
New Amino Naphthoquinone Derivatives as Anti-Trypanosoma cruzi Agents Targeting Trypanothione Reductase

 , , ,
, , ,
Abstract
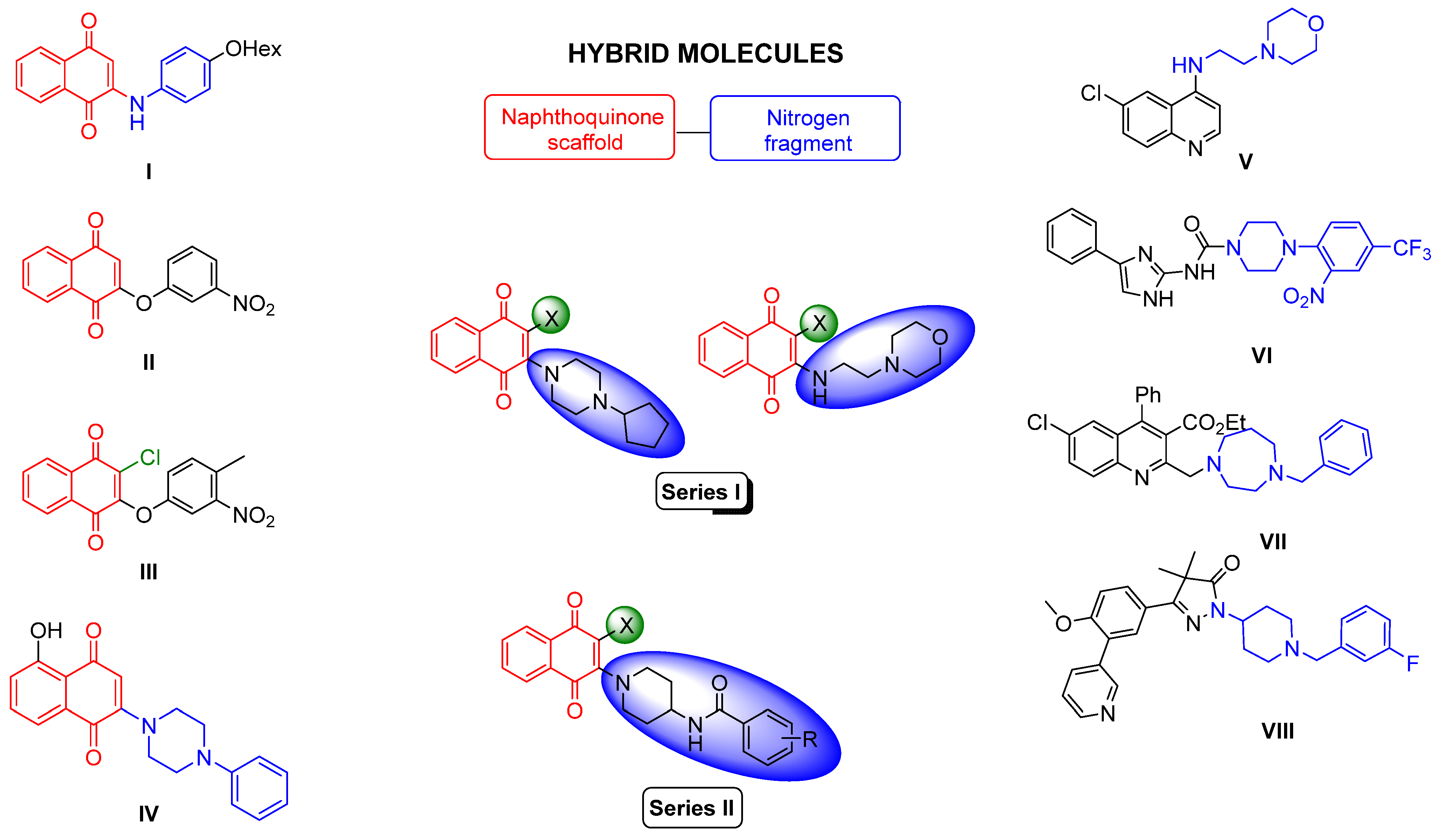
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Instrumentation
2.3. Synthesis
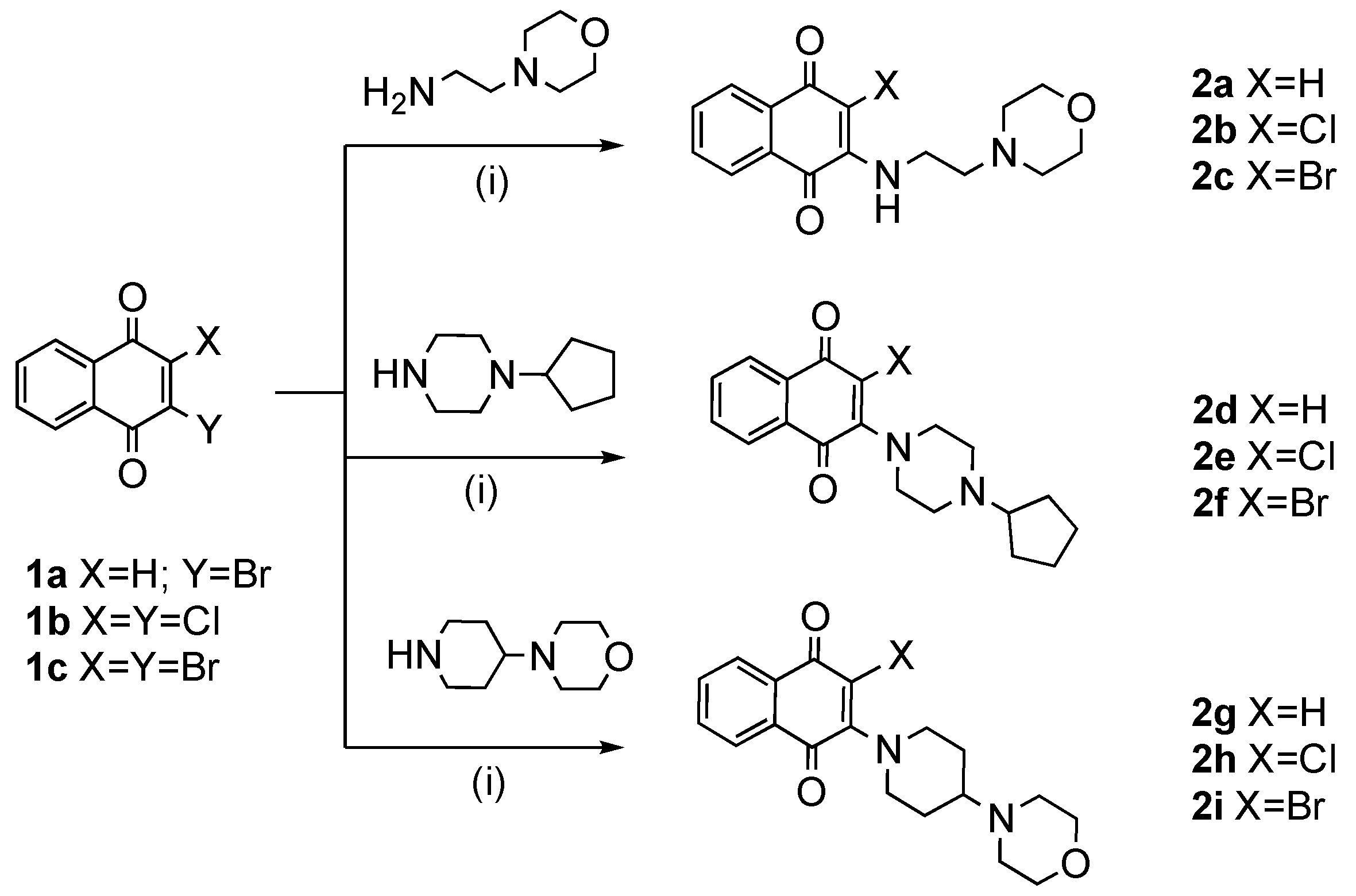
2.3.1. General Procedure for the Synthesis for Compounds 2a–i
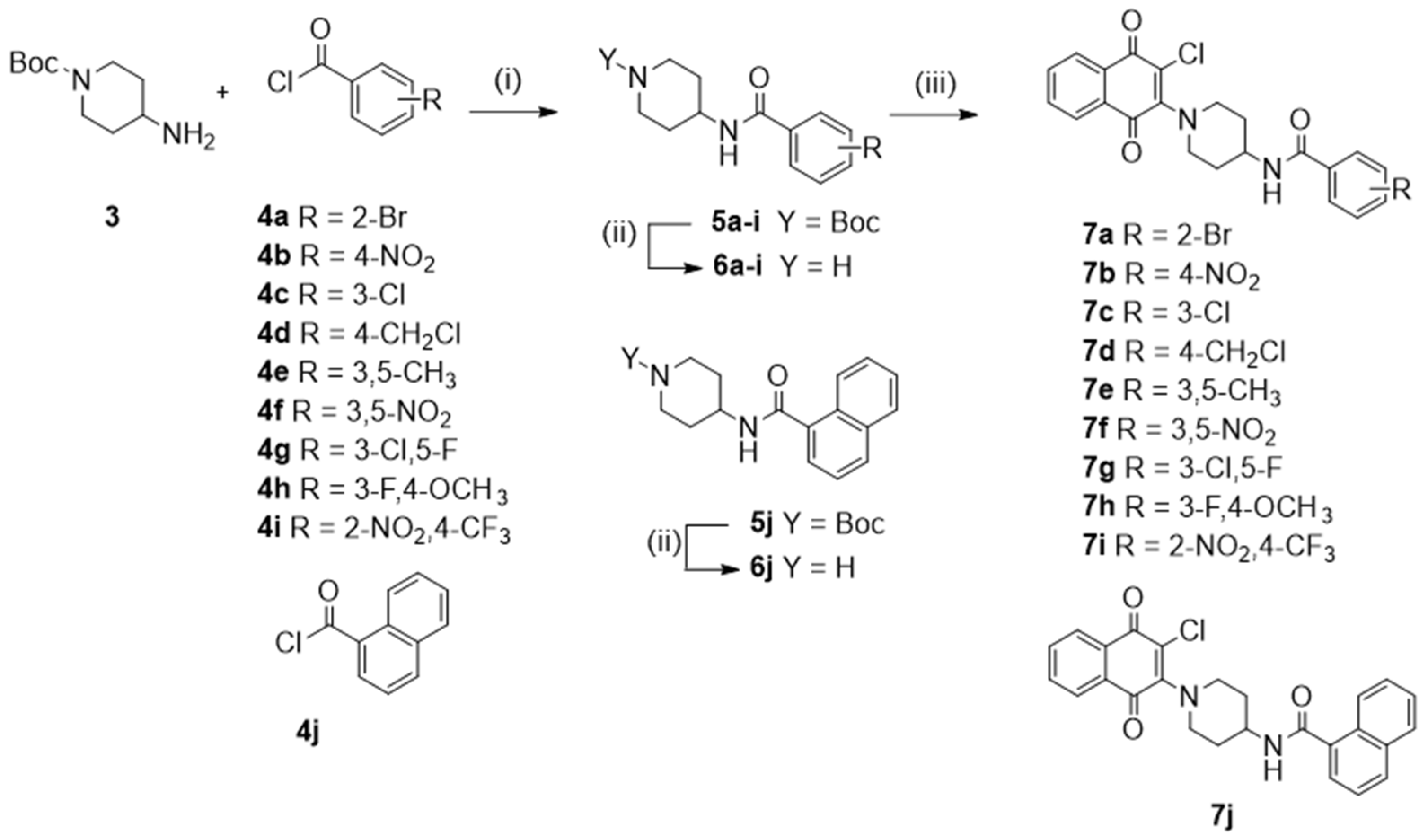
2.3.2. General Procedure for the Synthesis for Compounds 5a–j
2.3.3. General Procedure for the Synthesis for Compounds 6a–j
2.3.4. General Procedure for the Synthesis for Compounds 7a–j
2.4. Trypanocidal Activity
2.4.1. Trypanocidal Effect
2.4.2. Ex Vivo Evaluation of INC-5 and NINOA Strain Trypomastigotes
2.5. Cytotoxicity Assays
2.6. Docking
2.6.1. Ligand Preparation
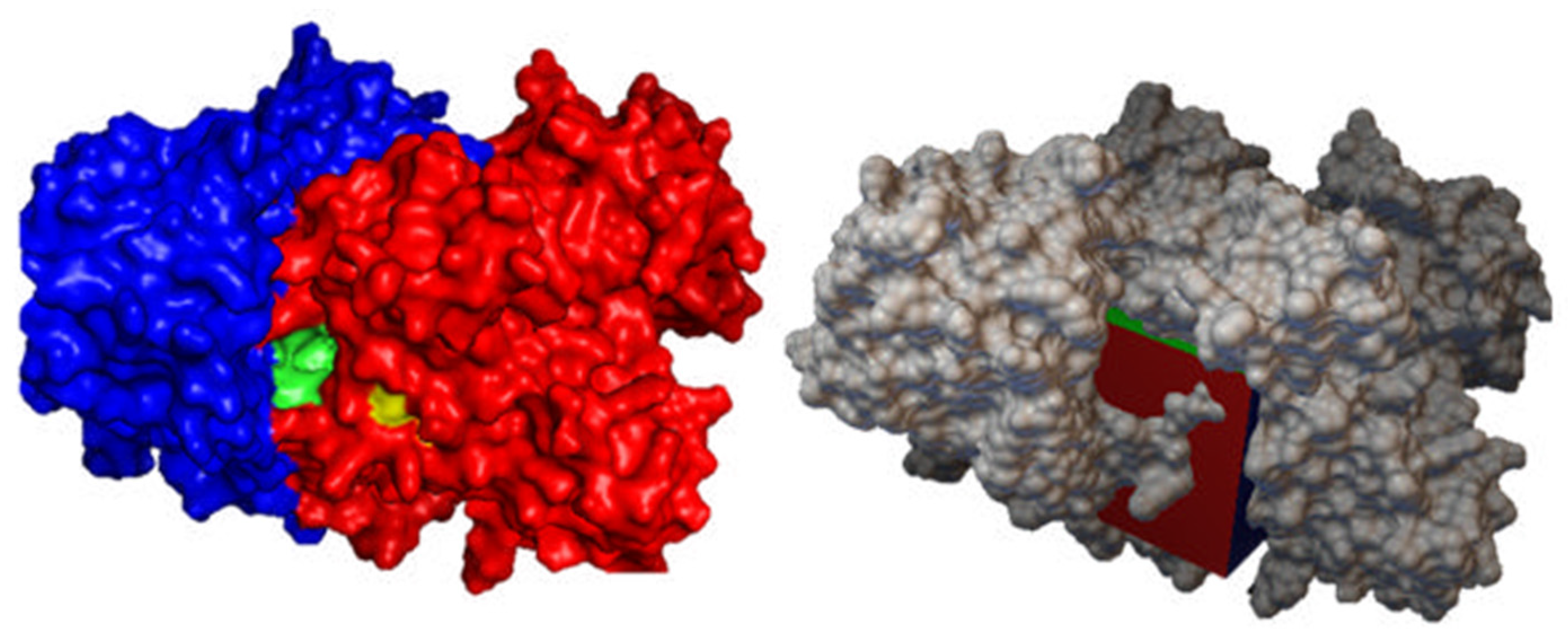
2.6.2. Predicted Bindings Site
2.6.3. Protein Preparation
2.6.4. Molecular Docking
2.7. Trypanothione Reductase Inhibition Assay
3. Results and Discussion
3.1. Chemistry
3.2. Trypanocidal Effects on Epimastigote and Trypomastigote Strains
3.3. Cytotoxicity in Murine Macrophages
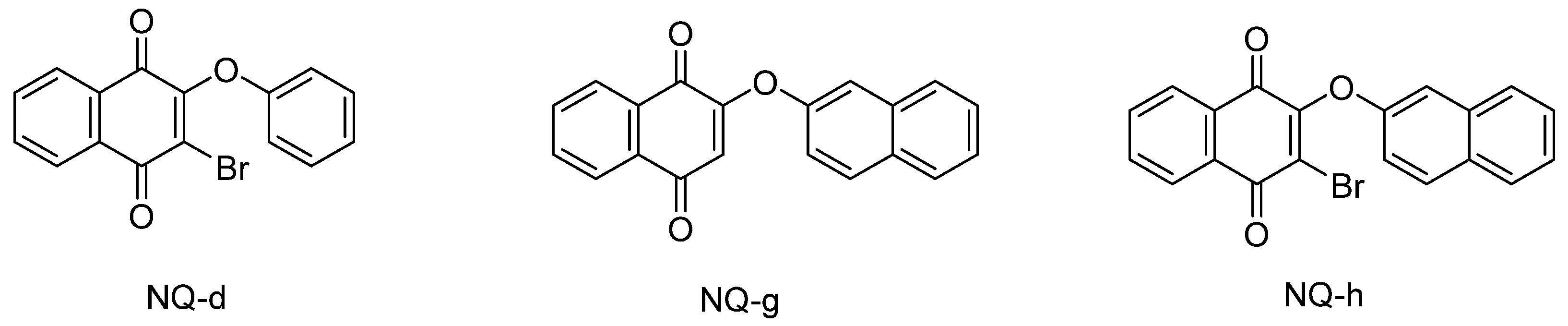
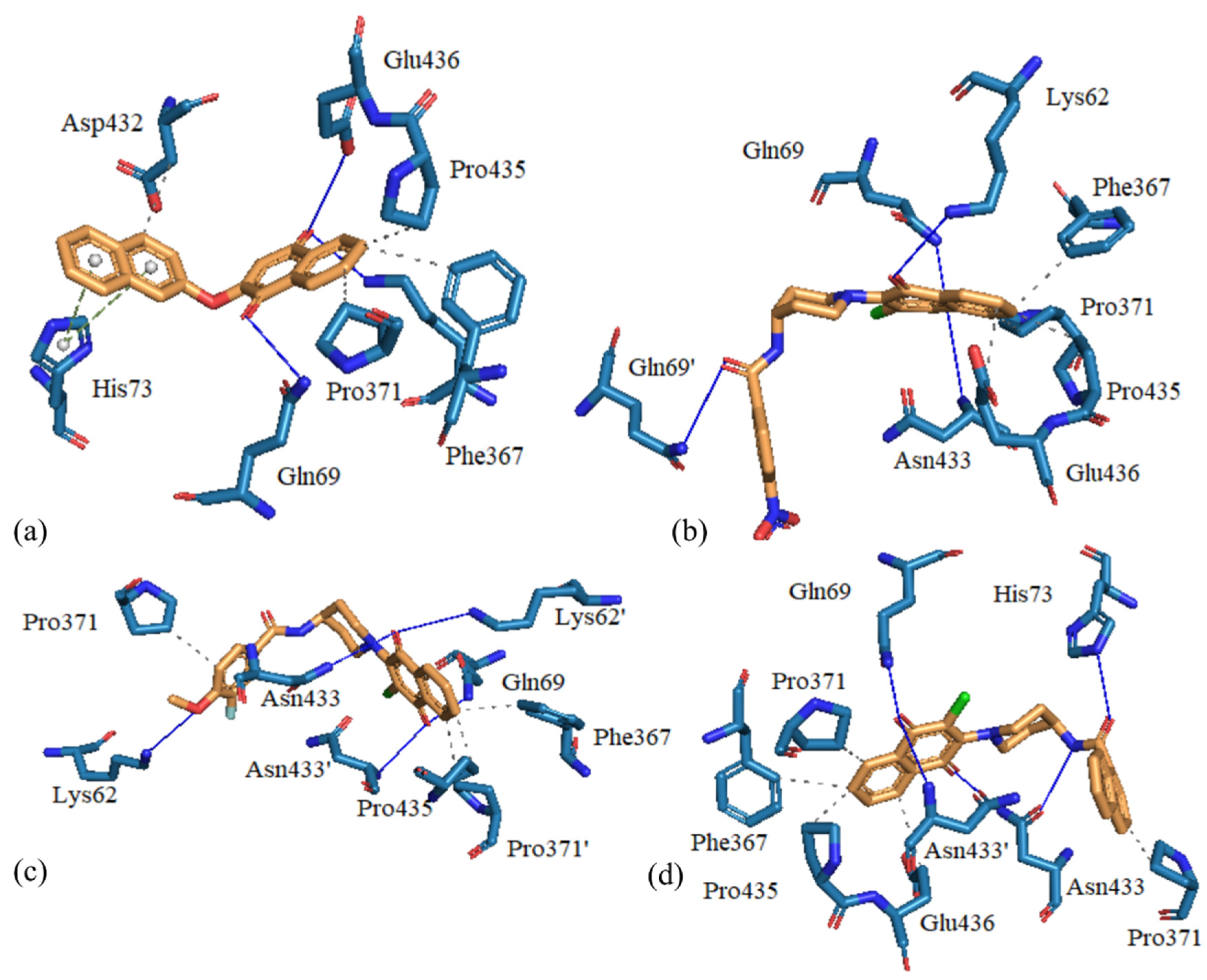
3.4. Molecular Docking Studies
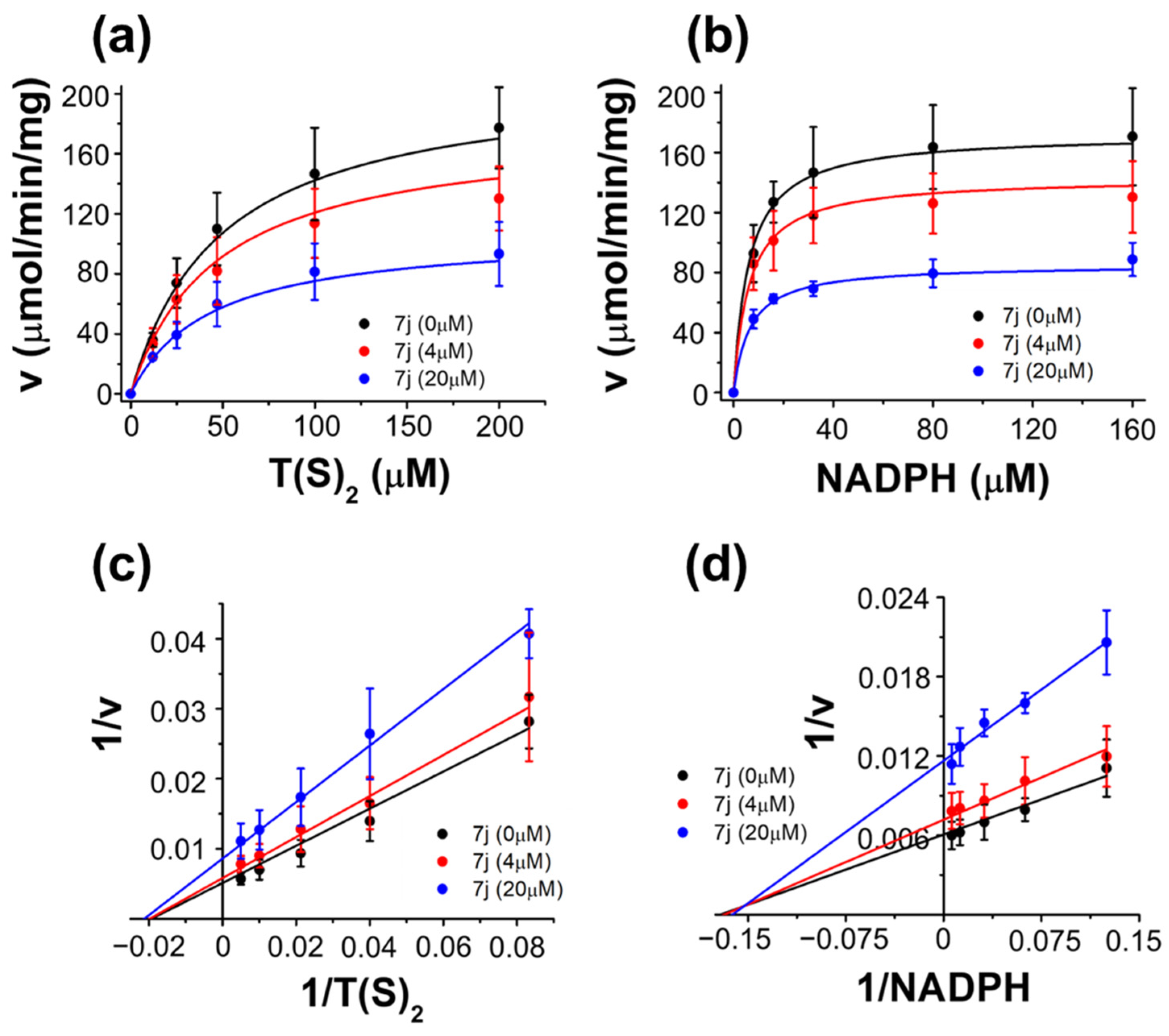
3.5. Evaluation of 7j on TcTR
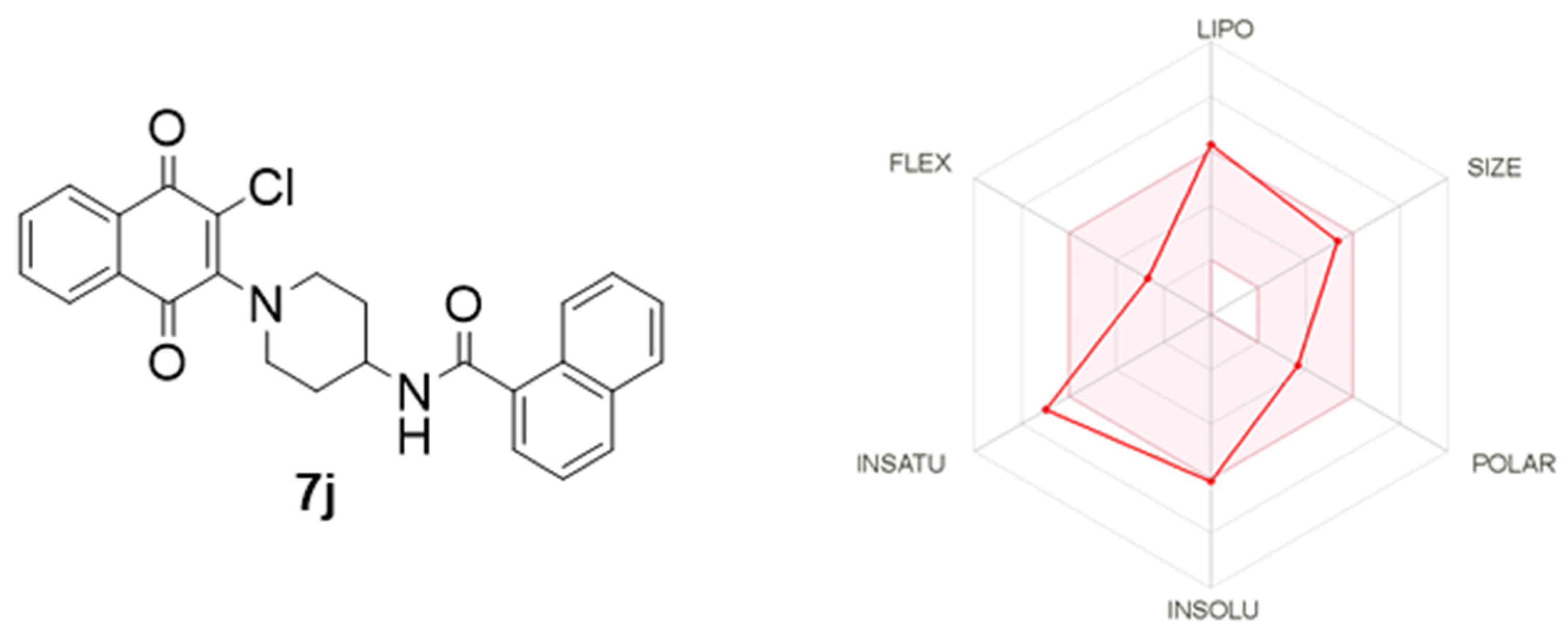
3.6. Calculated Physicochemical Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Heath Organization (WHO). 2020. Available online: https://www.who.int/neglected_diseases/diseases/en/ (accessed on 20 March 2020).
- Zuma, A.A.; de Souza, W. Chagas Disease Chemotherapy: What Do We Know So Far? Curr. Pharm. Des. 2021, 27, 3963–3995. [Google Scholar] [CrossRef] [PubMed]
- Bonney, K.M. Chagas disease in the 21st century: A public health success or an emerging threat? Parasite 2014, 21, 11. [Google Scholar] [CrossRef] [PubMed]
