
Therapeutic Approach to Alzheimer’s Disease: Current Treatments and New Perspectives


 , , , and
, , , and
Abstract
:1. Introduction
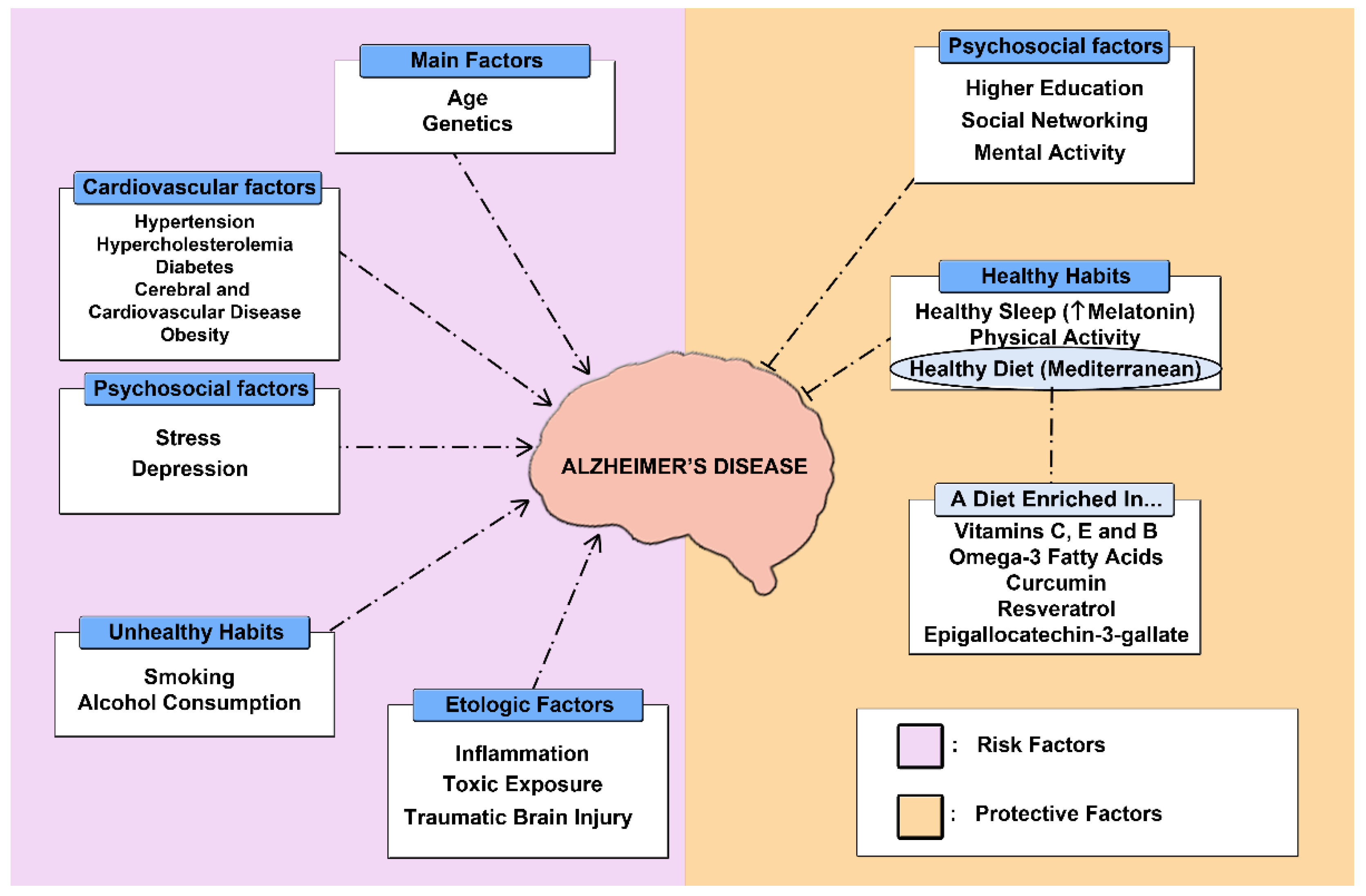
1.1. Epidemiology of Alzheimer’s Disease
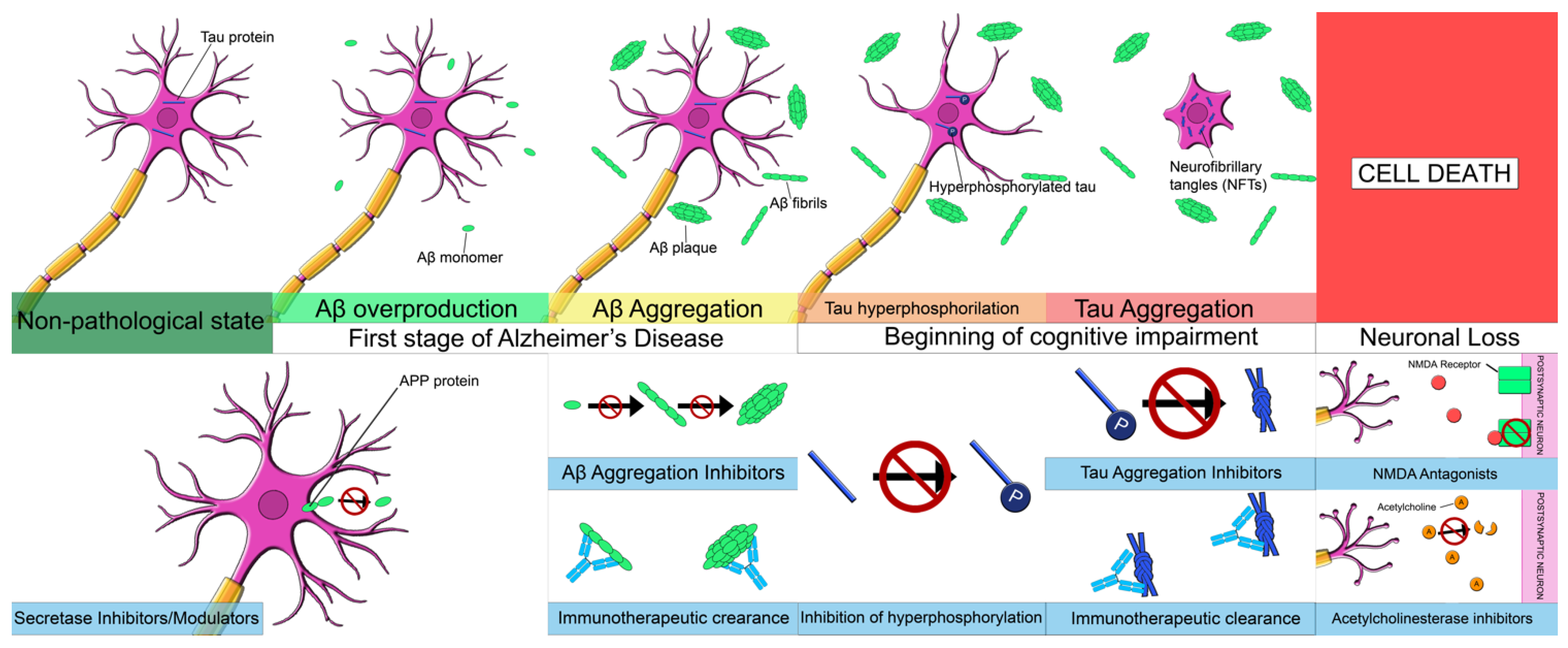
1.2. Physiopathology of Alzheimer’s Disease
1.2.1. Amyloid-β Pathology
1.2.2. Tau Pathology
1.2.3. Neuroinflammation and Neuronal Loss
2. Results
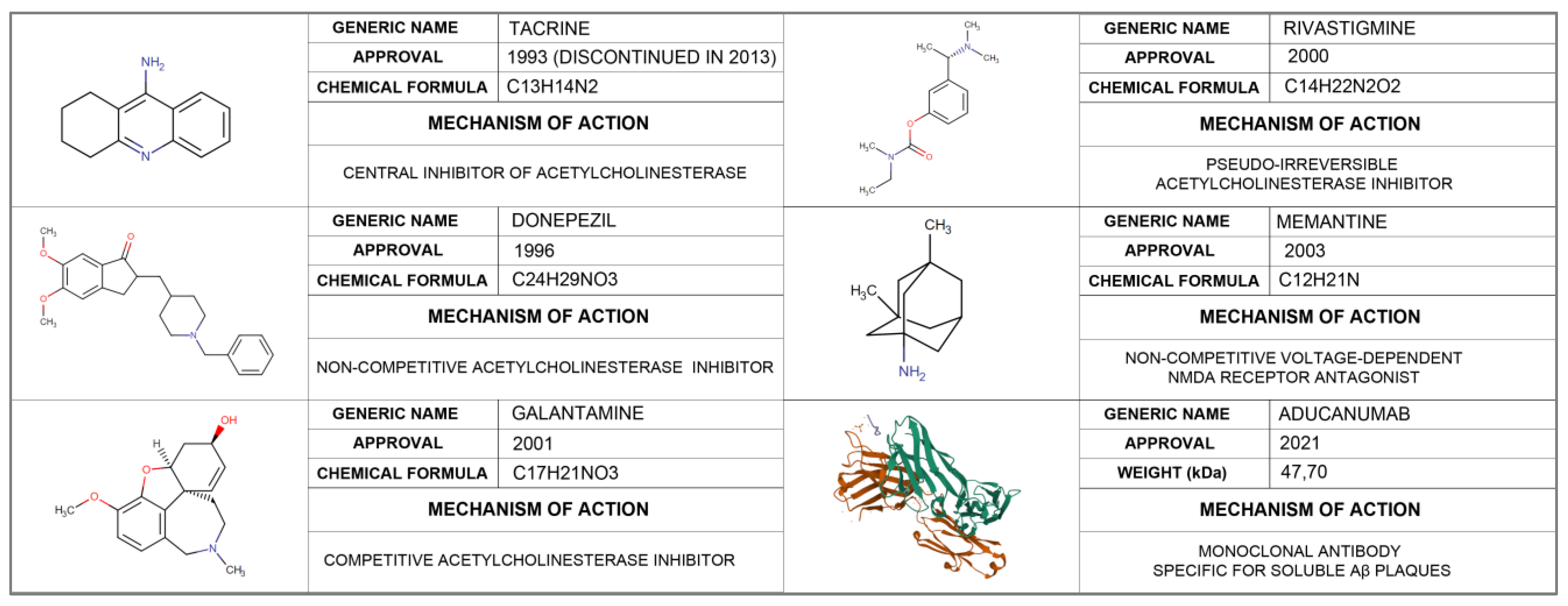
2.1. Approved Treatment for Alzheimer’s Disease
2.1.1. Aducanumab
2.1.2. Acetylcholinesterase Inhibitors (AChE)
- Tacrine
- 2.
- Donepezil
- 3.
- Galantamine
- 4.
- Rivastigmine
2.1.3. N-Methyl D-Aspartate (NMDA) Antagonists
- Memantine
2.2. Pharmacological Treatments under Investigation
2.2.1. Aβ Pathology
- γ-secretase inhibitors
- 2.
- β-secretase inhibitors
- 3.
- α-secretase modulators
- 4.
- Aggregation inhibitors
- 5.
- Metal interfering drugs
- 6.
- Drugs that enhance Aβ clearance (immunotherapy)
2.2.2. Tau Pathology
- Inhibitors of tau protein hyperphosphorylation
- 2.
- Tau protein aggregation inhibitors
- 3.
- Drugs that promote the clearance of tau (immunotherapy)
2.2.3. Other Treatments under Investigation
Nanomedicine Strategies
Others
2.3. Alternative Therapies
2.3.1. Physical Activity
2.3.2. Diet
2.3.3. Sleep Pattern
2.3.4. Complementary Therapies
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s Dement. 2013, 9, 63–75.e2. [Google Scholar] [CrossRef] [PubMed]
