs | Greater pain reduction 6 h after ibuprofen intake in CYP2C9 PMs compared with IMs/NMs |
| Weckwerth et al. (2020) [38] | Ibuprofen | Brazil | 200 patients with acute pain | Ibuprofen response | CYP2C8 CYP2C9 | CYP2C8*1/*1, *1/*2, *1/*3, *1/*4, *2/*3, *3/*4 CYP2C9*1/*1, *1/*2, *1/*3, *2/*2, *2/*3, *3/*3 | CYP2C9 and CYP2C8 IMs and PMs have lower levels of postoperative pain |
| Jaja et al. (2015) [39] | Ibuprofen, aspirin | African American | 50 patients with sickle cell disease | NSAIDs efficacy | CYP2C8, CYP2C9 | CYP2C8*1/*1, *1/*2, *1/*3, *1/*4, *2/*2, *2/*3, *2/*4 CYP2C9*1/*1, *1/*2, *1/*3, *1/*5, *1/*6, *1/*8, *1/*9, *1/*11, *2/*3, *5/*9, *6/*8, *8/*9, *9/*11 | CYP2C9 NMs visited the hospital more frequently due to severe pain |
| Hamilton et al. (2020) [49] | Celecoxib | N/A | 31 patients with postoperative pain | Celecoxib efficacy and safety | CYP2C9 | CYP2C9 NMs and IMs | Concomitant drug intake, no clear conclusion regarding a pharmacogenetic association |
| Murto et al. (2015) [50] | Celecoxib | Caucasian, African American Hispanic, South and East Asian | 93 patients with postoperative pain | Celecoxib efficacy | CYP2C9 | CYP2C9*1/*1, *1/*2, *1/*3, *2/*3, *2/*2, *3/*3 | Reduced pain recurrence in CYP2C9*3 allele carriers compared to wild-type carriers |
| Ustare et al. (2020) [51] | Celecoxib | Malay, Malay-Chinese, Malay-Polynesian, Filipinos | 99 patients with postoperative pain | Celecoxib efficacy | CYP2C9 | CYP2C9*1/*1, *1/*3 | Lower pain scores in CYP2C9 IMs after 24 and 48 h compared to NMs |
| Calvo et al. (2017) [54] | Piroxicam | Brazil | 102 patients with postoperative pain | Piroxicam efficacy and adverse effects | CYP2C8, CYP2C9 | CYP2C8*1/*1, *1/*3, *3/*3 CYP2C9*1/*1, *1/*2, *1/*3, *2/*2, *2/*3, *3/*3 | Postoperative pain scores and adverse effects were comparable between genotypes |
| Daly et al. (2007) [68] | Diclofenac | North European | Patients with and without diclofenac-induced hepatotoxicity (n = 28/48) Healthy volunteers (n = 112) | Diclofenac adverse effects | UGT2B7, CYP2C8, ABCC2 | UGT2B7*1/*1, *1/*2, *2/*2 CYP2C8*1/*1, *1/*2, *1/*3, *1/*4, 1/*5, *2/*3, *2/*2 ABCC2 C-24/C-24, C-24/T-24, T-24/T-24 | UGT2B7*2 allele was associated with a higher risk of diclofenac-induced hepatotoxicity compared with wild-type carriers |
| Aithal et al. (2000) [65] | Diclofenac | Caucasian | 124 patients with diclofenac-induced hepatotoxicity (n = 24); control group (n = 100) | Diclofenac adverse effects | CYP2C9 | CYP2C9*1/*1, *1/*2, *1/*3, *2/*3, *3/*3 | No association of CYP2C9*2 or CYP2C9*3 with diclofenac-induced hepatotoxicity |
| Shiotani et al. (2014) [79] | Aspirin | Japanese | 638 patients with peptic ulcer (n = 111); patients with GI bleeding (n = 45); control group (n = 482) | Aspirin adverse effects | SLCO1B1, CHST2 | SLCO1B1 388 A > G (rs2306283), 521 T > C (rs4149056) CHST2 2082 C > T (rs6664) | SLCO1B1*1b and CHST2 2082 T allele frequency was increased in patients with peptic ulcer and ulcer bleeding compared to the controls |
| Wang et al. (2019) [81] | Aspirin | N/A | 154 patients with coronary heart disease; with (n = 57) or without (n = 97) upper GI bleeding (UGIB) | Aspirin adverse effects | TNF-α gene | -1031T > C TT, TC, CC -863C > A CC, CA, AA -857C > T CC, CT, TT | -1031T > C: C allele and CC genotype carriers and -863C > A: A allele, CA, and CA + AA genotype carriers had increased risk of UGIB -857 C > T had no effect |
| Groza et al. (2017) [82] | Aspirin | N/A | Patients with UGIB (n = 154); control group (n = 178) | Aspirin adverse effects | VKORC1 | VKORC1 -1639 G > A GG, GA, AA | VKORC1 -1639 G > A: AA genotype is associated with an increased risk of UGIB |
| E. Piazuelo et al. (2008) [83] | Aspirin | White | Patients with UGIB (n = 88); control patients (n = 108) | Aspirin adverse effects | eNOS GP IIIa | eNOS 4b/4b, 4a/4b, 4a/4a GP IIIa PlA1/A1, PlA1/A2, PlA2/A1 | eNOS a allele carriers had reduced risk of UGIB |
| Figueiras et al. (2016) [80] | Multiple drugs | Caucasian | 1920 patients with hematemesis, melena; and hematochezia (n = 577); control group (n = 1343) | NSAIDs adverse effects | CYP2C9 | CYP2C9*1/*1, *1/*2, *1/*3, *2/*2, *2/*3, *3/*3 | Higher risk of upper GI bleeding in CYP2C9*3 allele carriers CYP2C9*2 allele had no such effect |
| Lee et al. (2014) [70] | Meloxicam | Korean | 22 healthy participants | Meloxicam adverse effects | CYP2C9 | CYP2C9*1/*1, *1/*3, *3/*3 | CYP2C9*3/*3 carriers have significantly greater TXB2 inhibition compared with CYP2C9*1/*1 and *1/3 (possible differences in the incidence of cardiovascular complications and bleeding) |
| Mejía-Abril et al. (2021) [58] | Dexketoprofen | Caucasian, Latin-American, Black, Asian | 85 healthy participants | Dexketoprofen adverse effects | CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2D6, CYP3A4, CYP3A5 ABCB1, ABCC2, SLCO1B, UGT1A1 | CYP1A2*1C, *1F, *1B CYP2A6*9 CYP2B6*9, *5, rs4803419, rs2279345, rs2279343 CYP2C8*2, *3, *4 CYP2C9*2, *3 CYP2C19*2, *3, *4, *17 CYP2D6*3, *4, *6, *7, *8, *9, *10, *14, *17, *41 CYP3A4*22, rs55785340, rs4646438 CYP3A5*3, *6 ABCB1 C3435T, G2677 T/A, C1236T ABCC2 rs2273697, rs717620 SLCO1B1*1B, *5, rs4149015, rs11045879 SLC22A1*2, *3, *5 UGT1A1*80 | No adverse effects after dexketoprofen intake were reported |
4.2. Antidepressants
4.2.1. Tricyclic Antidepressants
Amitriptyline
Nortriptyline
Imipramine
4.2.2. Selective Serotonin Reuptake Inhibitors
Citalopram and Escitalopram
