
Effects of Drug Physicochemical Properties on In-Situ Forming Implant Polymer Degradation and Drug Release Kinetics

 , ,
, ,
Abstract
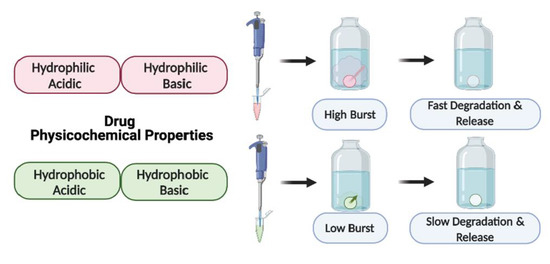
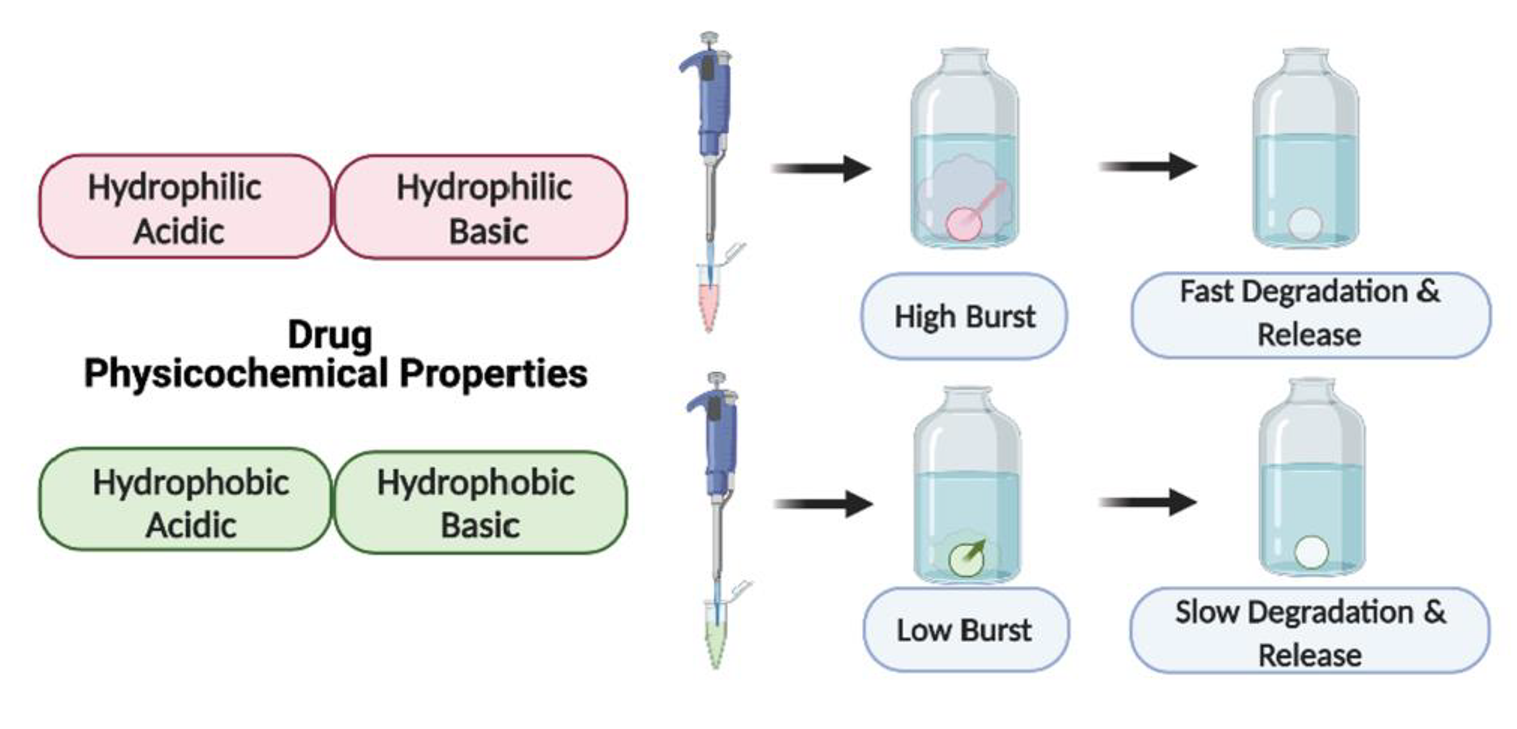
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. High-Performance Liquid Chromatography (HPLC)
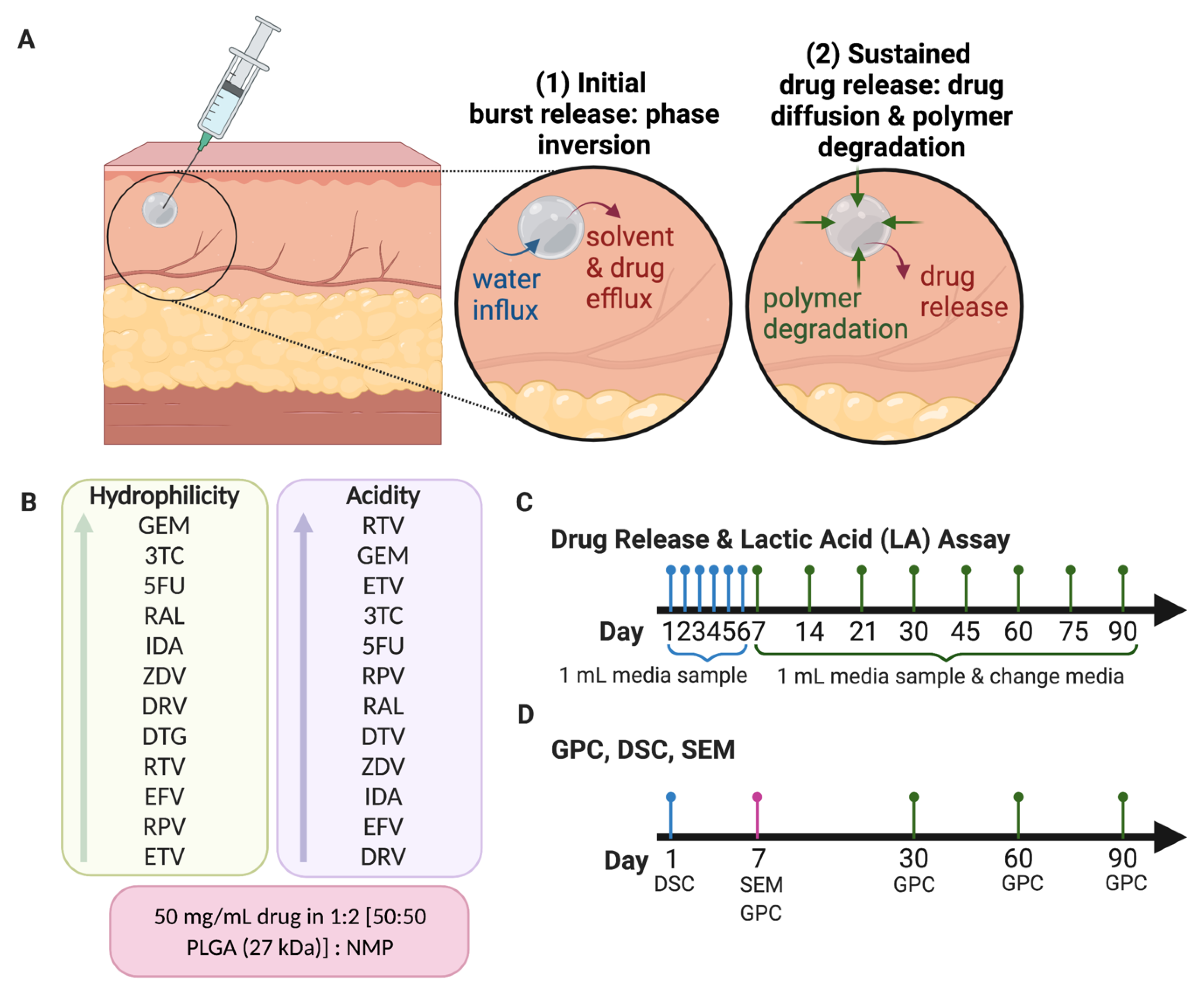
2.2.2. Preparation of ISFI Formulations
2.2.3. Differential Scanning Calorimetry (DSC)
2.2.4. In Vitro Cumulative Drug Release
2.2.5. Quantification of L-Lactic Acid (Lactic Acid Assay)
2.2.6. Gel Permeation Chromatography (GPC)
2.2.7. Scanning Electron Microscopy (SEM) Imaging and Analysis
3. Results
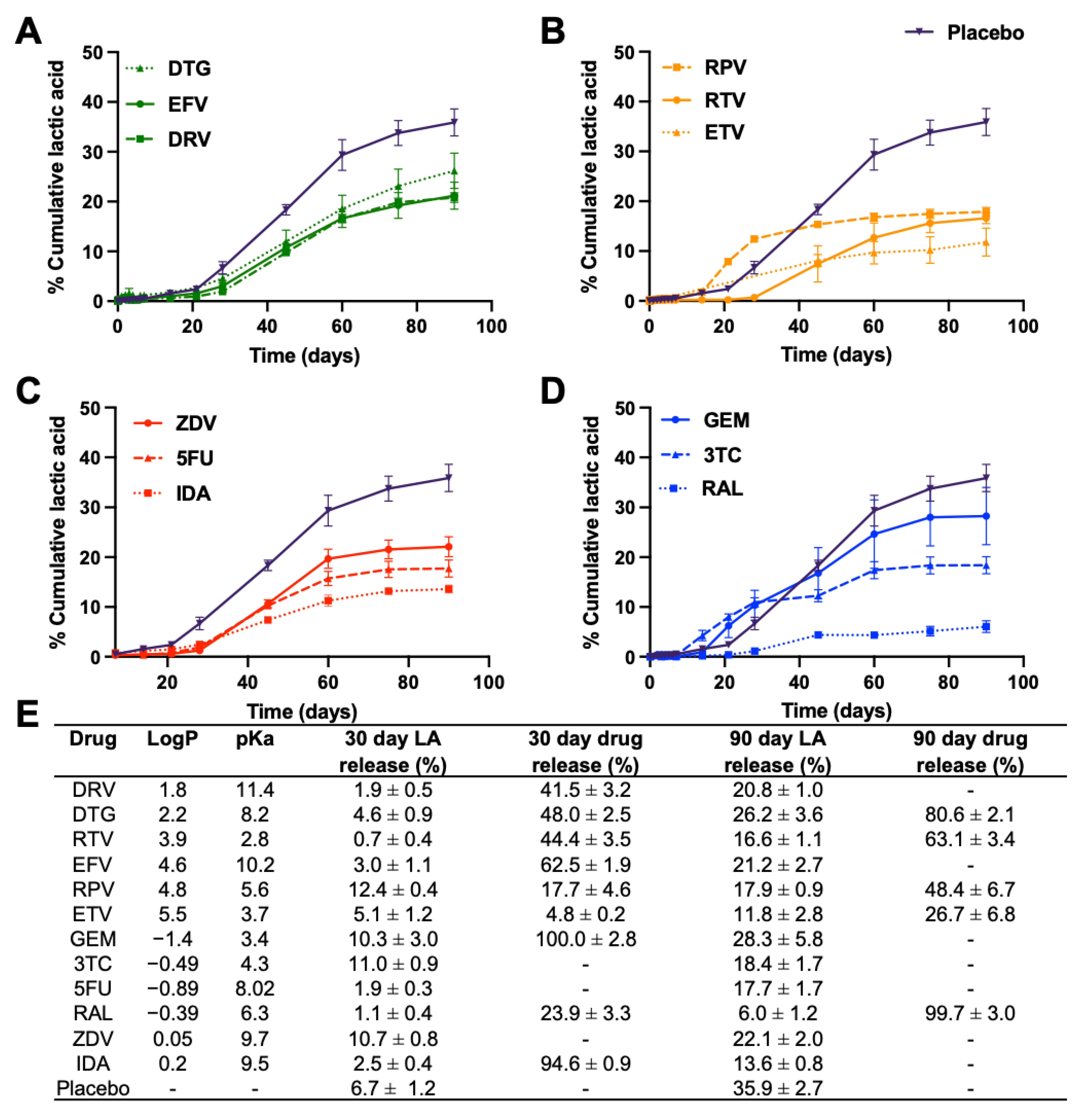
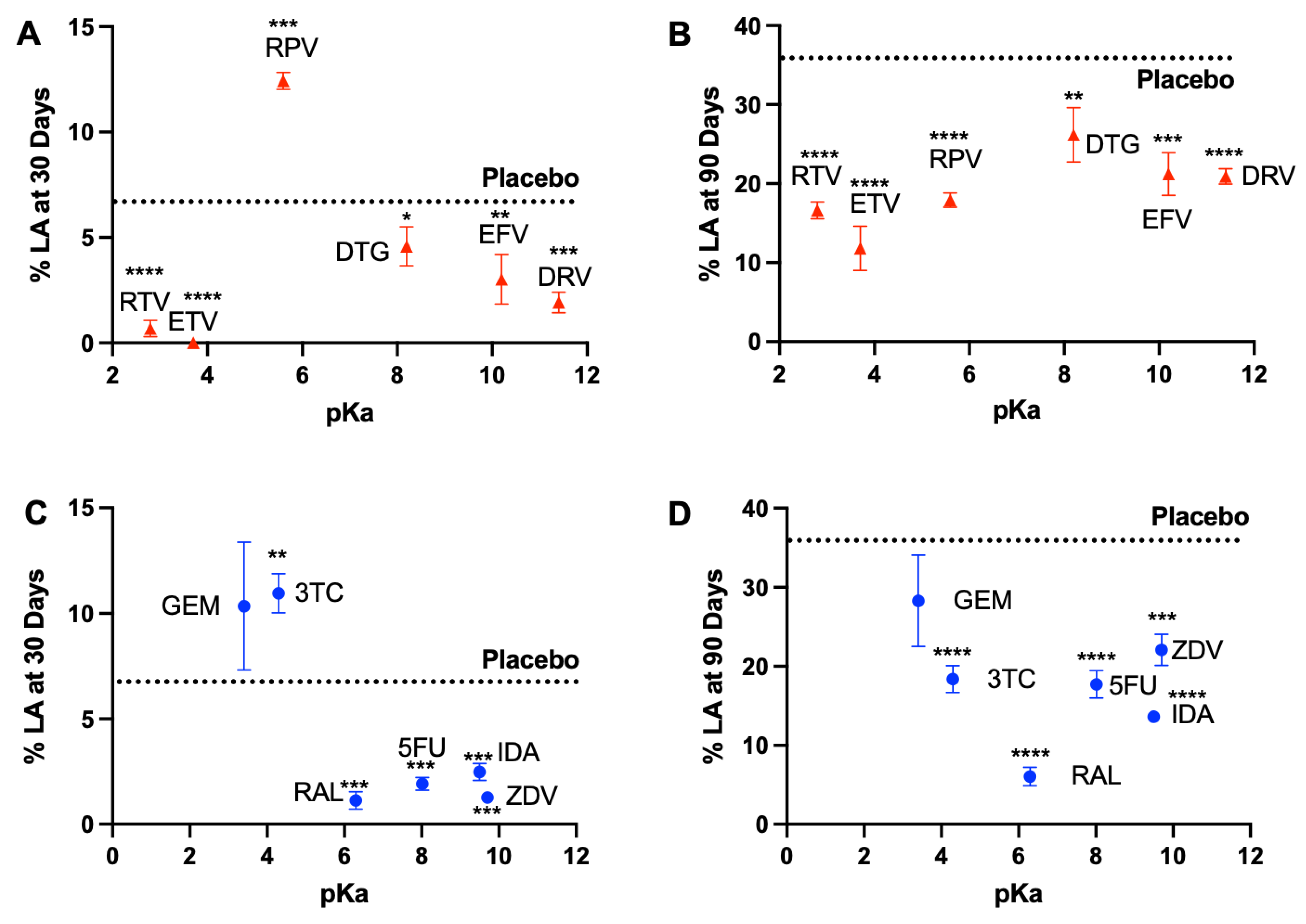
3.1. In Vitro Release Kinetics and Lactic Acid Release
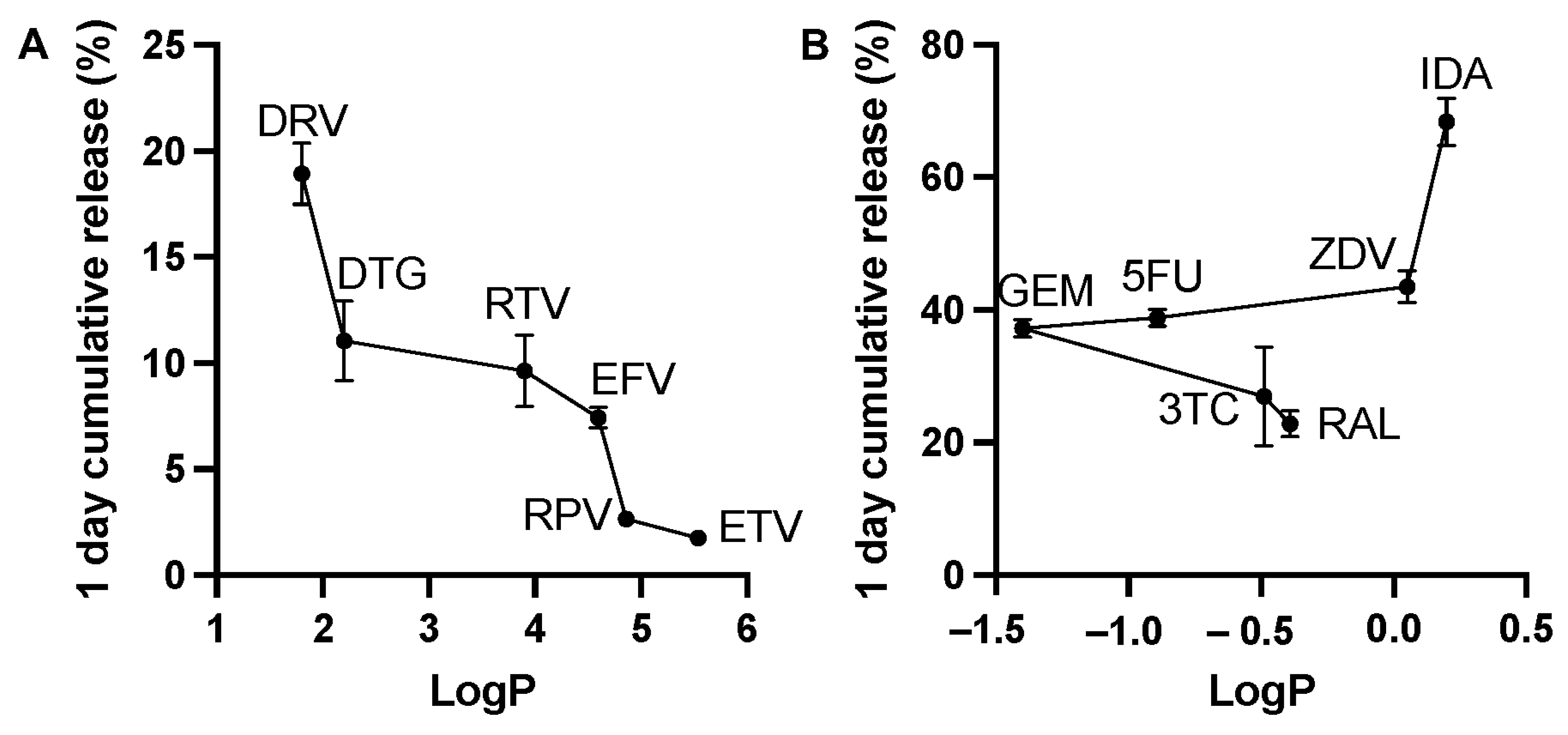
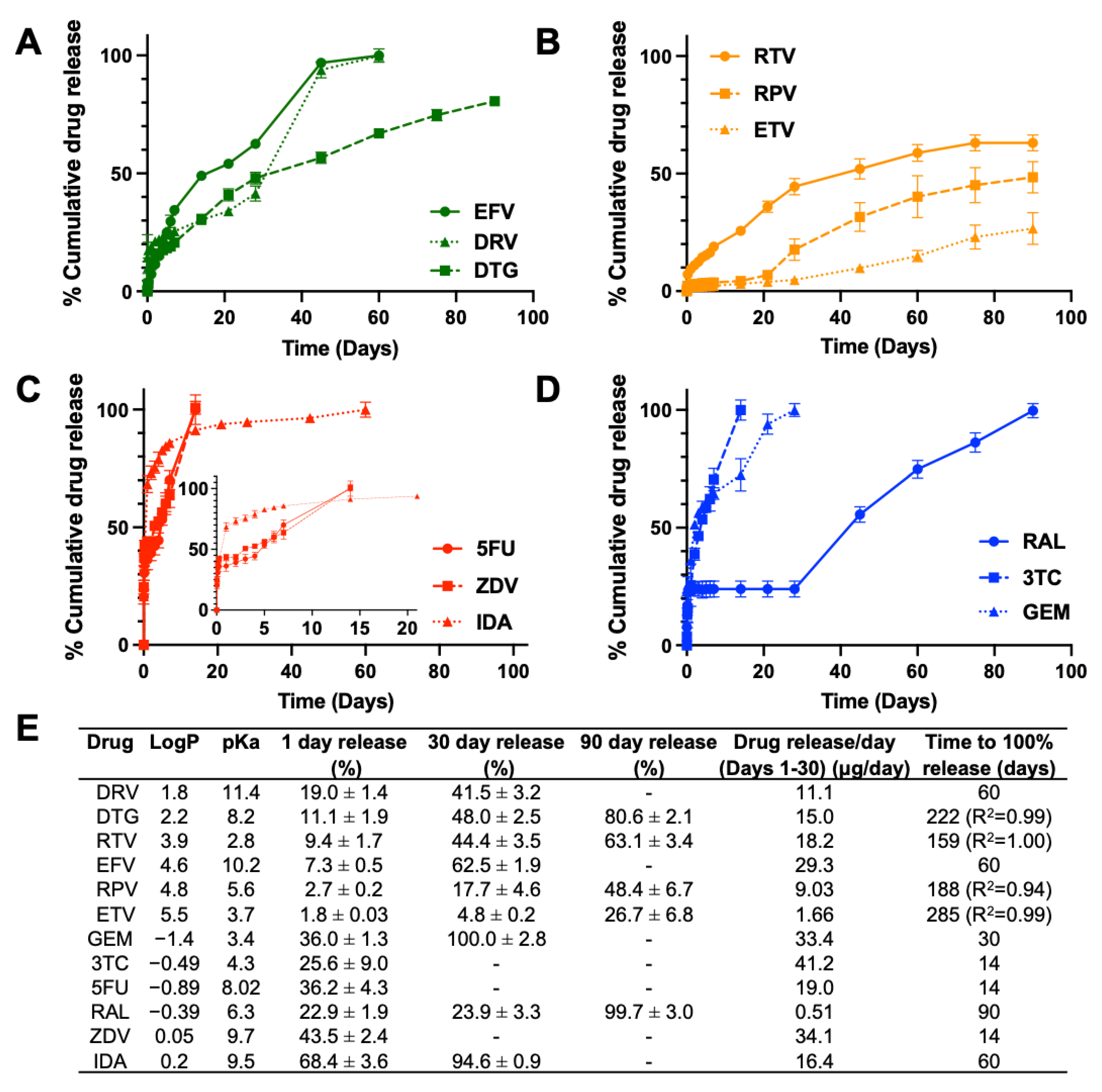
3.1.1. Drug Burst Release 1 Day Post-ISFI Injection
3.1.2. Overall Drug Release Kinetics
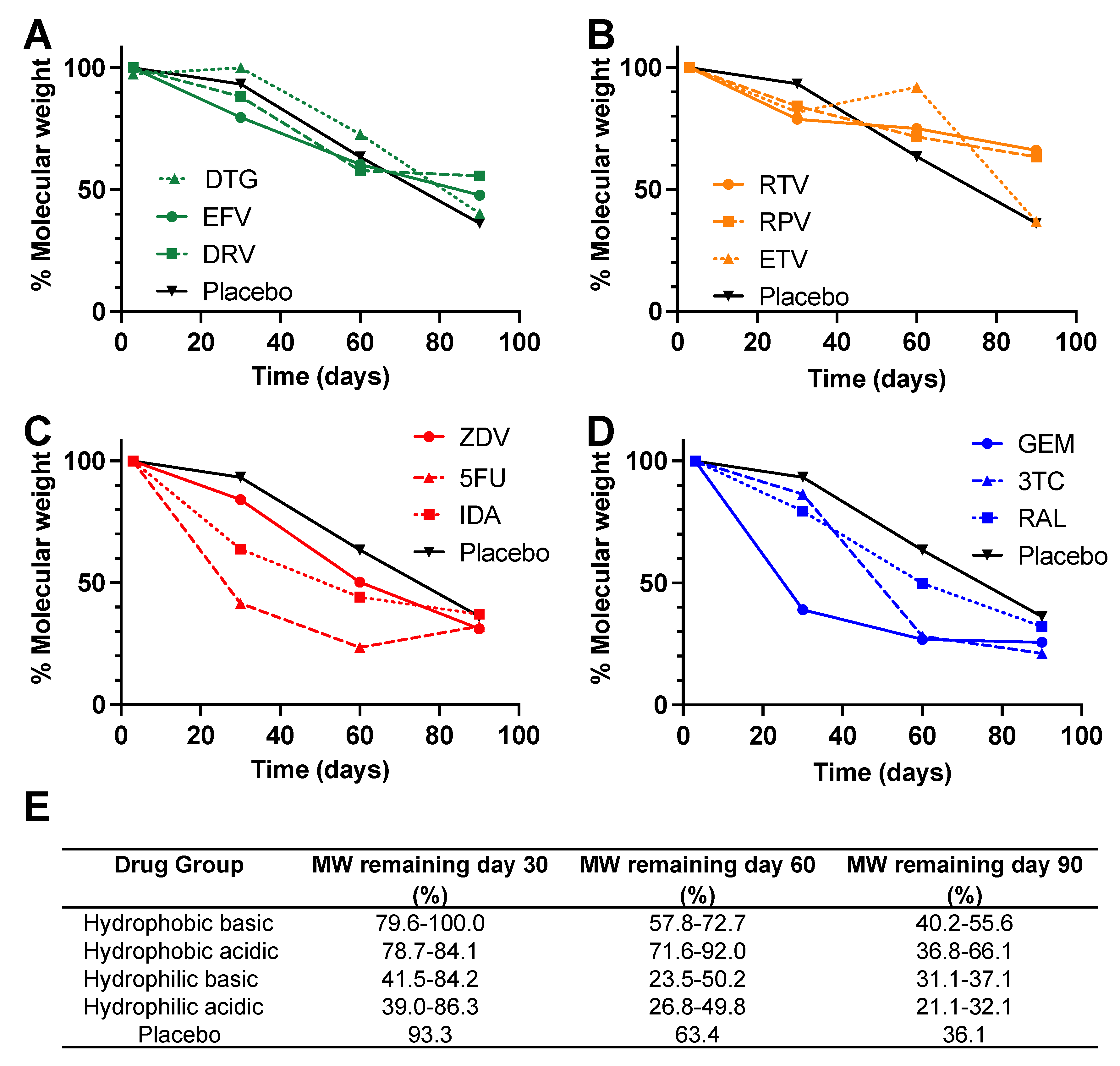
3.2. Gel Permeation Chromatography (GPC) Analysis
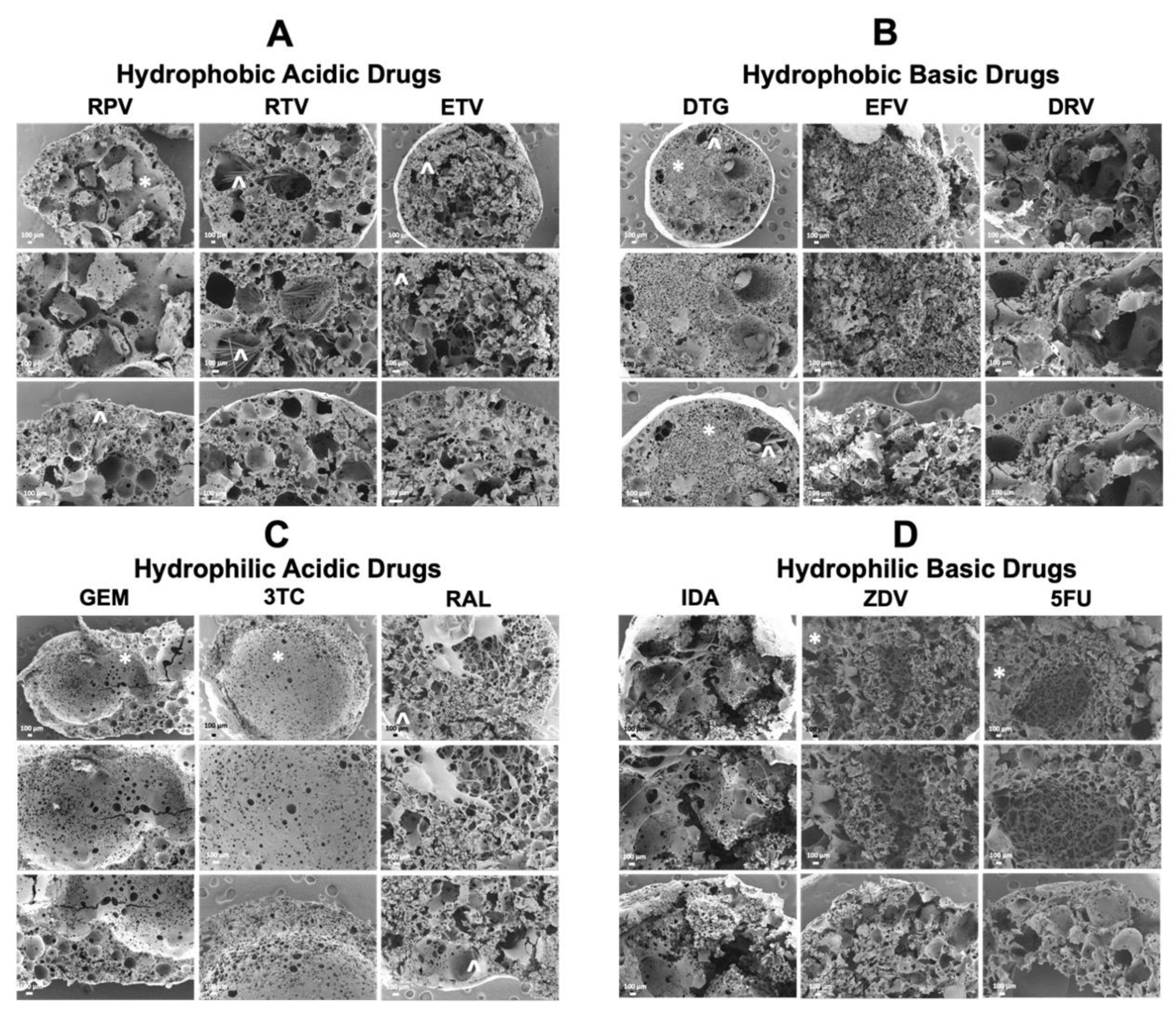
3.3. Scanning Electron Microscopy (SEM) Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ravivarapu, H.B.; Moyer, K.L.; Dunn, R.L. Sustained activity and release of leuprolide acetate from an in situ forming polymeric implant. AAPS PharmSciTech 2000, 1, 732–741. [Google Scholar]
