
Gene Targeting to the Cerebral Cortex Following Intranasal Administration of Polyplexes

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Acid Degradation of Glycol Chitosan (GC)
2.2. Characterization of Glycol Chitosan (GC)
2.3. Preparation of Glycol Chitosan Polyplexes and Lipoplexes with Lipofectamine (GCP 1 and LX 1)
2.4. Preparation of Hyaluronidase Coated Nanocomplexes (GCPH)
2.5. Dynamic Light Scattering (DLS)
2.6. Reversed-Phase High Performance Liquid Chromatography (RP-HPLC)
2.7. Cell Culture
2.8. In Vitro Transfection Experiments
2.9. β-Galactosidase Assay
2.10. MTS Assay
2.11. Hyaluronic Acid Digestion Assay
2.12. Intranasal Dosing
2.13. Brain Dissection and Homogenisation
2.14. Statistical Analysis
3. Results
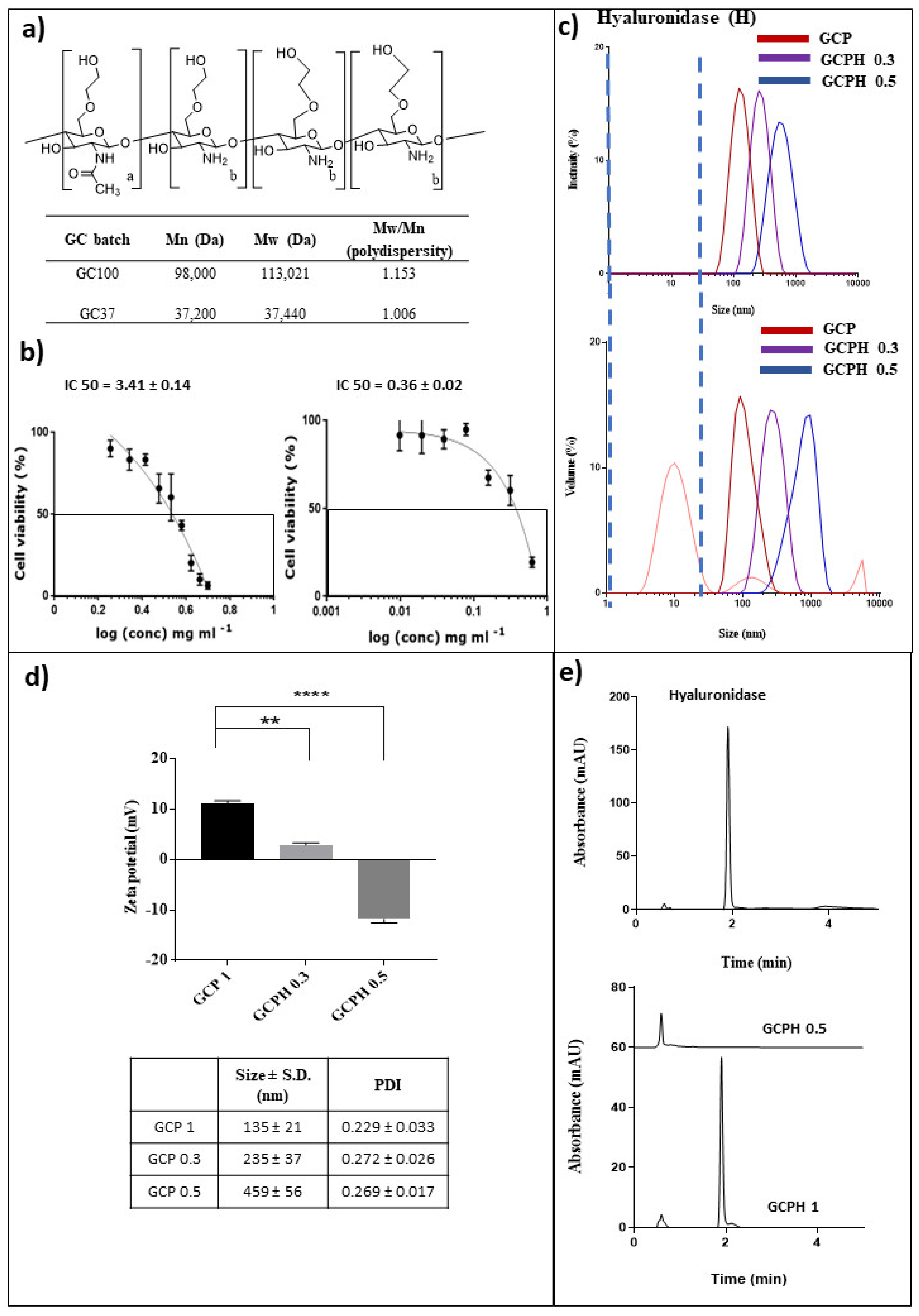
3.1. Acid Degradation of Glycol Chitosan
3.2. Toxicity of the Gene Carrier
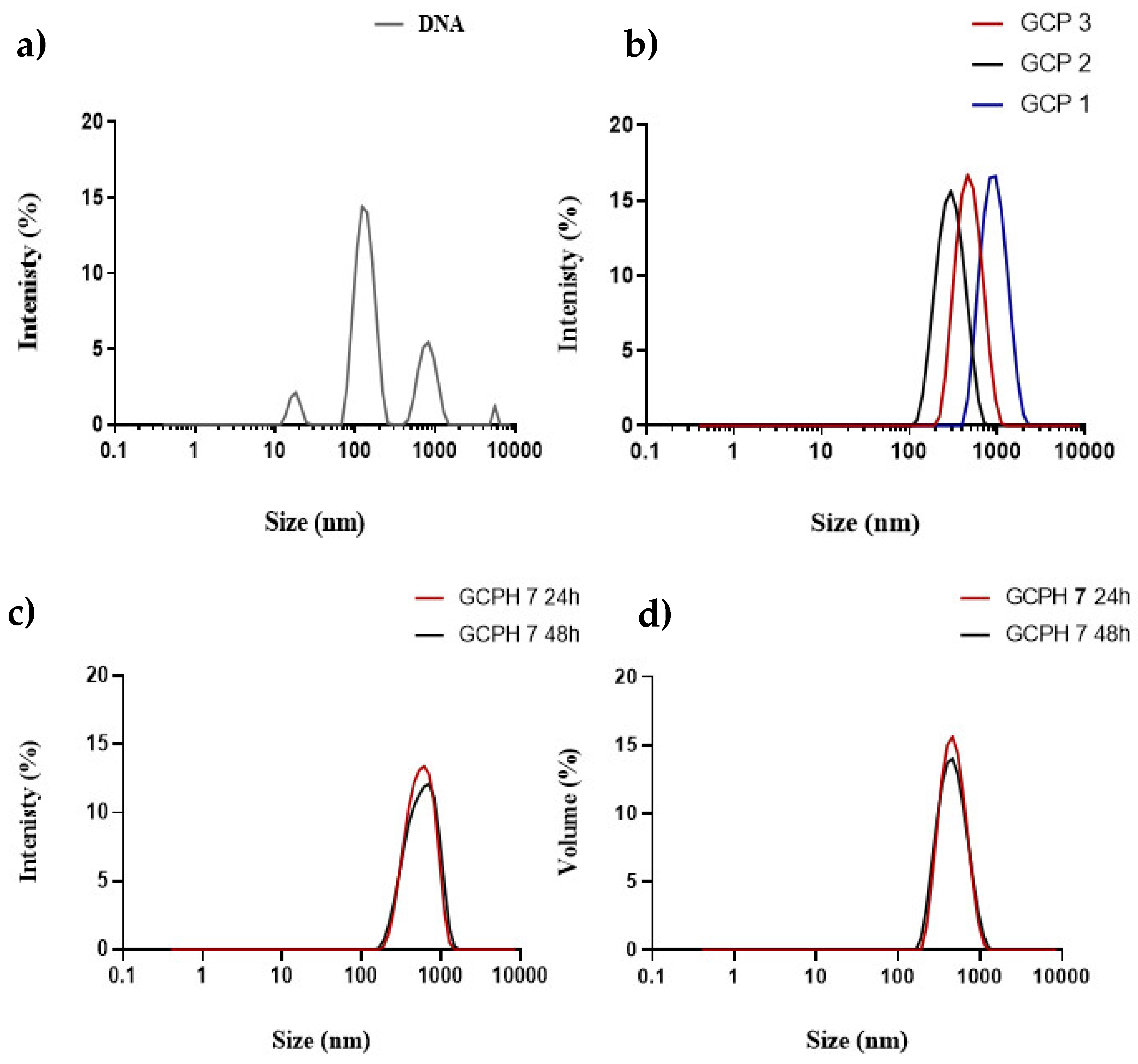
3.3. Polyplex Formation
3.4. HYD Coated Polyplexes
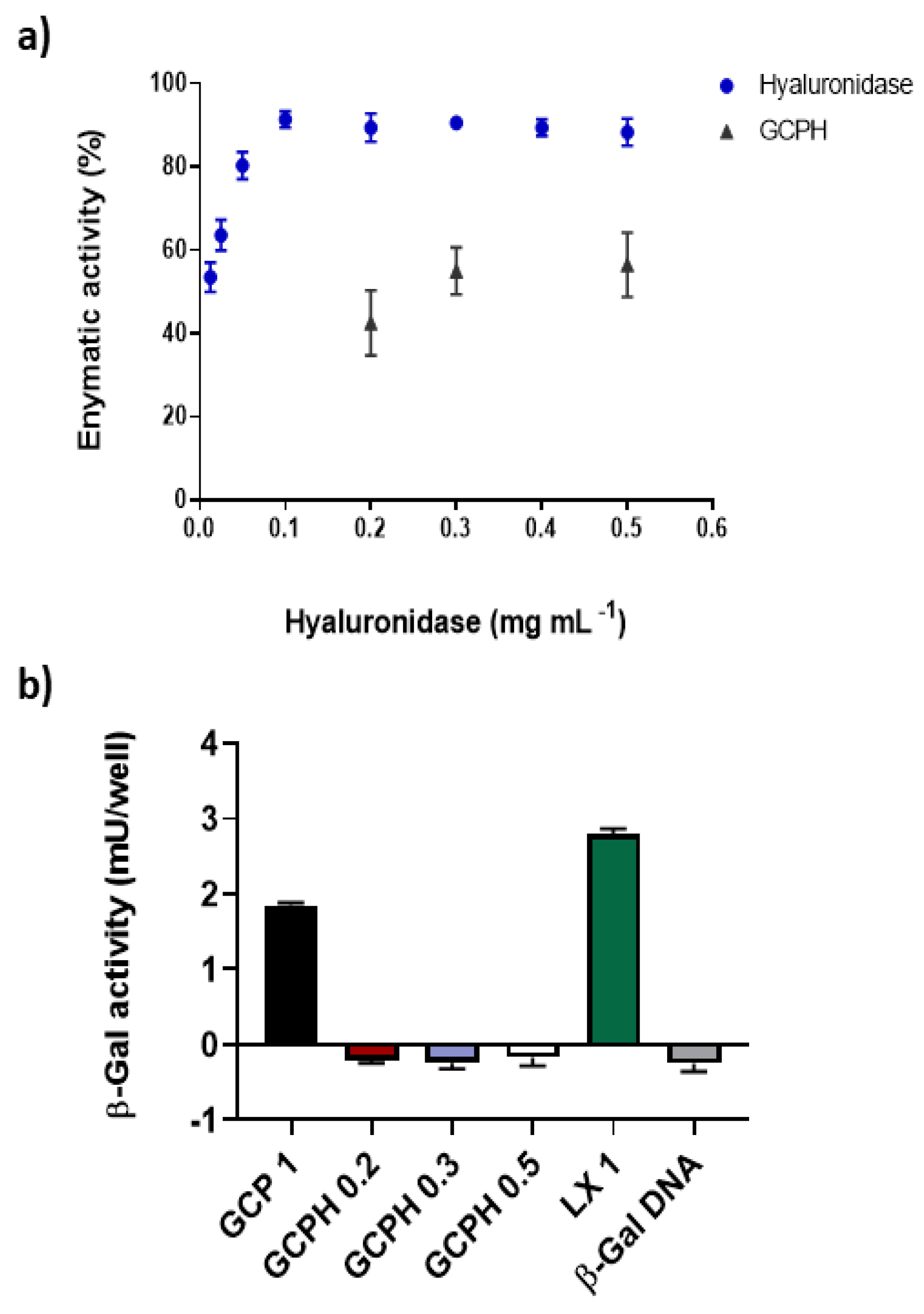
3.5. In Vitro Hyaluronic Acid Digestion Potential of Polyplexes Coated with Hyaluronidase
3.6. Transfection Efficiency of Polyplexes in U87 Glioma Cell Line
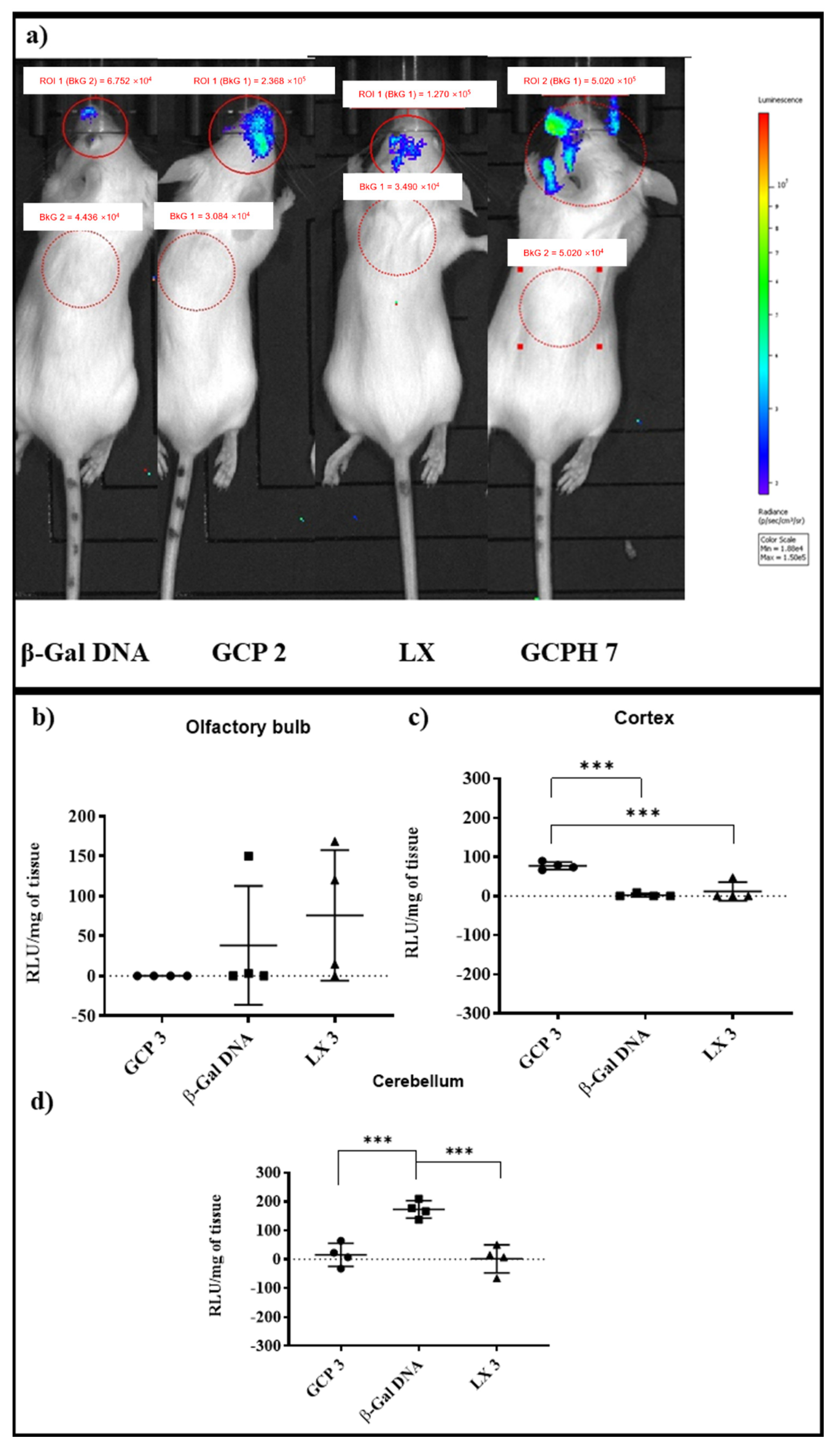
3.7. In Vivo Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, W.; Mehta, A.; Tong, Z.; Esser, L.; Voelcker, N.H. Development of Polymeric Nanoparticles for Blood–Brain Barrier Transfer—Strategies and Challenges. Adv. Sci. 2021, 8, 2003937. [Google Scholar] [CrossRef] [PubMed]