| Study | Drug Class | Drug Name | Ethnicity | Study Design | FDA/EMA Status (Indication for Pain) | Outcome Measured | Gene Assessed | Variants | Findings |
|---|---|---|---|---|---|---|---|---|---|
| Wilder-Smith et al. (2005) [93] | TCA | Amitriptyline | N/A | 30 patients with postamputation pain | Approved | Amitriptyline efficacy | CYP2D6 | PMs and UMs | Lower pain levels in CYP2D6 PMs |
| Chaudhry et al. (2017) [94] | TCA | Amitriptyline | Black African (n = 21), Caucasian (n = 9), Indian (n = 1) | 31 patients with diabetic peripheral neuropathy | Approved | Amitriptyline treatment response and adverse effects | CYP2D6 | *1/*1, *1/*1xN, *1/*45, *2/*2, *2M/*35, *1/*2, *35/*41, *1/*17, *2/*17, *2/*4, *2xN/*5, *2/*29, *1/*29, *17/*84 | No effect of CYP2D6 phenotype on amitriptyline efficacy. A trend towards more severe adverse effects in CYP2D6 IMs compared to NMs |
| Benavides et al. (2021) [98] | TCA | Nortriptyline | Caucasian | 25 neuropathic pain patients | Not approved | Nortriptyline treatment response and adverse effects | CYP2C19 CYP2D6 ABCB1 | CYP2C19 *2, *3, *4, *5, *6, *7, *8, *17 CYP2D6 rs1065852, *2A, *3, *4, *6, *7, *8, *9, *10, *11, *12, *14, *15, rs28371706, *17, *20, *29, *35, *41, rs1135840, *40, *58, *64 ABCB1 rs1045642, rs2032582 | ABCB1 rs1045642 C homozygotes showed an improved therapy response in pain conditions under a combined therapy with nortriptyline and morphine |
| Siegenthaler et al. (2015) [104] | TCA | Imipramine | N/A | 50 patients with chronic low-back pain | Not approved | Imipramine efficacy | CYP2D6 | *6, *7, *8, *10, *41, *3A, *4, *5, *2 | No significant effect of amitriptyline on low back pain reduction |
| Schliessbach et al. (2018) [105] | TCA | Imipramine | N/A | 50 patients with chronic low-back pain | Not approved | Imipramine response | CYP2D6 | *1, *3, *4, *5, *6, *8, *10, *41 | No overall reduction in low back pain with imipramine. No effect of CYP2D6 phenotype on pain tests results |
| Brasch-Andersen et al. (2011) [119] | SSRI | Escitalopram | N/A | 34 patients with peripheral neuropathic pain | Not approved | Escitalopram treatment response | HTR2A, HTR2C, ABCB1, CYP2C19, SLC6A4 | HTR2A rs6314 GG, GA, AA HTR2C rs6318 GG, GC, CC (women) HTR2C rs6318 G, C (men) ABCB1 rs2032582 GG, GT/AT, TT SLC6A4 5-HTTL polymorphic region L/L, L/S, S/S | Little evidence for decreased pain relief in HTR2C C allele carriers in male participants |
| Aldrich et al. (2019) [120] | SSRI | Escitalopram | White Black Other | 248 patients with depression and anxiety | Not approved | Escitalopram adverse effects | CYP2C19 | CYP2C19*1, *2, *3, *4, *5, *6, *7, *8, *17 | CYP2C19 PMs and IMs showed a higher total number of side effects compared with NMs and UMs |
| Kuo et al. (2013) [121] | SSRI | Escitalopram | Chinese | 158 patients with massive depressive disorder | Not approved | Escitalopram adverse effects | CYP1A2 | CYP1A2 rs2069521, *1K, *1F, rs4646425, rs35796837, rs34058039, rs2472304, rs3743484, rs4646427, rs2470890 | CYP1A2 SNPs rs2069521, rs4646425, and rs4646427 are associated with dry mouth, nausea, and vomiting at week 2, and fatigue at week 1 |
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CPIC | Clinical Pharmacogenetics Implementation Consortium |
| COX-1 | Cyclooxygenase 1 |
| COX-2 | Cyclooxygenase 2 |
| EM | Extensive metabolizer |
| GI | Gastrointestinal |
| IM | Intermediate metabolizer |
| NM | Normal metabolizer |
| NSAIDs | Nonsteroidal anti-inflammatory drugs |
| PM | Poor metabolizer |
| RM | Rapid metabolizer |
| SNPs | Single-nucleotide polymorphisms |
| SNRIs | Serotonin-norepinephrine reuptake inhibitors |
| SSRIs | Selective serotonin reuptake inhibitors |
| Tmax | Time of the maximum plasma concentration |
| TNF-α | Tumor necrosis factor α |
| TCAs | Tricyclic antidepressants |
| UGTs | Uridine 5′-diphospho-glucuronosyltransferase |
| UGIB | Upper gastrointestinal bleeding |
| UM | Ultra-rapid metabolizer |
| VKOR | Vitamin K epoxide reductase |
| ECG | Electrocardiography |
| Ads | Antidepressants |
References
- Henschke, N.; Kamper, S.J.; Maher, C.G. The epidemiology and economic consequences of pain. Mayo Clin. Proc. 2015, 90, 139–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woolf, C.J. What is this thing called pain? J. Clin. Investig. 2010, 120, 3742–3744. [Google Scholar] [CrossRef] [PubMed]
- Unruh, A.M. Gender variations in clinical pain experience. Pain 1996, 65, 123–167. [Google Scholar] [CrossRef]
- Luime, J.J.; Koes, B.W.; Hendriksen, I.J.M.; Burdorf, A.; Verhagen, A.P.; Miedema, H.S.; Verhaar, J.A.N. Prevalence and incidence of shoulder pain in the general population; a systematic review. Scand. J. Rheumatol. 2004, 33, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Mallen, C.; Peat, G.; Thomas, E.; Croft, P. Severely disabling chronic pain in young adults: Prevalence from a population-based postal survey in North Staffordshire. BMC Musculoskelet. Disord. 2005, 6, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, A.M.; Smith, B.H.; Penny, K.I.; Smith, W.C.; Chambers, W.A. The epidemiology of chronic pain in the community. Lancet Lond. Engl. 1999, 354, 1248–1252. [Google Scholar] [CrossRef]
- Sadhasivam, S.; Chidambaran, V.; Ngamprasertwong, P.; Esslinger, H.R.; Prows, C.; Zhang, X.; Martin, L.J.; McAuliffe, J. Race and unequal burden of perioperative pain and opioid related adverse effects in children. Pediatrics 2012, 129, 832–838. [Google Scholar] [CrossRef] [Green Version]