- Kapoor, D.N.; Katare, O.P.; Dhawan, S. In situ forming implant for controlled delivery of an anti-HIV fusion inhibitor. Int. J. Pharm. 2012, 426, 132–143. [Google Scholar] [CrossRef]
- Kempe, S.; Mader, K. In situ forming implants—An attractive formulation principle for parenteral depot formulations. J. Control. Release 2012, 161, 668–679. [Google Scholar] [CrossRef]
- Huang, C.C.; Chia, W.T.; Chung, M.F.; Lin, K.J.; Hsiao, C.W.; Jin, C.; Lim, W.H.; Chen, C.C.; Sung, H.W. An Implantable Depot That Can Generate Oxygen in Situ for Overcoming Hypoxia-Induced Resistance to Anticancer Drugs in Chemotherapy. J. Am. Chem. Soc. 2016, 138, 5222–5225. [Google Scholar] [CrossRef]
- Solorio, L.; Exner, A.A. Effect of the Subcutaneous Environment on Phase-Sensitive In Situ-Forming Implant Drug Release, Degradation, and Microstructure. J. Pharm. Sci. 2015, 104, 4322–4328. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zhang, X.; Mu, H.; Hua, H.; Duan, D.; Yan, X.; Wang, Y.; Meng, Q.; Lu, X.; Wang, A.; et al. Preparation and evaluation of injectable Rasagiline mesylate dual-controlled drug delivery system for the treatment of Parkinson’s disease. Drug Deliv. 