- Owensiii, D.; Peppas, N. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Pozzoli, M.; Rogueda, P.; Zhu, B.; Smith, T.; Young, P.M.; Traini, D.; Sonvico, F. Dry powder nasal drug delivery: Challenges, opportunities and a study of the commercial Teijin Puvlizer Rhinocort device and formulation. Drug Dev. Ind. Pharm. 2016, 42, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Pardeshi, C.V.; Belgamwar, V.S. Direct nose to brain drug delivery via integrated nerve pathways bypassing the blood–brain barrier: An excellent platform for brain targeting. Expert Opin. Drug Deliv. 2013, 10, 957–972. [Google Scholar] [CrossRef]
- Bahadur, S.; Pardhi, D.M.; Rautio, J.; Rosenholm, J.M.; Pathak, K. Intranasal nanoemulsions for direct nose-to-brain delivery of actives for cns disorders. Pharmaceutics 2020, 12, 1230. [Google Scholar] [CrossRef]
- Kim, I.D.; Shin, J.H.; Kim, S.W.; Choi, S.; Ahn, J.; Han, P.L.; Park, J.S.; Lee, J.K. Intranasal delivery of HMGB1 siRNA confers target gene knockdown and robust neuroprotection in the postischemic brain. Mol. Ther. 2012, 20, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Aly, A.E.E.; Waszczak, B.L. Intranasal gene delivery for treating Parkinsons disease: Overcoming the blood-brain barrier. Expert Opin. Drug Deliv. 2015, 12, 1923–1941. [Google Scholar] [CrossRef]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s Disease dementia. J. Alzheimer’s Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef] [Green Version]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimer’s Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K.; et al. Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 1099–1109. [Google Scholar] [CrossRef]