- Edlund, M.J.; Martin, B.C.; Russo, J.E.; DeVries, A.; Braden, J.B.; Sullivan, M.D. The role of opioid prescription in incident opioid abuse and dependence among individuals with chronic noncancer pain: The role of opioid prescription. Clin. J. Pain 2014, 30, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Carman, W.J.; Su, S.; Cook, S.F.; Wurzelmann, J.I.; McAfee, A. Coronary heart disease outcomes among chronic opioid and cyclooxygenase-2 users compared with a general population cohort. Pharmacoepidemiol. Drug Saf. 2011, 20, 754–762. [Google Scholar] [CrossRef]
- García Rodríguez, L.A.; Barreales Tolosa, L. Risk of upper gastrointestinal complications among users of traditional NSAIDs and COXIBs in the general population. Gastroenterology 2007, 132, 498–506. [Google Scholar] [CrossRef]
- Boyle, J.; Eriksson, M.E.V.; Gribble, L.; Gouni, R.; Johnsen, S.; Coppini, D.V.; Kerr, D. Randomized, placebo-controlled comparison of amitriptyline, duloxetine, and pregabalin in patients with chronic diabetic peripheral neuropathic pain: Impact on pain, polysomnographic sleep, daytime functioning, and quality of life. Diabetes Care 2012, 35, 2451–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, F.J.; Nebert, D.W. Evolution of the P450 gene superfamily: Animal-plant “warfare”, molecular drive and human genetic differences in drug oxidation. Trends Genet. TIG 1990, 6, 182–186. [Google Scholar] [CrossRef]
- Rodríguez-Vicente, A.E.; Lumbreras, E.; Hernández, J.M.; Martín, M.; Calles, A.; Otín, C.L.; Algarra, S.M.; Páez, D.; Taron, M. Pharmacogenetics and pharmacogenomics as tools in cancer therapy. Drug Metab. Pers. Ther. 2016, 31, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.G.; Patel, A.; Howard, R.F. Pharmacogenetics of codeine metabolism in an urban population of children and its implications for analgesic reliability. Br. J. Anaesth. 2002, 89, 839–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchheiner, J.; Schmidt, H.; Tzvetkov, M.; Keulen, J.-T.H.A.; Lötsch, J.; Roots, I.; Brockmöller, J. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J. 2007, 7, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Magarbeh, L.; Gorbovskaya, I.; Le Foll, B.; Jhirad, R.; Müller, D.J. Reviewing pharmacogenetics to advance precision medicine for opioids. Biomed. Pharmacother. Biomed. Pharmacother. 2021, 142, 112060. [Google Scholar] [CrossRef]
- Rodriguez Cairoli, F.; Appiani, F.; Sambade, J.M.; Comandé, D.; Camacho Arteaga, L.; Ciapponi, A. Efficacy and safety of opioid therapy guided by pharmacogenetics: A systematic review. Pharmacogenomics 2021, 22, 573–586. [Google Scholar] [CrossRef]
- Anekar, A.A.; Cascella, M. WHO Analgesic Ladder. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Wang, V.C.; Mullally, W.J. Pain Neurology. Am. J. Med. 2020, 133, 273–280. [Google Scholar] [CrossRef]
- Chou, R.; Qaseem, A.; Snow, V.; Casey, D.; Cross, J.T.; Shekelle, P.; Owens, D.K.; Clinical Efficacy Assessment Subcommittee of the American College of Physicians; American College of Physicians; American Pain Society Low Back Pain Guidelines Panel. Diagnosis and treatment of low back pain: A joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann. Intern. Med. 2007, 147, 478–491. [Google Scholar] [CrossRef]
- Tick, H.; Nielsen, A.; Pelletier, K.R.; Bonakdar, R.; Simmons, S.; Glick, R.; Ratner, E.; Lemmon, R.L.; Wayne, P.; Zador, V.; et al. Evidence-Based Nonpharmacologic Strategies for Comprehensive Pain Care: The Consortium Pain Task Force White Paper. Explore 2018, 14, 177–211. [Google Scholar] [CrossRef]
- Chou, R.; Fanciullo, G.J.; Fine, P.G.; Adler, J.A.; Ballantyne, J.C.; Davies, P.; Donovan, M.I.; Fishbain, D.A.; Foley, K.M.; Fudin, J.; et al. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J. Pain 2009, 10, 113–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiffen, P.J.; Wee, B.; Derry, S.; Bell, R.F.; Moore, R.A. Opioids for cancer pain-an overview of Cochrane reviews. Cochrane Database Syst. Rev. 2017, 7, CD012592. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.B.; Maher, C.G.; Pinto, R.Z.; Traeger, A.C.; Lin, C.-W.C.; Chenot, J.-F.; van Tulder, M.; Koes, B.W. Clinical practice guidelines for the management of non-specific low back pain in primary care: An updated overview. Eur. Spine J. 2018, 27, 2791–2803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnerup, N.B.; Kuner, R.; Jensen, T.S. Neuropathic Pain: From Mechanisms to Treatment. Physiol. Rev. 2021, 101, 259–301. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, E.; Mammana, S.; Nicoletti, F.; Bramanti, P.; Mazzon, E. The neuropathic pain: An overview of the current treatment and future therapeutic approaches. Int. J. Immunopathol. Pharmacol. 2019, 33. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.I.; Eisenberg, E.; Ahmedzai, S.H.; Bhaskar, A.; O’Brien, T.; Mercadante, S.; Krčevski Škvarč, N.; Vissers, K.; Wirz, S.; Wells, C.; et al. Standards for the management of cancer-related pain across Europe-A position paper from the EFIC Task Force on Cancer Pain. Eur. J. Pain Lond. Engl. 2019, 23, 660–668. [Google Scholar] [CrossRef]
- Hachimi-Idrissi, S.; Dobias, V.; Hautz, W.E.; Leach, R.; Sauter, T.C.; Sforzi, I.; Coffey, F. Approaching acute pain in emergency settings; European Society for Emergency Medicine (EUSEM) guidelines-part 2: Management and recommendations. Intern. Emerg. Med. 2020, 15, 1141–1155. [Google Scholar] [CrossRef]
- Martinez, L.; Ekman, E.; Nakhla, N. Perioperative Opioid-sparing Strategies: Utility of Conventional NSAIDs in Adults. Clin. Ther. 2019, 41, 2612–2628. [Google Scholar] [CrossRef]