2018, 25, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Joiner, J.B.; King, J.L.; Shrivastava, R.; Howard, S.A.; Cottrell, M.L.; Kashuba, A.D.M.; Dayton, P.A.; Benhabbour, S.R. Effects of Injection Volume and Route of Administration on Dolutegravir In Situ Forming Implant Pharmacokinetics. Pharmaceutics 2022, 14, 615. [Google Scholar] [CrossRef]
- Patel, R.B.; Carlson, A.N.; Solorio, L.; Exner, A.A. Characterization of formulation parameters affecting low molecular weight drug release from in situ forming drug delivery systems. J. Biomed. Mater. Res. A 2010, 94, 476–484. [Google Scholar]
- Solorio, L.; Olear, A.M.; Zhou, H.; Beiswenger, A.C.; Exner, A.A. Effect of cargo properties on in situ forming implant behavior determined by noninvasive ultrasound imaging. Drug Deliv. Transl. Res. 2012, 2, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Solorio, L.; Sundarapandiyan, D.; Olear, A.; Exner, A.A. The Effect of Additives on the Behavior of Phase Sensitive In Situ Forming Implants. J. Pharm. Sci. 2015, 104, 3471–3480. [Google Scholar] [CrossRef] [Green Version]
- McHugh, A.J. The role of polymer membrane formation in sustained release drug delivery systems. J Control. Release 2005, 109, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Lyu, S.; Untereker, D. Degradability of polymers for implantable biomedical devices. Int. J. Mol. Sci. 2009, 10, 