- Altcheh, J.; Moscatelli, G.; Moroni, S.; Garcia-Bournissen, F.; Freilij, H. Adverse Events After the Use of Benznidazole in Infants and Children With Chagas Disease. Pediatrics 2011, 127, e212–e218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apt, B.W.; Heitmann, G.I.; Jercic, L.M.; Jotre, M.L.; Munoz, C.D.V.P.; Noemi, H.I.; Martin, V.A.S.; Sapunar, P.J.; Torres, H.M.; Zulantay, A.I. Guidelines for chagas disease: Part IV. Chagas disease in immune compromised patients. Rev. Chil. Infectol. 2008, 25, 289–292. [Google Scholar]
- Scarim, C.B.; Chin, C.M. Current Approaches to Drug Discovery for Chagas Disease: Methodological Advances. Comb. Chem. High Throughput Screen. 2019, 22, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Villalta, F.; Rachakonda, G. Advances in preclinical approaches to Chagas disease drug discovery. Expert Opin. Drug Discov. 2019, 14, 1161–1174. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.V.; de Castro, S.L. The trypanocidal activity of naphthoquinones: A review. Molecules 2009, 14, 4570. [Google Scholar] [CrossRef]
- Salas, C.O.; Faúndez, M.; Morello, A.; Maya, J.D.; Tapia, R.A. Natural and synthetic naphthoquinones active against Trypanosoma cruzi: An initial step towards new drugs for Chagas disease. Curr. Med. Chem. 2011, 18, 144–161. [Google Scholar] [CrossRef]
- Dantas-Pereira, L.; Cunha-Junior, E.F.; Andrade-Neto, V.V.; Bower, J.F.; Jardim, G.A.M.; da Silva, E.N., Jr.; Torres-Santos, E.C.; Menna-Barreto, R.F.S. Naphthoquinones and Derivatives for Chemotherapy: Perspectives and Limitations of their Anti-trypanosomatids Activities. Curr. Pharm. Des. 2021, 27, 1807–1824. [Google Scholar] [CrossRef]
- Sieveking, I.; Thomas, P.; Estevez, J.C.; Quinones, N.; Cuellar, M.A.; Villena, J.; Espinosa-Bustos, C.; Fierro, A.; Tapia, R.A.; Maya, J.D.; et al. 2-Phenylaminonaphthoquinones and related compounds: Synthesis, trypanocidal and cytotoxic activities. Bioorg. Med. Chem. 2014, 22, 4609–4620. [Google Scholar] [CrossRef]