- Patel, J.; Lucas, C.J.; Margalit, M.; Martin, J.H. Laxative Use in Inpatients on Oxycodone/Naloxone Prolonged Release and Oxycodone Prolonged Release for Cancer and Non-cancer Pain. J. Pain Palliat. Care Pharmacother. 2018, 32, 116–123. [Google Scholar] [CrossRef]
- Vane, J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef]
- Grosser, T.; Theken, K.N.; FitzGerald, G.A. Cyclooxygenase Inhibition: Pain, Inflammation, and the Cardiovascular System. Clin. Pharmacol. Ther. 2017, 102, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Macías, Y.; Gómez Tabales, J.; García-Martín, E.; Agúndez, J.A.G. An update on the pharmacogenomics of NSAID metabolism and the risk of gastrointestinal bleeding. Expert Opin. Drug Metab. Toxicol. 2020, 16, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.J.; Björklund, E.; Lichtman, A.H.; Naidu, P.S.; Congiu, C.; Onnis, V. Inhibitory properties of ibuprofen and its amide analogues towards the hydrolysis and cyclooxygenation of the endocannabinoid anandamide. J. Enzyme Inhib. Med. Chem. 2013, 28, 172–182. [Google Scholar] [CrossRef] [Green Version]
- Hamman, M.A.; Thompson, G.A.; Hall, S.D. Regioselective and stereoselective metabolism of ibuprofen by human cytochrome P450 2C. Biochem. Pharmacol. 1997, 54, 33–41. [Google Scholar] [CrossRef]
- Ochoa, D.; Prieto-Pérez, R.; Román, M.; Talegón, M.; Rivas, A.; Galicia, I.; Abad-Santos, F.; Cabaleiro, T. Effect of gender and CYP2C9 and CYP2C8 polymorphisms on the pharmacokinetics of ibuprofen enantiomers. Pharmacogenomics 2015, 16, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Saiz-Rodríguez, M.; Valdez-Acosta, S.; Borobia, A.M.; Burgueño, M.; Gálvez-Múgica, M.Á.; Acero, J.; Cabaleiro, T.; Muñoz-Guerra, M.F.; Puerro, M.; Llanos, L.; et al. Influence of Genetic Polymorphisms on the Response to Tramadol, Ibuprofen, and the Combination in Patients With Moderate to Severe Pain After Dental Surgery. Clin. Ther. 2021, 43, e86–e102. [Google Scholar] [CrossRef]
- Weckwerth, G.M.; Dionísio, T.J.; Costa, Y.M.; Colombini-Ishiquiriama, B.L.; Oliveira, G.M.; Torres, E.A.; Bonjardim, L.R.; Calvo, A.M.; Moore, T.; Absher, D.M.; et al. CYP450 polymorphisms and clinical pharmacogenetics of ibuprofen after lower third molar extraction. Eur. J. Clin. Pharmacol. 2021, 77, 697–707. [Google Scholar] [CrossRef]
- Jaja, C.; Bowman, L.; Wells, L.; Patel, N.; Xu, H.; Lyon, M.; Kutlar, A. Preemptive Genotyping of CYP2C8 and CYP2C9 Allelic Variants Involved in NSAIDs Metabolism for Sickle Cell Disease Pain Management. Clin. Transl. Sci. 2015, 8, 272–280. [Google Scholar] [CrossRef]
- Martínez, C.; Blanco, G.; Ladero, J.M.; García-Martín, E.; Taxonera, C.; Gamito, F.G.; Diaz-Rubio, M.; Agúndez, J.A.G. Genetic predisposition to acute gastrointestinal bleeding after NSAIDs use. Br. J. Pharmacol. 2004, 141, 205–208. [Google Scholar] [CrossRef]
- Theken, K.N.; Lee, C.R.; Gong, L.; Caudle, K.E.; Formea, C.M.; Gaedigk, A.; Klein, T.E.; Agúndez, J.A.G.; Grosser, T. Clinical Pharmacogenetics Implementation Consortium Guideline (CPIC) for CYP2C9 and Nonsteroidal Anti-Inflammatory Drugs. Clin. Pharmacol. Ther. 2020, 108, 191–200. [Google Scholar] [CrossRef]
- Penning, T.D.; Talley, J.J.; Bertenshaw, S.R.; Carter, J.S.; Collins, P.W.; Docter, S.; Graneto, M.J.; Lee, L.F.; Malecha, J.W.; Miyashiro, J.M.; et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: Identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benze nesulfonamide (SC-58635, celecoxib). J. Med. Chem. 1997, 40, 1347–1365. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Shou, M.; Mei, Q.; Rushmore, T.H.; Rodrigues, A.D. Major role of human liver microsomal cytochrome P450 2C9 (CYP2C9) in the oxidative metabolism of celecoxib, a novel cyclooxygenase-II inhibitor. J. Pharmacol. Exp. Ther. 2000, 293, 453–459. [Google Scholar] [PubMed]
- Sandberg, M.; Yasar, U.; Strömberg, P.; Höög, J.-O.; Eliasson, E. Oxidation of celecoxib by polymorphic cytochrome P450 2C9 and alcohol dehydrogenase. Br. J. Clin. Pharmacol. 2002, 54, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M.; McLachlan, A.J.; Day, R.O.; Williams, K.M. Clinical pharmacokinetics and pharmacodynamics of celecoxib: A selective cyclo-oxygenase-2 inhibitor. Clin. Pharmacokinet. 2000, 38, 225–242. [Google Scholar] [CrossRef]
- Kirchheiner, J.; Störmer, E.; Meisel, C.; Steinbach, N.; Roots, I.; Brockmöller, J. Influence of CYP2C9 genetic polymorphisms on pharmacokinetics of celecoxib and its metabolites. Pharmacogenetics 2003, 13, 473–480. [Google Scholar] [CrossRef]
- Brenner, S.S.; Herrlinger, C.; Dilger, K.; Mürdter, T.E.; Hofmann, U.; Marx, C.; Klotz, U. Influence of age and cytochrome P450 2C9 genotype on the steady-state disposition of diclofenac and celecoxib. Clin. Pharmacokinet. 2003, 42, 283–292. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Kang, P.; Cho, C.-K.; Jung, E.H.; Park, H.-J.; Lee, Y.J.; Bae, J.-W.; Jang, C.-G.; Lee, S.-Y. Physiologically based pharmacokinetic (PBPK) modeling for prediction of celecoxib pharmacokinetics according to CYP2C9 genetic polymorphism. Arch. Pharm. Res. 2021, 44, 713–724. [Google Scholar] [CrossRef]
- Hamilton, W.G.; Gargiulo, J.M.; Parks, N.L. Using pharmacogenetics to structure individual pain management protocols in total knee arthroplasty. Bone Jt. J. 2020, 102, 73–78. [Google Scholar] [CrossRef]