4033–4065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodard, L.N.; Grunlan, M.A. Hydrolytic Degradation and Erosion of Polyester Biomaterials. ACS Macro Lett. 2018, 7, 976–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, T.W.J.; Huang, C.L.; Kumar, S.; Iskandar, A.; Baoxin, A.; Chiang Boey, F.Y.; Loo, J.S.C.; Venkatraman, S.S. Tuning drug release in polyester thin films: Terminal end-groups determine specific rates of additive-free controlled drug release. NPG Asia Mater. 2013, 5, e46. [Google Scholar] [CrossRef] [Green Version]
- Keles, H.; Naylor, A.; Clegg, F.; Sammon, C. Investigation of factors influencing the hydrolytic degradation of single PLGA microparticles. Polym. Degrad. Stab. 2015, 119, 228–241. [Google Scholar] [CrossRef] [Green Version]
- Benhabbour, S.R.; Kovarova, M.; Jones, C.; Copeland, D.J.; Shrivastava, R.; Swanson, M.D.; Sykes, C.; Ho, P.T.; Cottrell, M.L.; Sridharan, A.; et al. Ultra-long-acting tunable biodegradable and removable controlled release implants for drug delivery. Nat. Commun. 2019, 10, 4324. [Google Scholar] [CrossRef] [PubMed]
- Washington, M.A.; Swiner, D.J.; Bell, K.R.; Fedorchak, M.V.; Little, S.R.; Meyer, T.Y. The impact of monomer sequence and stereochemistry on the swelling and erosion of biodegradable poly(lactic-co-glycolic acid) matrices. Biomaterials 2017, 117, 66–76. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yang, L.; Zhang, C.; Liu, D.; Meng, S.; Zhang, W.; Meng, S. Effect of Polymer Permeability and Solvent Removal Rate on In Situ Forming Implants: Drug Burst Release and Microstructure. Pharmaceutics 2019, 11, 520. [Google Scholar] [CrossRef] [Green Version]
- Koocheki, S.; Madaeni, S.S.; Niroomandi, P. Development of an enhanced formulation for delivering sustained release of buprenorphine hydrochloride. Saudi Pharm. J. 2011, 19, 255–262. [Google Scholar] [CrossRef] [Green Version]
- Alexis, F. Factors affecting the degradation and drug-release mechanism of poly(lactic acid) and poly[(lactic acid)-co-(glycolic acid)]. Polym. Int. 2005, 54, 36–46. [Google Scholar] [CrossRef]
- Zolnik, B.S.; Burgess, D.J. Effect of acidic pH on PLGA microsphere degradation and release. J. Control. Release 2007, 122, 338–344. [Google Scholar] [CrossRef]
- Li, J.; Jiang, G.; Ding, F. The effect of pH on the polymer degradation and drug release from PLGA-mPEG microparticles. J. Appl. Polym. Sci. 2008, 109, 475–482. [Google Scholar] [CrossRef]
- Zeng, L.; An, L.; Wu, X. Modeling drug-carrier interaction in the drug release from nanocarriers. J. Drug Deliv. 2011, 2011, 370308. [Google Scholar] [CrossRef] [PubMed]