- Vázquez, K.; Espinosa-Bustos, C.; Soto-Delgado, J.; Tapia, R.A.; Varela, J.; Birriel, E.; Segura, R.; Pizarro, J.; Cerecetto, H.; González, M.; et al. New aryloxy-quinone derivatives as potential anti-Chagasic agents: Synthesis, trypanocidal activity, electrochemical properties, pharmacophore elucidation and 3D-QSAR analysis. RSC Adv. 2015, 5, 65153–65166. [Google Scholar] [CrossRef]
- Espinosa-Bustos, C.; Vazquez, K.; Varela, J.; Cerecetto, H.; Paulino, M.; Segura, R.; Pizarro, J.; Vera, B.; Gonzalez, M.; Zarate, A.M.; et al. New aryloxy-quinone derivatives with promising activity on Trypanosoma cruzi. Arch. Pharm. 2020, 353, e1900213. [Google Scholar] [CrossRef] [PubMed]
- El Hage, S.; Ane, M.; Stigliani, J.L.; Marjorie, M.; Vial, H.; Baziard-Mouysset, G.; Payard, M. Synthesis and antimalarial activity of new atovaquone derivatives. Eur. J. Med. Chem. 2009, 44, 4778–4782. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Duan, L.; Chen, H.; Ren, X.; Zhang, Z.; Zhou, F.; Liu, J.; Pei, D.; Ding, K. Atovaquone derivatives as potent cytotoxic and apoptosis inducing agents. Bioorg. Med. Chem. Lett. 2009, 19, 5091–5094. [Google Scholar] [CrossRef]
- Albino, S.L.; Da Silva, J.M.; Nobre, M.S.D.C.; Silva, Y.M.S.D.M.E.; Santos, M.B.; De Araújo, R.S.A.; De Lima, M.D.C.A.; Schmitt, M.; De Moura, R.O. Bioprospecting of Nitrogenous Heterocyclic Scaffolds with Potential Action for Neglected Parasitosis: A Review. Curr. Pharm. Des. 2020, 26, 4112–4150. [Google Scholar] [CrossRef]
- Braga, S.F.; Martins, L.C.; da Silva, E.B.; Sales, P.A., Jr.; Murta, S.M.; Romanha, A.J.; Soh, W.T.; Brandstetter, H.; Ferreira, R.S.; de Oliveira, R.B. Synthesis and biological evaluation of potential inhibitors of the cysteine proteases cruzain and rhodesain designed by molecular simplification. Bioorg. Med. Chem. 2017, 25, 1889–1900. [Google Scholar] [CrossRef]
- de Araujo, J.S.; Garcia-Rubia, A.; Sebastian-Perez, V.; Kalejaiye, T.D.; da Silva, P.B.; Fonseca-Berzal, C.R.; Maes, L.; De Koning, H.P.; Soeiro, M.N.C.; Gil, C. Imidazole Derivatives as Promising Agents for the Treatment of Chagas Disease. Antimicrob. Agents Chemother. 2019, 63, e02156-18. [Google Scholar] [CrossRef] [Green Version]