- Murto, K.; Lamontagne, C.; McFaul, C.; MacCormick, J.; Ramakko, K.-A.; Aglipay, M.; Rosen, D.; Vaillancourt, R. Celecoxib pharmacogenetics and pediatric adenotonsillectomy: A double-blinded randomized controlled study. Can. J. Anesth./J. Can. D’anesthésie 2015, 62, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Ustare, L.A.T.; Reyes, K.G.; Lasac, M.A.G.; Brodit, S.E.; Baclig, M.O. Single nucleotide polymorphisms on CYP2C9 gene among Filipinos and its association with post-operative pain relief via COX-2 inhibitors. Int. J. Mol. Epidemiol. Genet. 2020, 11, 31–38. [Google Scholar]
- Gupta, A.; Zheng, L.; Ramanujam, V.; Gallagher, J. Novel Use of Pharmacogenetic Testing in the Identification of CYP2C9 Polymorphisms Related to NSAID-Induced Gastropathy. Pain Med. 2015, 16, 866–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perini, J.A.; Vianna-Jorge, R.; Brogliato, A.R.; Suarez-Kurtz, G. Influence of CYP2C9 genotypes on the pharmacokinetics and pharmacodynamics of piroxicam. Clin. Pharmacol. Ther. 2005, 78, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Calvo, A.M.; Zupelari-Gonçalves, P.; Dionísio, T.J.; Brozoski, D.T.; Faria, F.A.; Santos, C.F. Efficacy of piroxicam for postoperative pain after lower third molar surgery associated with CYP2C8*3 and CYP2C9. J. Pain Res. 2017, 10, 1581–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbanoj, M.J.; Antonijoan, R.M.; Gich, I. Clinical pharmacokinetics of dexketoprofen. Clin. Pharmacokinet. 2001, 40, 245–262. [Google Scholar] [CrossRef]
- Gaskell, H.; Derry, S.; Wiffen, P.J.; Moore, R.A. Single dose oral ketoprofen or dexketoprofen for acute postoperative pain in adults. Cochrane Database Syst. Rev. 2017, 5, CD007355. [Google Scholar] [CrossRef]
- Zhou, S.-F.; Zhou, Z.-W.; Yang, L.-P.; Cai, J.-P. Substrates, inducers, inhibitors and structure-activity relationships of human Cytochrome P450 2C9 and implications in drug development. Curr. Med. Chem. 2009, 16, 3480–3675. [Google Scholar] [CrossRef]
- Mejía-Abril, G.; Zubiaur, P.; Navares-Gómez, M.; Villapalos-García, G.; Román, M.; Ochoa, D.; Abad-Santos, F. Dexketoprofen Pharmacokinetics is not Significantly Altered by Genetic Polymorphism. Front. Pharmacol. 2021, 12, 660639. [Google Scholar] [CrossRef]
- Kuehl, G.E.; Lampe, J.W.; Potter, J.D.; Bigler, J. Glucuronidation of nonsteroidal anti-inflammatory drugs: Identifying the enzymes responsible in human liver microsomes. Drug Metab. Dispos. 2005, 33, 1027–1035. [Google Scholar] [CrossRef] [Green Version]
- Bort, R.; Macé, K.; Boobis, A.; Gómez-Lechón, M.J.; Pfeifer, A.; Castell, J. Hepatic metabolism of diclofenac: Role of human CYP in the minor oxidative pathways. Biochem. Pharmacol. 1999, 58, 787–796. [Google Scholar] [CrossRef]
- Schwarz, U.I. Clinical relevance of genetic polymorphisms in the human CYP2C9 gene. Eur. J. Clin. Investig. 2003, 33 (Suppl. 2), 23–30. [Google Scholar] [CrossRef] [Green Version]
- Todd, P.A.; Sorkin, E.M. Diclofenac sodium. A reappraisal of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy. Drugs 1988, 35, 244–285. [Google Scholar] [CrossRef] [PubMed]
- Dorado, P.; Cavaco, I.; Cáceres, M.C.; Piedade, R.; Ribeiro, V.; Llerena, A. Relationship between CYP2C8 genotypes and diclofenac 5-hydroxylation in healthy Spanish volunteers. Eur. J. Clin. Pharmacol. 2008, 64, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Llerena, A.; Alvarez, M.; Dorado, P.; González, I.; Peñas-LLedó, E.; Pérez, B.; Cobaleda, J.; Calzadilla, L.R. Interethnic differences in the relevance of CYP2C9 genotype and environmental factors for diclofenac metabolism in Hispanics from Cuba and Spain. Pharm. J. 2014, 14, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.P.; Day, C.P.; Leathart, J.B.; Daly, A.K. Relationship of polymorphism in CYP2C9 to genetic susceptibility to diclofenac-induced hepatitis. Pharmacogenetics 2000, 10, 511–518. [Google Scholar] [CrossRef]
- Björnsson, E.; Jerlstad, P.; Bergqvist, A.; Olsson, R. Fulminant drug-induced hepatic failure leading to death or liver transplantation in Sweden. Scand. J. Gastroenterol. 2005, 40, 1095–1101. [Google Scholar] [CrossRef]
- Björnsson, E.; Olsson, R. Outcome and prognostic markers in severe drug-induced liver disease. Hepatol. Baltim. Md 2005, 42, 481–489. [Google Scholar] [CrossRef]
- Daly, A.K.; Aithal, G.P.; Leathart, J.B.S.; Swainsbury, R.A.; Dang, T.S.; Day, C.P. Genetic susceptibility to diclofenac-induced hepatotoxicity: Contribution of UGT2B7, CYP2C8, and ABCC2 genotypes. Gastroenterology 2007, 132, 272–281. [Google Scholar] [CrossRef]
- Chesné, C.; Guyomard, C.; Guillouzo, A.; Schmid, J.; Ludwig, E.; Sauter, T. Metabolism of Meloxicam in human liver involves cytochromes P4502C9 and 3A4. Xenobiotica Fate Foreign Compd. Biol. Syst. 1998, 28, 1–13. [Google Scholar] [CrossRef]
- Lee, H.-I.; Bae, J.-W.; Choi, C.-I.; Lee, Y.-J.; Byeon, J.-Y.; Jang, C.-G.; Lee, S.-Y. Strongly increased exposure of meloxicam in CYP2C9*3/*3 individuals. Pharm. Genom. 2014, 24, 113–117. [Google Scholar] [CrossRef]
- Aoyama, T.; Ishida, Y.; Kaneko, M.; Miyamoto, A.; Saito, Y.; Tohkin, M.; Kawai, S.; Matsumoto, Y. Pharmacokinetics and Pharmacodynamics of Meloxicam in East Asian Populations: The Role of Ethnicity on Drug Response. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 823–832. [Google Scholar] [CrossRef]