- Brambilla, M.; Manenti, R.; de Girolamo, G.; Adenzato, M.; Bocchio-Chiavetto, L.; Cotelli, M. Effects of Intranasal Oxytocin on Long-Term Memory in Healthy Humans: A Systematic Review. Drug Dev. Res. 2016, 77, 479–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliedner, S.; Schulz, C.; Lehnert, H. Brain uptake of intranasally applied radioiodinated leptin in Wistar rats. Endocrinology 2006, 147, 2088–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.R.; Shnitko, T.A.; Blue, S.W.; Kaucher, A.V.; Winchell, A.J.; Erikson, D.W.; Grant, K.A.; Leggio, L. Labeled oxytocin administered via the intranasal route reaches the brain in rhesus macaques. Nat. Commun. 2020, 11, 2783. [Google Scholar] [CrossRef]
- Berger, S.; Pho, H.; Fleury-Curado, T.; Bevans-Fonti, S.; Younas, H.; Shin, M.K.; Jun, J.C.; Anokye-Danso, F.; Ahima, R.S.; Enquist, L.W.; et al. Intranasal leptin relieves sleep-disordered breathing in mice with diet-induced obesity. Am. J. Respir. Crit. Care Med. 2019, 199, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Harmon, B.T.; Aly, A.E.; Padegimas, L.; Sesenoglu-Laird, O.; Cooper, M.J.; Waszczak, B.L. Intranasal administration of plasmid DNA nanoparticles yields successful transfection and expression of a reporter protein in rat brain. Gene Ther. 2014, 21, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Aly, A.E.E.; Harmon, B.T.; Padegimas, L.; Sesenoglu-Laird, O.; Cooper, M.J.; Waszczak, B.L. Intranasal Delivery of pGDNF DNA Nanoparticles Provides Neuroprotection in the Rat 6-Hydroxydopamine Model of Parkinson’s Disease. Mol. Neurobiol. 2019, 56, 688–701. [Google Scholar] [CrossRef]