- Muscia, G.C.; Pacheco, F.J.R.; Asis, S.E.; Buldain, G.Y.; Frank, F.M. Hit-to-lead optimization of novel 2-alkylaminomethylquinoline derivatives as anti-chagas agents. Eur. J. Med. Chem. 2020, 186, 111877. [Google Scholar] [CrossRef]
- Sijm, M.; Maes, L.; de Esch, I.J.P.; Caljon, G.; Sterk, G.J.; Leurs, R. Structure Activity Relationship of N-Substituted Phenyldihydropyrazolones Against Trypanosoma cruzi Amastigotes. Front. Chem. 2021, 9, 608438. [Google Scholar] [CrossRef]
- Vera, B.; Vazquez, K.; Mascayano, C.; Tapia, R.A.; Espinosa, V.; Soto-Delgado, J.; Salas, C.O.; Paulino, M. Structural analysis and molecular docking of trypanocidal aryloxy-quinones in trypanothione and glutathione reductases: A comparison with biochemical data. J. Biomol. Struct. Dyn. 2017, 35, 1785–1803. [Google Scholar] [CrossRef]
- Vazquez, K.; Paulino, M.; Salas, C.O.; Zarate-Ramos, J.J.; Vera, B.; Rivera, G. Trypanothione Reductase: A Target for the Development of Anti-Trypanosoma cruzi Drugs. Mini-Rev. Med. Chem. 2017, 17, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Porter, T.H.; Skelton, F.S.; Bowman, C.M.; Folkers, K. Coenzyme Q. 147. Synthesis of new 2-alkylamino-1,4-naphthoquinones as inhibitors of coenzyme Q and as antimalarials. J. Med. Chem. 1972, 15, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Brun, M.-P.; Braud, E.; Angotti, D.; Mondésert, O.; Quaranta, M.; Montes, M.; Miteva, M.; Gresh, N.; Ducommun, B.; Garbay, C. Design, synthesis, and biological evaluation of novel naphthoquinone derivatives with CDC25 phosphatase inhibitory activity. Bioorg. Med. Chem. 2005, 13, 4871–4879. [Google Scholar] [CrossRef] [PubMed]
- Villegas, A.; Satheeshkumar, R.; Ballesteros-Casallas, A.; Paulino, M.; Castro, A.; Espinosa-Bustos, C.; Salas, C.O. Convergent synthesis, drug target prediction, and docking studies of new 2,6,9-trisubstituted purine derivatives. J. Heterocyclic. Chem. 2022, 59, 97–111. [Google Scholar] [CrossRef]