- Hasunuma, T.; Tohkin, M.; Kaniwa, N.; Jang, I.-J.; Yimin, C.; Kaneko, M.; Saito, Y.; Takeuchi, M.; Watanabe, H.; Yamazoe, Y.; et al. Absence of ethnic differences in the pharmacokinetics of moxifloxacin, simvastatin, and meloxicam among three East Asian populations and Caucasians. Br. J. Clin. Pharmacol. 2016, 81, 1078–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eikelboom, J.W.; Hirsh, J.; Spencer, F.A.; Baglin, T.P.; Weitz, J.I. Antiplatelet drugs: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed.: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141, e89S–e119S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurth, T.; Hennekens, C.H.; Buring, J.E.; Gaziano, J.M. Aspirin, NSAIDs, and COX-2 inhibitors in cardiovascular disease: Possible interactions and implications for treatment of rheumatoid arthritis. Curr. Rheumatol. Rep. 2004, 6, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, G.E.; Bigler, J.; Potter, J.D.; Lampe, J.W. Glucuronidation of the aspirin metabolite salicylic acid by expressed UDP-glucuronosyltransferases and human liver microsomes. Drug Metab. Dispos. Biol. Fate Chem. 2006, 34, 199–202. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Kuehl, G.E.; Bigler, J.; Rimorin, C.F.; Schwarz, Y.; Shen, D.D.; Lampe, J.W. UGT1A6 polymorphism and salicylic acid glucuronidation following aspirin. Pharm. Genom. 2007, 17, 571–579. [Google Scholar] [CrossRef]
- Van Oijen, M.G.H.; Barthélémy, C.; Janssen, M.J.R.; Joiris, E.; Peters, W.H.M.; Laheij, R.J.F.; Smits, P.; Odou, P.; Jansen, J.B.M.J. Effect of genetic polymorphisms in UDP-glucuronosyltransferase 1A6 (UGT1A6) on acetylsalicylic acid metabolism in healthy female volunteers. Pharmacology 2009, 83, 237–242. [Google Scholar] [CrossRef]
- Lai, K.C.; Lam, S.K.; Chu, K.M.; Wong, B.C.Y.; Hui, W.M.; Hu, W.H.C.; Lau, G.K.K.; Wong, W.M.; Yuen, M.F.; Chan, A.O.O.; et al. Lansoprazole for the prevention of recurrences of ulcer complications from long-term low-dose aspirin use. N. Engl. J. Med. 2002, 346, 2033–2038. [Google Scholar] [CrossRef] [Green Version]
- Shiotani, A.; Murao, T.; Fujita, Y.; Fujimura, Y.; Sakakibara, T.; Nishio, K.; Haruma, K. Single nucleotide polymorphism markers for low-dose aspirin-associated peptic ulcer and ulcer bleeding. J. Gastroenterol. Hepatol. 2014, 29 (Suppl. 4), 47–52. [Google Scholar] [CrossRef] [Green Version]
- Figueiras, A.; Estany-Gestal, A.; Aguirre, C.; Ruiz, B.; Vidal, X.; Carvajal, A.; Salado, I.; Salgado-Barreira, A.; Rodella, L.; Moretti, U.; et al. CYP2C9 variants as a risk modifier of NSAID-related gastrointestinal bleeding: A case-control study. Pharm. Genom. 2016, 26, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.-P. Association between TNF-α polymorphisms and the risk of upper gastrointestinal bleeding induced by aspirin in patients with coronary heart disease. Ann. Hum. Genet. 2019, 83, 124–133. [Google Scholar] [CrossRef]
- Groza, I.; Matei, D.; Tantau, M.; Trifa, A.P.; Crisan, S.; Vesa, S.C.; Bocsan, C.; Buzoianu, A.D.; Acalovschi, M. VKORC1-1639 G>A Polymorphism and the Risk of Non-Variceal Upper Gastrointestinal Bleeding. J. Gastrointest. Liver Dis. 2017, 26, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Piazuelo, E.; Fuentes, J.; García-González, M.A.; Jiménez, P.; Lanas, A. A case-control study of the association between polymorphisms of the endothelial nitric oxide synthase and glycoprotein IIIa genes and upper gastrointestinal bleeding in users of low-dose aspirin. Clin. Ther. 2008, 30, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Breivik, H.; Collett, B.; Ventafridda, V.; Cohen, R.; Gallacher, D. Survey of chronic pain in Europe: Prevalence, impact on daily life, and treatment. Eur. J. Pain Lond. Engl. 2006, 10, 287–333. [Google Scholar] [CrossRef] [PubMed]
- Häuser, W.; Bernardy, K.; Uçeyler, N.; Sommer, C. Treatment of fibromyalgia syndrome with antidepressants: A meta-analysis. JAMA 2009, 301, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.P. The treatment of neuropathic pain: Antidepressants and opioids. Clin. J. Pain 2000, 16, S49–S55. [Google Scholar] [CrossRef]
- Max, M.B.; Culnane, M.; Schafer, S.C.; Gracely, R.H.; Walther, D.J.; Smoller, B.; Dubner, R. Amitriptyline relieves diabetic neuropathy pain in patients with normal or depressed mood. Neurology 1987, 37, 589–596. [Google Scholar] [CrossRef]
- Ryu, S.; Park, S.; Lee, J.H.; Kim, Y.R.; Na, H.S.; Lim, H.S.; Choi, H.Y.; Hwang, I.Y.; Lee, J.G.; Park, Z.W.; et al. A Study on CYP2C19 and CYP2D6 Polymorphic Effects on Pharmacokinetics and Pharmacodynamics of Amitriptyline in Healthy Koreans. Clin. Transl. Sci. 2017, 10, 93–101. [Google Scholar] [CrossRef]
- Hicks, J.K.; Sangkuhl, K.; Swen, J.J.; Ellingrod, V.L.; Müller, D.J.; Shimoda, K.; Bishop, J.R.; Kharasch, E.D.; Skaar, T.C.; Gaedigk, A.; et al. Clinical pharmacogenetics implementation consortium guideline (CPIC) for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants: 2016 update. Clin. Pharmacol. Ther. 2017, 102, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Olesen, O.V.; Linnet, K. Metabolism of the tricyclic antidepressant amitriptyline by cDNA-expressed human cytochrome P450 enzymes. Pharmacology 1997, 55, 235–243. [Google Scholar] [CrossRef]
- Shimoda, K.; Someya, T.; Yokono, A.; Morita, S.; Hirokane, G.; Takahashi, S.; Okawa, M. The impact of CYP2C19 and CYP2D6 genotypes on metabolism of amitriptyline in Japanese psychiatric patients. J. Clin. Psychopharmacol. 2002, 22, 371–378. [Google Scholar] [CrossRef]
- Matthaei, J.; Brockmöller, J.; Steimer, W.; Pischa, K.; Leucht, S.; Kullmann, M.; Jensen, O.; Ouethy, T.; Tzvetkov, M.V.; Rafehi, M. Effects of Genetic Polymorphism in CYP2D6, CYP2C19, and the Organic Cation Transporter OCT1 on Amitriptyline Pharmacokinetics in Healthy Volunteers and Depressive Disorder Patients. Front. Pharmacol. 2021, 12, 688950. [Google Scholar] [CrossRef] [PubMed]