- Ayodele, T.; Rogaeva, E.; Kurup, J.T.; Beecham, G.; Reitz, C. Early-Onset Alzheimer’s Disease: What Is Missing in Research? Curr. Neurol. Neurosci. Rep. 2021, 21, 4. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, A.; Aztiria, A.; Basarab, A. On the early diagnosis of Alzheimer’s Disease from multimodal signals: A survey. Artif. Intell. Med. 2016, 71, 1–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, M.A.; Bickel, H.; Prince, M.; Fratiglioni, L.; von Strauss, E.; Frydecka, D.; Kiejna, A.; Georges, J.; Reynish, E. Estimating the burden of early onset dementia; systematic review of disease prevalence. Eur. J. Neurol. 2014, 21, 563–569. [Google Scholar] [CrossRef]
- García-Morales, V.; González-Acedo, A.; Melguizo-Rodríguez, L.; Pardo-Moreno, T.; Costela-Ruiz, V.J.; Montiel-Troya, M.; Ramos-Rodríguez, J.J. Current Understanding of the Physiopathology, Diagnosis and Therapeutic Approach to Alzheimer’s Disease. Biomedicines 2021, 9, 1910. [Google Scholar] [CrossRef]
- Baranello, R.J.; Bharani, K.; Padmaraju, V.; Chopra, N.; Lahiri, D.K.; Greig, N.H.; Pappolla, M.A.; Sambamurti, K. Amyloid-Beta Protein Clearance and Degradation (ABCD) Pathways and their Role in Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 32–46. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Rodriguez, J.J.; Spires-Jones, T.; Pooler, A.M.; Lechuga-Sancho, A.; Bacskai, B.J.; Garcia-Alloza, M. Progressive Neuronal Pathology and Synaptic Loss Induced by Prediabetes and Type 2 Diabetes in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 3428–3438. [Google Scholar] [CrossRef]