- Alvarez, G.; Varela, J.; Marquez, P.; Gabay, M.; Rivas, C.E.A.; Cuchilla, K.; Echeverria, G.A.; Piro, O.E.; Chorilli, M.; Leal, S.M.; et al. Optimization of antitrypanosomatid agents: Identification of nonmutagenic drug candidates with in vivo activity. J. Med. Chem. 2014, 57, 3984–3999. [Google Scholar] [CrossRef]
- Becerra, N.A.; Espinosa-Bustos, C.; Vázquez, K.; Rivera, G.; Paulino, M.; Cantero, J.; Nogueda, B.; Chacón-Vargas, F.; Castillo-Velazquez, U.; Rodríguez, A.F.E.; et al. Expanding the chemical space of aryloxy-naphthoquinones as potential anti-Chagasic agents: Synthesis and trypanocidal activity. Med. Chem. Res. 2021, 30, 2256–2265. [Google Scholar] [CrossRef]
- Faundez, M.; Pino, L.; Letelier, P.; Ortiz, C.; Lopez, R.; Seguel, C.; Ferreira, J.; Pavani, M.; Morello, A.; Maya, J.D. Buthionine sulfoximine increases the toxicity of nifurtimox and benznidazole to Trypanosoma cruzi. Antimicrob. Agents Chemother. 2005, 49, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Chavez, Z.; Vazquez, C.; Mejia-Tlachi, M.; Marquez-Duenas, C.; Manning-Cela, R.; Encalada, R.; Rodriguez-Enriquez, S.; Michels, P.A.M.; Moreno-Sanchez, R.; Saavedra, E. Gamma-glutamylcysteine synthetase and tryparedoxin 1 exert high control on the antioxidant system in Trypanosoma cruzi contributing to drug resistance and infectivity. Redox Biol. 2019, 26, 101231. [Google Scholar] [CrossRef]
- Tapia, R.A.; Salas, C.; Morello, A.; Maya, J.D.; Toro-Labbe, A. Synthesis of dihydronaphthofurandiones and dihydrofuroquinolinediones with trypanocidal activity and analysis of their stereoelectronic properties. Bioorg. Med. Chem. 2004, 12, 2451–2458. [Google Scholar] [CrossRef]
- Moretti, N.S.; Mortara, R.A.; Schenkman, S. Trypanosoma cruzi. Trends Parasitol. 2020, 36, 404–405. [Google Scholar] [CrossRef]
- De Souza, W.; Barrias, E.S. May the epimastigote form of Trypanosoma cruzi be infective? Acta Trop. 2020, 212, 105688. [Google Scholar] [CrossRef] [PubMed]