- Ha, J.; Kim, M.; Lee, Y.; Lee, M. Intranasal delivery of self-assembled nanoparticles of therapeutic peptides and antagomirs elicits anti-tumor effects in an intracranial glioblastoma model. Nanoscale 2021, 13, 14745–14759. [Google Scholar] [CrossRef]
- Chapman, C.D.; Frey, W.H.; Craft, S.; Danielyan, L.; Hallschmid, M.; Schiöth, H.B.; Benedict, C. Intranasal treatment of central nervous system dysfunction in humans. Pharm. Res. 2013, 30, 2475–2484. [Google Scholar] [CrossRef]
- Wang, Z.; Xiong, G.; Tsang, W.C.; Schätzlein, A.G.; Uchegbu, I.F. Nose-to-brain delivery. J. Pharmacol. Exp. Ther. 2019, 370, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Alexander, A.; Saraf, S. Nose-to-brain drug delivery approach: A key to easily accessing the brain for the treatment of Alzheimer’s disease. Neural Regen. Res. 2018, 13, 2102. [Google Scholar] [CrossRef]
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018, 195, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H. Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Ozawa, T.; Gryaznov, S.M.; Bollen, A.W.; Lamborn, K.R.; Frey, W.H.; Deen, D.F. New therapeutic approach for brain tumors: Intranasal delivery of telomerase inhibitor GRN163. Neuro. Oncol. 2008, 10, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonaka, N.; Farr, S.A.; Kageyama, H.; Shioda, S.; Banks, W.A. Delivery of Galanin-Like Peptide to the Brain: Targeting with Intranasal Delivery and Cyclodextrins. J. Pharmacol. Exp. Ther. 2008, 325, 