- Kartalou, G.-I.; Endres, T.; Lessmann, V.; Gottmann, K. Golgi-Cox impregnation combined with fluorescence staining of amyloid plaques reveals local spine loss in an Alzheimer mouse model. J. Neurosci. Methods 2020, 341, 108797. [Google Scholar] [CrossRef]
- Pluta, R.; Ouyang, L.; Januszewski, S.; Li, Y.; Czuczwar, S. Participation of Amyloid and Tau Protein in Post-Ischemic Neurodegeneration of the Hippocampus of a Nature Identical to Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2460. [Google Scholar] [CrossRef]
- Yamada, K.; Patel, T.K.; Hochgräfe, K.; Mahan, T.E.; Jiang, H.; Stewart, F.R.; Mandelkow, E.-M.; Holtzman, D.M. Analysis of in vivo turnover of tau in a mouse model of tauopathy. Mol. Neurodegener. 2015, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- Van Opstal, A.M.; van Rooden, S.; van Harten, T.; Ghariq, E.; Labadie, G.; Fotiadis, P.; Gurol, M.E.; Terwindt, G.M.; Wermer, M.J.H.; van Buchem, M.A.; et al. Cerebrovascular function in presymptomatic and symptomatic individuals with hereditary cerebral amyloid angiopathy: A case-control study. Lancet Neurol. 2017, 16, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Toombs, J.; Zetterberg, H. Untangling the tau microtubule-binding region. Brain 2021, 144, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Venkatramani, A.; Panda, D. Regulation of neuronal microtubule dynamics by tau: Implications for tauopathies. Int. J. Biol. Macromol. 2019, 133, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Maury, E.A.; Kirk, M.J.; Saqran, L.; Roe, A.D.; Devos, S.L.; Nicholls, S.B.; Fan, Z.; Takeda, S.; Cagsal-Getkin, O.; et al. Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J. 2015, 34, 3028–3041. [Google Scholar] [CrossRef] [Green Version]