- Battista, T.; Colotti, G.; Ilari, A.; Fiorillo, A. Targeting Trypanothione Reductase, a Key Enzyme in the Redox Trypanosomatid Metabolism, to Develop New Drugs against Leishmaniasis and Trypanosomiases. Molecules 2020, 25, 1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran-Hortelano, I.; Alcolea, V.; Font, M.; Pérez-Silanes, S. Examination of multiple Trypanosoma cruzi targets in a new drug discovery approach for Chagas disease. Bioorg. Med. Chem. 2022, 58, 116577. [Google Scholar] [CrossRef] [PubMed]
- El-Waer, A.; Douglas, K.T.; Smith, K.; Fairlamb, A.H. Synthesis of N-benzyloxycarbonyl-L-cysteinylglycine 3-dimethylaminopropylamide disulfide: A cheap and convenient new assay for trypanothione reductase. Anal. Biochem. 1991, 198, 212–216. [Google Scholar] [CrossRef]
- Meanwell, N.A. Improving drug candidates by design: A focus on physicochemical properties as a means of improving compound disposition and safety. Chem. Res. Toxicol. 2011, 24, 1420–1456. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Epi NINOA IC50 (µM) a,b | Epi INC-5 IC50 (µM) a,b | Trypo NINOA LC50 (µM) a,c | Trypo INC-5 LC50 (µM) a,c |
|---|---|---|---|---|
| 2a | 1.10 ± 0.12 | 3.06 ± 0.01 | >100 | >100 |
| 2b | 1.10 ± 0.05 | 2.98 ± 0.2 | >100 | >100 |
| 2c | 1.14 ± 0.06 | 2.22 ± 0.04 | >100 | >100 |
| 2d | 1.12 ± 0.04 | 5.04 ± 0.26 | >100 | 87.34 ± 5.32 |
| 2e | 0.43 ± 0.15 | 0.19 ± 0.07 | >100 | >100 |
| 2f | 1.15 ± 0.09 | 1.46 ± 0.03 | 97.86 ± 2.13 | >100 |
| 2g | 7.05 ± 0.95 | 8.45 ± 0.35 | NT d | NT d |
| 2h | 6.40 ± 0.02 | 6.60 ± 0.08 | NT d | NT d |
| 2i | 10.1 ± 0.05 | 1.74 ± 0.02 | NT d | NT d |
| 7a | 6.41 ± 0.02 | 2.46 ± 0.08 | NT d | NT d |
| 7b | 0.77 ± 0.21 | 2.23 ± 0.06 | 80.32 ± 4.03 | >100 |
| 7c | 0.52 ± 0.04 | 1.14 ± 0.07 | 68.35 ± 6.30 | >100 |
| 7d | 1.25 ± 0.06 | 2.30 ± 0.11 | >100 | >100 |
| 7e | 4.45 ± 0.06 | 5.60 ± 0.03 | NT d | NT d |
| 7f | 0.99 ± 0.01 | 1.84 ± 0.06 | 70.86 ± 7.31 | 98.56 ± 8.2 |