513–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlton, S.T.; Whetstone, J.; Fayinka, S.T.; Read, K.D.; Illum, L.; Davis, S.S. Evaluation of Direct Transport Pathways of Glycine Receptor Antagonists and an Angiotensin Antagonist from the Nasal Cavity to the Central Nervous System in the Rat Model. Pharm. Res. 2008, 25, 1531–1543. [Google Scholar] [CrossRef]
- Hada, N.; Netzer, W.J.; Belhassan, F.; Wennogle, L.P.; Gizurarson, S. Nose-to-brain transport of imatinib mesylate: A pharmacokinetic evaluation. Eur. J. Pharm. Sci. 2017, 102, 46–54. [Google Scholar] [CrossRef]
- Godfrey, L.; Iannitelli, A.; Garrett, N.L.; Moger, J.; Imbert, I.; King, T.; Porreca, F.; Soundararajan, R.; Lalatsa, A.; Schätzlein, A.G.; et al. Nanoparticulate peptide delivery exclusively to the brain produces tolerance free analgesia. J. Control. Release 2018, 270, 135–144. [Google Scholar] [CrossRef]
- Lochhead, J.J.; Wolak, D.J.; Pizzo, M.E.; Thorne, R.G. Rapid transport within cerebral perivascular spaces underlies widespread tracer distribution in the brain after intranasal administration. J. Perinatol. 2015, 35, 371–381. [Google Scholar] [CrossRef]
- Thorne, R.G.; Pronk, G.J.; Padmanabhan, V.; Frey, W.H. Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 2004, 127, 481–496. [Google Scholar] [CrossRef]
- Aly, A.E.E.; Harmon, B.; Padegimas, L.; Sesenoglu-Laird, O.; Cooper, M.J.; Yurek, D.M.; Waszczak, B.L. Intranasal delivery of hGDNF plasmid DNA nanoparticles results in long-term and widespread transfection of perivascular cells in rat brain. Nanomed. Nanotechnol. Biol. Med. 2019, 16, 20–33. [Google Scholar] [CrossRef]