- Yapar, E.A.; Baykara, T. Thermal Analysis on Phase Sensitive Granisetron In Situ Forming Implants. J. Appl. Pharm. Sci. 2014, 4, 010–013. [Google Scholar]
- Saber, R.A.; Attia, A.K.; Salem, W.M. Thermal analysis study of antihypertensive drugs telmisartan and cilazapril. Adv. Pharm. Bull. 2014, 4, 283–287. [Google Scholar]
- Ding, A.; Schwendeman, S.P. Determination of Water-Soluble Acid Distribution in Poly(lactide-co-glycolide). J. Pharm. Sci. 2004, 93, 322–331. [Google Scholar] [CrossRef] [Green Version]
- Manaspon, C.; Hernandez, C.; Nittayacharn, P.; Jeganathan, S.; Nasongkla, N.; Exner, A.A. Increasing Distribution of Drugs Released from In Situ Forming PLGA Implants Using Therapeutic Ultrasound. Ann. Biomed. Eng. 2017, 45, 2879–2887. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef]
- Prabhu, S.; Tran, L.P.; Betageri, G.V. Effect of co-solvents on the controlled release of calcitonin polypeptide from in situ biodegradable polymer implants. Drug Deliv. 2005, 12, 393–398. [Google Scholar] [CrossRef]
- Zare, M.; Mobedi, H.; Barzin, J.; Mivehchi, H.; Jamshidi, A.; Mashayekhi, R. Effect of additives on release profile of leuprolide acetate in an in situ forming controlled-release system: In vitro study. J. Appl. Polym. Sci. 2008, 107, 3781–3787. [Google Scholar] [CrossRef]