| 7g | 3.55 ± 0.02 | 2.57 ± 0.04 | >100 | >100 |
| 7h | 0.84 ± 0.11 | 1.35 ± 0.01 | 78.57 ± 3.91 | 94.65 ± 5.21 |
| 7i | 1.09 ± 0.01 | 3.35 ± 0.05 | 90.45 ± 6.58 | >100 |
| 7j | 0.43 ± 0.03 | 0.92 ± 0.01 | 98.06 ± 8.9 | 59.73 ± 3.72 |
| Bzn | 8.21 ± 1.80 | 42.3 ± 5.80 | >100 | 85.62 ± 4.23 |
| Compound | J774 IC50 (µM) a | NINOA Epi SI b | INC-5 Epi SI b | NINOA Trypo SI b | INC-5 Trypo SI b |
|---|---|---|---|---|---|
| 7c | 25.0 | 48.0 | 21.9 | 0.37 | ND c |
| 7f | 25.0 | 25.2 | 13.6 | 0.32 | 0.25 |
| 7h | 22.0 | 26.2 | 16.3 | 0.28 | 0.23 |
| 7j | 33.0 | 76.7 | 35.9 | 0.34 | 0.55 |
| Bzn | 352 | 42.9 | 8.38 | 2.36 | 4.11 |
| Putative Binding Z-Site | |
|---|---|
| Chain A | Lys62 Leu63 Val65 Thr66 Gln69 Tyr70 His73 Glu76 Gln242 Phe367 Ser368 Ile369 Pro370 Pro371 Pro398 Leu399 Met400 His401 Lys409 Thr410 Phe411 Leu430 Gly431 Asp432 Asn433 Pro435 Glu436 Pro462 Thr463 Ser464 |
| Chain B | Lys62 Leu63 Val65 Thr66 Gln69 Tyr70 His73 Glu76 Phe367 Ser368 Ile369 Pro370 Pro371 Pro398 Leu399 Met400 His401 Phe411 Gly431 Asp432 Asn433 Pro435 Glu436 Pro462 Thr463 Ser464 |
| Compound | FEB TR (Kcal/Mol) | Compound | FEB TR (Kcal/Mol) |
|---|---|---|---|
| 2a | −7.2 | 7c | −8.2 |
| 2b | −7.5 | 7d | −9.5 |
| 2c | −7.4 | 7e | −8.5 |
| 2d | −7.8 | 7f | −8.9 |
| 2e | −7.7 | 7g | −8.7 |
| 2f | −7.8 | 7h | −9.7 |
| 2g | −7.7 | 7i | −10.5 |
| 2h | −7.8 | 7j | −9.5 |
| 2i | −7.8 | NQ-d | −7.8 |
| 7a | −8.8 | NQ-g | −8.4 |
| 7b | −9.7 | NQ-h | −8.0 |
| Compound | Interactions |
|---|---|
| 7a | HI: Gln69, Glu72, Phe367, Pro435, Glu436. HB: Gln69, Asn433. HalB: Asn433. |
| 7b | HI: Phe367, Pro371, Pro435, Glu436. HB: Lys62, Gln69, Asn433. |
| 7d | HI: Phe367, Pro371, Pro435. HB: Lys62, Gln69, Asn433. |
| 7e | HI: Gln69, Met400, Asp432. HB: Lys62, Asn433, Glu436 |
| 7f | HI: Phe367, Pro371, Asp432, Pro435, Glu436. HB: Gln69, Asn433. |
| 7g | HI: Thr66, Gln69, Glu72, Pro371. HB: Lys62, Tyr70, His73, His401. Π-c: His73. |
| 7h | HI: Phe367, Pro371, Pro435. HB: Lys62, Gln69, Asn433. |