- Das, M.; Wang, C.; Bedi, R.; Mohapatra, S.S.; Mohapatra, S. Magnetic micelles for DNA delivery to rat brains after mild traumatic brain injury. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 1539–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scodeller, P.; Catalano, P.N.; Salguero, N.; Duran, H.; Wolosiuk, A.; Soler-Illia, G.J.A.A. Hyaluronan degrading silica nanoparticles for skin cancer therapy. Nanoscale 2013, 5, 9690–9698. [Google Scholar] [CrossRef] [PubMed]
- Monroe Freeman, R.E.; Anderson, P.; Wezster, M.E.; Dorfman, A. Ethanolic fractionation of bovine testicular hyaluronidase. J. Biol. Chem. 1950, 186, 201–206. [Google Scholar] [CrossRef]
- Wang, S.S.; Yan, Y.; Ho, K. US FDA-approved therapeutic antibodies with high-concentration formulation: Summaries and perspectives. Antib. Ther. 2021, 4, 262–272. [Google Scholar] [CrossRef]
- Soundararajan, R.; Wang, G.; Petkova, A.; Uchegbu, I.F.; Schä, A.G. Hyaluronidase Coated Molecular Envelope Technology Nanoparticles Enhance Drug Absorption via the Subcutaneous Route. Mol. Pharm. 2020, 17, 2599–2611. [Google Scholar] [CrossRef]
- Wang, W.; Mcconaghy, A.M.; Tetley, L.; Uchegbu, I.F. Controls on Polymer Molecular Weight May Be Used To Control the Size of Palmitoyl Glycol Chitosan Polymeric Vesicles. Langmuir 2001, 17, 631–636. [Google Scholar] [CrossRef]
- Enzymatic Assay of Hyaluronidase. Available online: https://www.sigmaaldrich.com/BG/en/technical-documents/protocol/protein-biology/enzyme-activity-assays/enzymatic-assay-of-hyaluronidase (accessed on 5 May 2022).