- Parent, M.; Nouvel, C.; Koerber, M.; Sapin, A.; Maincent, P.; Boudier, A. PLGA in situ implants formed by phase inversion: Critical physicochemical parameters to modulate drug release. J. Control. Release 2013, 172, 292–304. [Google Scholar] [CrossRef]
- Siegel, S.J.; Kahn, J.B.; Metzger, K.; Winey, K.I.; Werner, K.; Dan, N. Effect of drug type on the degradation rate of PLGA matrices. Eur. J. Pharm. Biopharm. 2006, 64, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, M.; Koshika, A.; Okada, J.I.; Ikeda, M. Effect of polymer/basic drug interactions on the two-stage diffusion-controlled release from a poly(l-lactic acid) matrix. J. Control. Release 1999, 61, 295–304. [Google Scholar] [CrossRef]
- D’Souza, S.; Faraj, J.A.; Dorati, R.; DeLuca, P.P. Enhanced degradation of lactide-co-glycolide polymer with basic nucleophilic drugs. Adv. Pharm. 2015, 2015, 154239. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Chemical Structure | MW (g/mol) | LogP * | pKa * | Class |
|---|---|---|---|---|---|
| Rilpivirine (RPV) |  | 366.42 | 4.86 | 5.6 | ARV-NNRTI |
| Ritonavir (RTV) |  | 720.95 | 3.90 | 2.8 | Booster |
| Etravirine (ETV) |  | 435.28 | 5.54 | 3.7 | ARV-NNRTI |
| Efavirenz (EFV) |  | 315.6 | 4.6 | 10.2 | ARV-NNRTI |
| Darunavir (DRV) |  | 547.6 | 1.8 | 11.4 | ARV-PI |
| Dolutegravir (DTG) |  | 419.3 | 2.2 | 8.2 | ARV-ISTI |
| Gemcitabine (GEM) |  | 263.2 | −1.4 | 3.6 | Antimetabolite |