- Sankar, S.B.; García, C.I.; Weinstock, L.D.; Ramos-Rodriguez, J.J.; Hierro-Bujalance, C.; Fernandez-Ponce, C.; Wood, L.B.; Garcia-Alloza, M. Amyloid beta and diabetic pathology cooperatively stimulate cytokine expression in an Alzheimer’s mouse model. J. Neuroinflammation 2020, 17, 38. [Google Scholar] [CrossRef]
- Sita, G.; Hrelia, P.; Tarozzi, A.; Morroni, F. Isothiocyanates Are Promising Compounds against Oxidative Stress, Neuroinflammation and Cell Death that May Benefit Neurodegeneration in Parkinson’s Disease. Int. J. Mol. Sci. 2016, 17, 1454. [Google Scholar] [CrossRef]
- Ionescu-Tucker, A.; Cotman, C.W. Emerging roles of oxidative stress in brain aging and Alzheimer’s disease. Neurobiol. Aging 2021, 107, 86–95. [Google Scholar] [CrossRef]
- Kim, T.-S.; Pae, C.-U.; Yoon, S.-J.; Jang, W.-Y.; Lee, N.J.; Kim, J.-J.; Lee, S.-J.; Lee, C.; Paik, I.-H.; Lee, C.-U. Decreased plasma antioxidants in patients with Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2006, 21, 344–348. [Google Scholar] [CrossRef]
- Biogen. Highlights of Prescribing Information: Aduhelmtm (Aducanumab-Avwa) Injection, for Intravenous Use Initial U.S. Approval: 2021; Biogen: Cambridge, MA, USA, 2021. [Google Scholar]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Biogen A Phase 3 Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study to Evaluate the Efficacy and Safety of Aducanumab (BIIB037) in Subjects With Early Alzheimer’s Disease. 2021. Available online: clinicaltrials.gov (accessed on 5 March 2022).