- Hanson, L.R.; Fine, J.M.; Svitak, A.L.; Faltesek, K.A. Intranasal administration of CNS therapeutics to awake mice. J. Vis. Exp. 2013, 74, e4440. [Google Scholar] [CrossRef] [Green Version]
- Lončarević, A.; Ivanković, M.; Rogina, A. Lysozyme-Induced Degradation of Chitosan: The Characterisation of Degraded Chitosan Scaffolds. J. Tissue Repair Regen. 2017, 1, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-K.; Shen, C.-R.; Liu, C.-L. N-acetylglucosamine: Production and applications. Mar. Drugs 2010, 8, 2493–2516. [Google Scholar] [CrossRef] [Green Version]
- Sohaebuddin, S.K.; Thevenot, P.T.; Baker, D.; Eaton, J.W.; Tang, L. Nanomaterial cytotoxicity is composition, size, and cell type dependent. Part. Fibre Toxicol. 2010, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Mann, A.; Richa, R.; Ganguli, M. DNA condensation by poly-l-lysine at the single molecule level: Role of DNA concentration and polymer length. J. Control. Release 2008, 125, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Cherng, J.Y.; Talsma, H.; Verrijk, R.; Crommelin, D.J.; Hennink, W.E. The effect of formulation parameters on the size of poly-((2-dimethylamino)ethyl methacrylate)-plasmid complexes. Eur. J. Pharm. Biopharm. 1999, 47, 215–224. [Google Scholar] [CrossRef]
- Ogris, M.; Steinlein, P.; Kursa, M.; Mechtler, K.; Kircheis, R.; Wagner, E. The size of DNA/transferrin-PEI complexes is an important factor for gene expression in cultured cells. Gene Ther. 1998, 5, 1425–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, A.; Khan, M.A.; Shukla, V.; Ganguli, M. Atomic force microscopy reveals the assembly of potential DNA “nanocarriers” by poly-l-ornithine. Biophys. Chem. 2007, 129, 126–136. [Google Scholar] [CrossRef]
- Dai, J.; Han, S.; Ju, F.; Han, M.; Xu, L.; Zhang, R.; Sun, Y. Preparation and evaluation of tumour microenvironment response multistage nanoparticles for epirubicin delivery and deep tumour penetration. Artif. Cells Nanomed. Biotechnol. 2018, 46, 860–873. [Google Scholar] [CrossRef] [Green Version]
- Uchegbu, I.F. Pharmaceutical nanotechnology: Polymeric vesicles for drug and gene delivery. Expert Opin. Drug Deliv. 2006, 3, 629–640. [Google Scholar] [CrossRef]
- Hanefeld, U.; Cao, L.; Magner, E. Enzyme immobilisation: Fundamentals and application. Chem. Soc. Rev. 2013, 42, 6211. [Google Scholar] [CrossRef]
- Fernandez-Lafuente, R. Editorial Special Issue: Enzyme Immobilization. Molecules 2014, 19, 20671–20674. [Google Scholar] [CrossRef] [Green Version]
- Karmakar, S. Enzyme Activity—An overview|ScienceDirect Topics. In Handbook of Nanomaterials for Industrial Applications; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Nyamay’antu, A.; Dumont, M.; Kedinger, V.; Erbacher, P. Non-Viral Vector Mediated Gene Delivery: The Outsider to Watch out for in Gene Therapy. Cell Gene Ther. Insights 2019, 5, 51–57. [Google Scholar] [CrossRef]
- Villate-Beitia, I.; Puras, G.; Zarate, J.; Agirre, M.; Ojeda, E.; Pedraz, J.L. First Insights into Non-invasive Administration Routes for Non-viral Gene Therapy. In Gene Therapy—Principles and Challenges; InTech: Vienna, Austria, 2015. [Google Scholar]
- Hadaczek, P.; Yamashita, Y.; Mirek, H.; Tamas, L.; Bohn, M.C.; Noble, C.; Park, J.W.; Bankiewicz, K. The ‘Perivascular Pump’ Driven by Arterial Pulsation Is a Powerful Mechanism for the Distribution of Therapeutic Molecules within the Brain. Mol. Ther. 2006, 14, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Paldor, I.; Pearce, F.C.; Drummond, K.J.; Kaye, A.H. Frontal glioblastoma multiforme may be biologically distinct from non-frontal and multilobar tumors. J. Clin. Neurosci. 2016, 34, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [Green Version]
- Pluen, A.; Boucher, Y.; Ramanujan, S.; McKee, T.D.; Gohongi, T.; di Tomaso, E.; Brown, E.B.; Izumi, Y.; Campbell, R.B.; Berk, D.A.; et al. Role of tumor-host interactions in interstitial diffusion of macromolecules: Cranial vs. subcutaneous tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 4628–4633. [Google Scholar] [CrossRef] [Green Version]