- Wilder-Smith, C.H.; Hill, L.T.; Laurent, S. Postamputation pain and sensory changes in treatment-naive patients: Characteristics and responses to treatment with tramadol, amitriptyline, and placebo. Anesthesiology 2005, 103, 619–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, M.; Alessandrini, M.; Rademan, J.; Dodgen, T.M.; Steffens, F.E.; van Zyl, D.G.; Gaedigk, A.; Pepper, M.S. Impact of CYP2D6 genotype on amitriptyline efficacy for the treatment of diabetic peripheral neuropathy: A pilot study. Pharmacogenomics 2017, 18, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Steimer, W.; Zöpf, K.; von Amelunxen, S.; Pfeiffer, H.; Bachofer, J.; Popp, J.; Messner, B.; Kissling, W.; Leucht, S. Amitriptyline or not, that is the question: Pharmacogenetic testing of CYP2D6 and CYP2C19 identifies patients with low or high risk for side effects in amitriptyline therapy. Clin. Chem. 2005, 51, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Swen, J.J.; Nijenhuis, M.; de Boer, A.; Grandia, L.; Maitland-van der Zee, A.H.; Mulder, H.; Rongen, G.A.P.J.M.; van Schaik, R.H.N.; Schalekamp, T.; Touw, D.J.; et al. Pharmacogenetics: From bench to byte—An update of guidelines. Clin. Pharmacol. Ther. 2011, 89, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Merwar, G.; Gibbons, J.R.; Hosseini, S.A.; Saadabadi, A. Nortriptyline. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Benavides, R.; Vsevolozhskaya, O.; Cattaneo, S.; Zaykin, D.; Brenton, A.; Parisien, M.; Verma, V.; Khoury, S.; Gilron, I.; Diatchenko, L. A functional polymorphism in the ATP-Binding Cassette B1 transporter predicts pharmacologic response to combination of nortriptyline and morphine in neuropathic pain patients. Pain 2020, 161, 619–629. [Google Scholar] [CrossRef]
- Gardiner, S.J.; Begg, E.J. Pharmacogenetics, drug-metabolizing enzymes, and clinical practice. Pharmacol. Rev. 2006, 58, 521–590. [Google Scholar] [CrossRef]
- Hicks, J.K.; Swen, J.J.; Thorn, C.F.; Sangkuhl, K.; Kharasch, E.D.; Ellingrod, V.L.; Skaar, T.C.; Müller, D.J.; Gaedigk, A.; Stingl, J.C.; et al. Clinical Pharmacogenetics Implementation Consortium guideline for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants. Clin. Pharmacol. Ther. 2013, 93, 402–408. [Google Scholar] [CrossRef] [Green Version]
- Koyama, E.; Tanaka, T.; Chiba, K.; Kawakatsu, S.; Morinobu, S.; Totsuka, S.; Ishizaki, T. Steady-state plasma concentrations of imipramine and desipramine in relation to S-mephenytoin 4’-hydroxylation status in Japanese depressive patients. J. Clin. Psychopharmacol. 1996, 16, 286–293. [Google Scholar] [CrossRef]
- Brøsen, K.; Klysner, R.; Gram, L.F.; Otton, S.V.; Bech, P.; Bertilsson, L. Steady-state concentrations of imipramine and its metabolites in relation to the sparteine/debrisoquine polymorphism. Eur. J. Clin. Pharmacol. 1986, 30, 679–684. [Google Scholar] [CrossRef]
- Morinobu, S.; Tanaka, T.; Kawakatsu, S.; Totsuka, S.; Koyama, E.; Chiba, K.; Ishizaki, T.; Kubota, T. Effects of genetic defects in the CYP2C19 gene on the N-demethylation of imipramine, and clinical outcome of imipramine therapy. Psychiatry Clin. Neurosci. 1997, 51, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Siegenthaler, A.; Schliessbach, J.; Vuilleumier, P.H.; Juni, P.; Zeilhofer, H.U.; Arendt-Nielsen, L.; Curatolo, M. Linking altered central pain processing and genetic polymorphism to drug efficacy in chronic low back pain. BMC Pharmacol. Toxicol. 2015, 16, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schliessbach, J.; Siegenthaler, A.; Bütikofer, L.; Limacher, A.; Juni, P.; Vuilleumier, P.H.; Stamer, U.; Arendt-Nielsen, L.; Curatolo, M. Effect of single-dose imipramine on chronic low-back and experimental pain. A randomized controlled trial. PLoS ONE 2018, 13, e0195776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sindrup, S.H.; Bach, F.W.; Madsen, C.; Gram, L.F.; Jensen, T.S. Venlafaxine versus imipramine in painful polyneuropathy: A randomized, controlled trial. Neurology 2003, 60, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Banzi, R.; Cusi, C.; Randazzo, C.; Sterzi, R.; Tedesco, D.; Moja, L. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of migraine in adults. Cochrane Database Syst. Rev. 2015, 4, CD002919. [Google Scholar] [CrossRef]
- Walitt, B.; Urrútia, G.; Nishishinya, M.B.; Cantrell, S.E.; Häuser, W. Selective serotonin reuptake inhibitors for fibromyalgia syndrome. Cochrane Database Syst. Rev. 2015, 6, CD011735. [Google Scholar] [CrossRef]
- Atluri, D.K.; Chandar, A.K.; Fass, R.; Falck-Ytter, Y. Systematic review with meta-analysis: Selective serotonin reuptake inhibitors for noncardiac chest pain. Aliment. Pharmacol. Ther. 2015, 41, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Saarto, T.; Wiffen, P.J. Antidepressants for neuropathic pain. Cochrane Database Syst. Rev. 2005, 4, CD005454. [Google Scholar] [CrossRef]
- Wang, L.; Tobe, J.; Au, E.; Tran, C.; Jomy, J.; Oparin, Y.; Couban, R.J.; Paul, J. Selective serotonin reuptake inhibitors and serotonin-norepinephrine reuptake inhibitors as adjuncts for postoperative pain management: Systematic review and meta-analysis of randomised controlled trials. Br. J. Anaesth. 2022, 128, 118–134. [Google Scholar] [CrossRef]