- Biogen. 221AD302 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer’s Disease—Tabular View—ClinicalTrials.Gov. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT02484547 (accessed on 3 February 2022).
- Salloway, S.; Chalkias, S.; Barkhof, F.; Burkett, P.; Barakos, J.; Purcell, D.; Suhy, J.; Forrestal, F.; Tian, Y.; Umans, K.; et al. Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients With Early Alzheimer Disease. JAMA Neurol. 2022, 79, 1–10. [Google Scholar] [CrossRef]
- Wagstaff, A.J.; McTavish, D. Tacrine: A Review of Its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Efficacy in Alzheimer’s Disease. Drugs Aging 1994, 4, 510–540. [Google Scholar] [CrossRef] [PubMed]
- Almkvist, O.; Jelic, V.; Amberla, K.; Hellström-Lindahl, E.; Meurling, L.; Nordberg, A. Responder Characteristics to a Single Oral Dose of Cholinesterase Inhibitor: A Double-Blind Placebo-Controlled Study withTacrine in Alzheimer P atients. Dement. Geriatr. Cogn. Disord. 2010, 12, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Gutzmann, H.; Kühl, K.-P.; Hadler, D.; Rapp, M.A. Safety and Efficacy of Idebenone versus Tacrine in Patients with Alzheimer’s Disease: Results of a Randomized, Double-Blind, Parallel-Group Multicenter Study. Pharm. 2002, 35, 12–18. [Google Scholar] [CrossRef]
- Raskind, M.A.; Sadowsky, C.H.; Sigmund, W.R.; Beitler, P.J.; Auster, S.B. Effect of tacrine on language, praxis, and noncognitive behavioral problems in Alzheimer disease. Arch. Neurol. 1997, 54, 836–840. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.-J.; Liu, H.-C.; Fuh, J.-L.; Wang, S.-J.; Hsu, L.-C.; Wang, P.-N.; Sheng, W.-Y. A Double-Blind, Placebo-Controlled Study of Tacrine in Chinese Patients with Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 1999, 10, 289–294. [Google Scholar] [CrossRef]
- Jarrott, B. Tacrine: In vivo veritas. Pharmacol. Res. 2017, 116, 29–31. [Google Scholar] [CrossRef]
- Lou, Y.-H.; Wang, J.-S.; Dong, G.; Guo, P.-P.; Wei, D.-D.; Xie, S.-S.; Yang, M.-H.; Kong, L.-Y. The acute hepatotoxicity of tacrine explained by 1H NMR based metabolomic profiling. Toxicol. Res. 2015, 4, 1465–1478. [Google Scholar] [CrossRef]
- Eagger, S.A.; Levy, R.; Sahakian, B.J. Tacrine in Alzheimer’s Disease. Lancet 1991, 337, 989–992. [Google Scholar] [CrossRef]
- A Jacobson, S.; Sabbagh, M.N. Donepezil: Potential neuroprotective and disease-modifying effects. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1363–1369. [Google Scholar] [CrossRef]
- Kumar, A.; Gupta, V.; Sharma, S. Donepezil. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Homma, A.; Imai, Y.; Tago, H.; Asada, T.; Shigeta, M.; Iwamoto, T.; Takita, M.; Arimoto, I.; Koma, H.; Ohbayashi, T. Donepezil Treatment of Patients with Severe Alzheimer’s Disease in a Japanese Population: Results from a 24-Week, Double-Blind, Placebo-Controlled, Randomized Trial. Dement. Geriatr. Cogn. Disord. 2008, 25, 399–407. [Google Scholar] [CrossRef]
- Howard, R.J.; Juszczak, E.; Ballard, C.; Bentham, P.; Brown, R.; Bullock, R.; Burns, A.; Holmes, C.; Jacoby, R.; Johnson, T.; et al. Donepezil for the Treatment of Agitation in Alzheimer’s Disease. N. Engl. J. Med. 2007, 357, 1382–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, J.; Wei, C.; Jia, L.; Tang, Y.; Liang, J.; Zhou, A.; Li, F.; Shi, L.; Doody, R.S. Efficacy and Safety of Donepezil in Chinese Patients with Severe Alzheimer’s Disease: A Randomized Controlled Trial. J. Alzheimer’s Dis. 2017, 56, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Maher-Edwards, G.; Dixon, R.; Hunter, J.; Gold, M.; Hopton, G.; Jacobs, G.; Hunter, J.; Williams, P. SB-742457 and donepezil in Alzheimer disease: A randomized, placebo-controlled study. Int. J. Geriatr. Psychiatry 2011, 26, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Homma, A.; Atarashi, H.; Kubota, N.; Nakai, K.; Takase, T. Efficacy and Safety of Sustained Release Donepezil High Dose versus Immediate Release Donepezil Standard Dose in Japanese Patients with Severe Alzheimer’s Disease: A Randomized, Double-Blind Trial. J. Alzheimer’s Dis. 2016, 52, 345–357. [Google Scholar] [CrossRef]