| Lamivudine (3TC) |  | 229.2 | −0.49 | 4.8 | ARV-NRTI |
| Raltegravir (RAL) |  | 444.4 | −0.39 | 6.3 | ARV-ISTI |
| Idarubicin (IDA) |  | 497.5 | 0.2 | 9.5 | Anthracycline |
| Zidovudine (ZDV) |  | 267.24 | 0.05 | 9.5 | ARV-NRTI |
| 5-Fluorouracil (5FU) |  | 130.27 | −0.89 | 8.02 | Antimetabolite |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joiner, J.B.; Prasher, A.; Young, I.C.; Kim, J.; Shrivastava, R.; Maturavongsadit, P.; Benhabbour, S.R. Effects of Drug Physicochemical Properties on In-Situ Forming Implant Polymer Degradation and Drug Release Kinetics. Pharmaceutics 2022, 14, 1188. https://doi.org/10.3390/pharmaceutics14061188
Joiner JB, Prasher A, Young IC, Kim J, Shrivastava R, Maturavongsadit P, Benhabbour SR. Effects of Drug Physicochemical Properties on In-Situ Forming Implant Polymer Degradation and Drug Release Kinetics. Pharmaceutics. 2022; 14(6):1188. https://doi.org/10.3390/pharmaceutics14061188
Chicago/Turabian StyleJoiner, Jordan B., Alka Prasher, Isabella C. Young, Jessie Kim, Roopali Shrivastava, Panita Maturavongsadit, and Soumya Rahima Benhabbour. 2022. "Effects of Drug Physicochemical Properties on In-Situ Forming Implant Polymer Degradation and Drug Release Kinetics" Pharmaceutics 14, no. 6: 1188. https://doi.org/10.3390/pharmaceutics14061188