- Hicks, J.K.; Bishop, J.R.; Sangkuhl, K.; Müller, D.J.; Ji, Y.; Leckband, S.G.; Leeder, J.S.; Graham, R.L.; Chiulli, D.L.; LLerena, A.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 Genotypes and Dosing of Selective Serotonin Reuptake Inhibitors. Clin. Pharmacol. Ther. 2015, 98, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Otto, M.; Bach, F.W.; Jensen, T.S.; Brøsen, K.; Sindrup, S.H. Escitalopram in painful polyneuropathy: A randomized, placebo-controlled, cross-over trial. Pain 2008, 139, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Huezo-Diaz, P.; Perroud, N.; Spencer, E.P.; Smith, R.; Sim, S.; Virding, S.; Uher, R.; Gunasinghe, C.; Gray, J.; Campbell, D.; et al. CYP2C19 genotype predicts steady state escitalopram concentration in GENDEP. J. Psychopharmacol. Oxf. Engl. 2012, 26, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Rudberg, I.; Mohebi, B.; Hermann, M.; Refsum, H.; Molden, E. Impact of the ultrarapid CYP2C19*17 allele on serum concentration of escitalopram in psychiatric patients. Clin. Pharmacol. Ther. 2008, 83, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Xu, Y.; Jiang, T.; Feng, R.; Sun, J.; Zhang, W.; Yang, W.; Li, J.; Adeniyi, O.; Chen, H. Estimation of CYP2D6*10 genotypes on citalopram disposition in Chinese subjects by population pharmacokinetic assay. J. Clin. Pharm. Ther. 2013, 38, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Fudio, S.; Borobia, A.M.; Piñana, E.; Ramírez, E.; Tabarés, B.; Guerra, P.; Carcas, A.; Frías, J. Evaluation of the influence of sex and CYP2C19 and CYP2D6 polymorphisms in the disposition of citalopram. Eur. J. Pharmacol. 2010, 626, 200–204. [Google Scholar] [CrossRef]
- Stingl, J.C.; Brockmöller, J.; Viviani, R. Genetic variability of drug-metabolizing enzymes: The dual impact on psychiatric therapy and regulation of brain function. Mol. Psychiatry 2013, 18, 273–287. [Google Scholar] [CrossRef]
- Brasch-Andersen, C.; Møller, M.U.; Christiansen, L.; Thinggaard, M.; Otto, M.; Brøsen, K.; Sindrup, S.H. A candidate gene study of serotonergic pathway genes and pain relief during treatment with escitalopram in patients with neuropathic pain shows significant association to serotonin receptor2C (HTR2C). Eur. J. Clin. Pharmacol. 2011, 67, 1131–1137. [Google Scholar] [CrossRef]
- Aldrich, S.L.; Poweleit, E.A.; Prows, C.A.; Martin, L.J.; Strawn, J.R.; Ramsey, L.B. Influence of CYP2C19 Metabolizer Status on Escitalopram/Citalopram Tolerability and Response in Youth With Anxiety and Depressive Disorders. Front. Pharmacol. 2019, 10, 99. [Google Scholar] [CrossRef]
- Kuo, H.-W.; Liu, S.C.; Tsou, H.-H.; Liu, S.-W.; Lin, K.-M.; Lu, S.-C.; Hsiao, M.-C.; Hsiao, C.-F.; Liu, C.-Y.; Chen, C.-H.; et al. CYP1A2 genetic polymorphisms are associated with early antidepressant escitalopram metabolism and adverse reactions. Pharmacogenomics 2013, 14, 1191–1201. [Google Scholar] [CrossRef]
- Schult, R.F.; Morris, A.J.; Picard, L.; Wiegand, T.J. Citalopram overdose and severe serotonin syndrome in an intermediate metabolizing patient. Am. J. Emerg. Med. 2019, 37, 1993.e5–1993.e6. [Google Scholar] [CrossRef]
- Castro, V.M.; Clements, C.C.; Murphy, S.N.; Gainer, V.S.; Fava, M.; Weilburg, J.B.; Erb, J.L.; Churchill, S.E.; Kohane, I.S.; Iosifescu, D.V.; et al. QT interval and antidepressant use: A cross sectional study of electronic health records. BMJ 2013, 346, f288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwer, J.M.J.L.; Nijenhuis, M.; Soree, B.; Guchelaar, H.-J.; Swen, J.J.; van Schaik, R.H.N.; Weide, J.V.D.; Rongen, G.A.P.J.M.; Buunk, A.-M.; de Boer-Veger, N.J.; et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction between CYP2C19 and CYP2D6 and SSRIs. Eur. J. Hum. Genet. 2021, 28, 508–517. [Google Scholar] [CrossRef] [PubMed]


| Drug Class | Drug Name |
|---|---|
| Non-steroidal anti-inflammatory drugs (NSAIDS) | Aspirin, celecoxib, etoricoxib, parecoxib, diclofenac, aceclofenac, ibuprofen, dexibuprofen, indomethacin, acemetacin, ketoprofen, dexketoprofen, meloxicam, piroxicam, naproxen, oxaprozine, ketorolac, nabumetone, metamizole, phenazone, propyphenazone, tiaprofenic acid |
| Tricyclic antidepressants (TCAs) | Amitriptyline, amoxapine, clomipramine, desipramine, doxepin, imipramine, nortriptyline, protriptyline, trimipramine |
| Serotonin-norepinephrine reuptake inhibitors (SNRIs) | Duloxetine, venlafaxine, desvenlafaxine, levomilnacipran |
| Selective serotonin reuptake inhibitors (SSRIs) | Citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, sertraline |
| Tetracyclic antidepressants | Maprotiline, mianserin, mirtazapine, setiptiline |
| Monoamine oxidase (MAO) inhibitors | Isocarboxazid, phenelzine, selegiline, tranylcypromine |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zobdeh, F.; Eremenko, I.I.; Akan, M.A.; Tarasov, V.V.; Chubarev, V.N.; Schiöth, H.B.; Mwinyi, J. Pharmacogenetics and Pain Treatment with a Focus on Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) and Antidepressants: A Systematic Review. Pharmaceutics 2022, 14, 1190. https://doi.org/10.3390/pharmaceutics14061190
Zobdeh F, Eremenko II, Akan MA, Tarasov VV, Chubarev VN, Schiöth HB, Mwinyi J. Pharmacogenetics and Pain Treatment with a Focus on Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) and Antidepressants: A Systematic Review. Pharmaceutics. 2022; 14(6):1190. https://doi.org/10.3390/pharmaceutics14061190
Chicago/Turabian StyleZobdeh, Farzin, Ivan I. Eremenko, Mikail A. Akan, Vadim V. Tarasov, Vladimir N. Chubarev, Helgi B. Schiöth, and Jessica Mwinyi. 2022. "Pharmacogenetics and Pain Treatment with a Focus on Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) and Antidepressants: A Systematic Review" Pharmaceutics 14, no. 6: 1190. https://doi.org/10.3390/pharmaceutics14061190