- Wenning, L.A.; Murphy, R.M. Coupled cellular trafficking and diffusional limitations in delivery of immunotoxins to multicell tumor spheroids. Biotechnol. Bioeng. 1999, 62, 562–575. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Ni, S.; Yang, X.; Kiviat, N.; Lieber, A. Xenograft models for liver metastasis: Relationship between tumor morphology and adenovirus vector transduction. Mol. Ther. 2004, 9, 650–657. [Google Scholar] [CrossRef]
- Huang, M.; Khor, E.; Lim, L.-Y. Uptake and Cytotoxicity of Chitosan Molecules and Nanoparticles: Effects of Molecular Weight and Degree of Deacetylation. Pharm Res. 2004, 21, 344–353. [Google Scholar] [CrossRef]
- Yilmaz, E. Chitosan: A Versatile Biomaterial. In Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 2004; pp. 59–68. [Google Scholar]
- Zhang, W.; Yang, H.; Kong, X.; Mohapatra, S.; Juan-Vergara, H.S.; Hellermann, G.; Behera, S.; Singam, R.; Lockey, R.F.; Mohapatra, S.S. Inhibition of respiratory syncytial virus infection with intranasal siRNA nanoparticles targeting the viral NS1 gene. Nat. Med. 2005, 11, 56–62. [Google Scholar] [CrossRef]
- Simão Carlos, M.I.; Zheng, K.; Garrett, N.; Arifin, N.; Workman, D.G.; Kubajewska, I.; Halwani, A.A.; Moger, J.; Zhang, Q.; Schätzlein, A.G.; et al. Limiting the level of tertiary amines on polyamines leads to biocompatible nucleic acid vectors. Int. J. Pharm. 2017, 526, 106–124. [Google Scholar] [CrossRef]
- Sanchez-Ramos, J.; Song, S.; Kong, X.; Foroutan, P.; Martinez, G.; Dominguez-Viqueria, W.; Mohapatra, S.; Mohapatra, S.; Haraszti, R.A.; Khvorova, A.; et al. Chitosan-Mangafodipir nanoparticles designed for intranasal delivery of siRNA and DNA to brain. J. Drug Deliv. Sci. Technol. 2018, 43, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yurek, D.M.; Fletcher, A.M.; McShane, M.; Kowalczyk, T.H.; Padegimas, L.; Weatherspoon, M.R.; Kaytor, M.D.; Cooper, M.J.; Ziady, A.G. DNA nanoparticles: Detection of long-term transgene activity in brain using bioluminescence imaging. Mol. Imaging 2011, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gänger, S.; Schindowski, K. Tailoring Formulations for Intranasal Nose-to-Brain Delivery: A Review on Architecture, Physico-Chemical Characteristics and Mucociliary Clearance of the Nasal Olfactory Mucosa. Pharmaceutics 2018, 10, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaraj, K.; Gowthamarajan, K.; Karri, V.V.S.R. Nose to brain transport pathways an overview: Potential of nanostructured lipid carriers in nose to brain targeting. Artif. Cells Nanomed. Biotechnol. 2018, 46, 2088–2095. [Google Scholar] [CrossRef]
- Liu, X.-F.; Fawcett, J.R.; Hanson, L.R.; Frey, W.H. The window of opportunity for treatment of focal cerebral ischemic damage with noninvasive intranasal insulin-like growth factor-I in rats. J. Stroke Cerebrovasc. Dis. 2004, 13, 16–23. [Google Scholar] [CrossRef]
- Sava, V.; Fihurka, O.; Khvorova, A.; Sanchez-Ramos, J. Enriched chitosan nanoparticles loaded with siRNA are effective in lowering Huntington’s disease gene expression following intranasal administration. Nanomed. Nanotechnol. Biol. Med. 2020, 24, 102119. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polyplex | Size (nm) | PDI | Zeta Potential (mV) |
|---|---|---|---|
| GCP 1 | 135 ± 21 | 0.229 ± 0.033 | +7 ± 3 |
| GCP 2 | 482 ± 36 | 0.113 ± 0.023 | +9 ± 4 |
| GCP 3 | 863 ± 28 | 0.161 ± 0.021 | +10 ± 3 |
| GCPH 7 | 526 ± 14 | 0.578 ± 0.053 | −10 ± 2 |
| DNA | 140 ± 31 (65%) | 0.589 ± 0.077 | −26 ± 5 |
| 950 ± 61 (23%) | |||
| 20 ± 9 (7%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petkova, A.I.; Kubajewska, I.; Vaideanu, A.; Schätzlein, A.G.; Uchegbu, I.F. Gene Targeting to the Cerebral Cortex Following Intranasal Administration of Polyplexes. Pharmaceutics 2022, 14, 1136. https://doi.org/10.3390/pharmaceutics14061136
Petkova AI, Kubajewska I, Vaideanu A, Schätzlein AG, Uchegbu IF. Gene Targeting to the Cerebral Cortex Following Intranasal Administration of Polyplexes. Pharmaceutics. 2022; 14(6):1136. https://doi.org/10.3390/pharmaceutics14061136
Chicago/Turabian StylePetkova, Asya I., Ilona Kubajewska, Alexandra Vaideanu, Andreas G. Schätzlein, and Ijeoma F. Uchegbu. 2022. "Gene Targeting to the Cerebral Cortex Following Intranasal Administration of Polyplexes" Pharmaceutics 14, no. 6: 1136. https://doi.org/10.3390/pharmaceutics14061136