| 7i | HI: Phe367, Pro371, Asp432, Pro435, Glu436. HB: Lys62, Gln69, Asn433. HalB: Glu436. |
| 7j | HI: Phe367, Pro371, Pro435, Glu436. HB: Gln69, His73, Asn433. |
| NQ-d | HI: Thr66, Gln69, Tyr70, Phe367, Pro371, Pro435, Glu436. HB. Gln69, Asn433. Π-s: His73. |
| NQ-g | HI: Phe367, Pro371, Asp432, Pro435. HB: Lys62, Gln69, Glu436. Π-s: His73. |
| NQ-h | HI: Phe367, Pro371, Asp432, Pro435. HB: Asp432. |
| Kinetic Parameter | Value |
| Vmax (µmol/min/mg) a | 215 ± 38 |
| Km TS2 (µM) a | 50 ± 4 |
| Km NADPH (µM) a | 6.0 ± 0.2 |
| Ki7j vs. TS2 (µM) a | 28 ± 4.5 |
| Ki 7j vs. NADPH (µM) a | 18 ± 3.5 |
| α TS2 b | 0.77 |
| α NADPH b | 1.12 |
| Compound | MW (Da) | HBA | HBD | cLogP | TPSA (A2) | NRB |
|---|---|---|---|---|---|---|
| Desirable value | ≤500 | ≤10 | ≤5 | ≤5 | ≤140 | ≤10 |
| 7j | 444.91 | 3 | 1 | 3.99 | 66.48 | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espinosa-Bustos, C.; Ortiz Pérez, M.; Gonzalez-Gonzalez, A.; Zarate, A.M.; Rivera, G.; Belmont-Díaz, J.A.; Saavedra, E.; Cuellar, M.A.; Vázquez, K.; Salas, C.O. New Amino Naphthoquinone Derivatives as Anti-Trypanosoma cruzi Agents Targeting Trypanothione Reductase. Pharmaceutics 2022, 14, 1121. https://doi.org/10.3390/pharmaceutics14061121
Espinosa-Bustos C, Ortiz Pérez M, Gonzalez-Gonzalez A, Zarate AM, Rivera G, Belmont-Díaz JA, Saavedra E, Cuellar MA, Vázquez K, Salas CO. New Amino Naphthoquinone Derivatives as Anti-Trypanosoma cruzi Agents Targeting Trypanothione Reductase. Pharmaceutics. 2022; 14(6):1121. https://doi.org/10.3390/pharmaceutics14061121
Chicago/Turabian StyleEspinosa-Bustos, Christian, Mariana Ortiz Pérez, Alonzo Gonzalez-Gonzalez, Ana María Zarate, Gildardo Rivera, Javier A. Belmont-Díaz, Emma Saavedra, Mauricio A. Cuellar, Karina Vázquez, and Cristian O. Salas. 2022. "New Amino Naphthoquinone Derivatives as Anti-Trypanosoma cruzi Agents Targeting Trypanothione Reductase" Pharmaceutics 14, no. 6: 1121. https://doi.org/10.3390/pharmaceutics14061121