- Hong, Y.J.; Han, H.J.; Youn, Y.C.; Park, K.W.; Yang, D.W.; Kim, S.; Kim, H.J.; Kim, J.E.; Lee, J.-H. Safety and tolerability of donepezil 23 mg with or without intermediate dose titration in patients with Alzheimer’s disease taking donepezil 10 mg: A multicenter, randomized, open-label, parallel-design, three-arm, prospective trial. Alzheimer’s Res. Ther. 2019, 11, 37. [Google Scholar] [CrossRef] [Green Version]
- Cacabelos, R. Donepezil in Alzheimer’s disease: From conventional trials to pharmacogenetics. Neuropsychiatr. Dis. Treat. 2007, 3, 303–333. [Google Scholar]
- Seltzer, B. Donepezil: An update. Expert Opin. Pharmacother. 2007, 8, 1011–1023. [Google Scholar] [CrossRef]
- Farlow, M.R. Clinical Pharmacokinetics of Galantamine. Clin. Pharmacokinet. 2003, 42, 1383–1392. [Google Scholar] [CrossRef]
- Olin, J.T.; Schneider, L.S. Galantamine for dementia due to Alzheimer’s disease. Cochrane Database Syst. Rev. 2002, 3, CD001747. [Google Scholar] [CrossRef]
- Seltzer, B. Galantamine-ER for the treatment of mild-to-moderate Alzheimer’s disease. Clin. Interv. Aging 2010, 5, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Hanafy, A.S.; Farid, R.M.; Helmy, M.W.; ElGamal, S.S. Pharmacological, toxicological and neuronal localization assessment of galantamine/chitosan complex nanoparticles in rats: Future potential contribution in Alzheimer’s disease management. Drug Deliv. 2016, 23, 3111–3122. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Chopra, K.; Sinha, V.R.; Medhi, B. Galantamine-loaded solid–lipid nanoparticles for enhanced brain delivery: Preparation, characterization, in vitro and in vivo evaluations. Drug Deliv. 2016, 23, 1434–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahba, S.M.; Darwish, A.S.; Kamal, S.M. Ceria-containing uncoated and coated hydroxyapatite-based galantamine nanocomposites for formidable treatment of Alzheimer’s disease in ovariectomized albino-rat model. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 65, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, S.; Gaudig, M.; Van Baelen, B.; Adami, M.; Delgado, A.; Guzman, C.; Jedenius, E.; Schäuble, B. Galantamine and behavior in Alzheimer disease: Analysis of four trials. Acta Neurol. Scand. 2011, 124, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Tariot, P.; Solomon, P.; Morris, J.; Kershaw, P.; Lilienfeld, S.; Ding, C.; the Galantamine USA-Study Group. A 5-month, randomized, placebo-controlled trial of galantamine in AD. Neurology 2000, 54, 2269–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.-D.; Zhang, Y.-H.; Zhang, W.; Zhao, P. Meta-Analysis of Randomized Controlled Trials on the Efficacy and Safety of Donepezil, Galantamine, Rivastigmine, and Memantine for the Treatment of Alzheimer’s Disease. Front. Neurosci. 2019, 13, 472. [Google Scholar] [CrossRef]
- Haake, A.; Nguyen, K.; Friedman, L.; Chakkamparambil, B.; Grossberg, G.T. An update on the utility and safety of cholinesterase inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Drug Saf. 2020, 19, 147–157. [Google Scholar] [CrossRef]
- Desai, A.K.; Grossberg, G.T. Rivastigmine for Alzheimer’s disease. Expert Rev. Neurother. 2005, 5, 563–580. [Google Scholar] [CrossRef]
- Birks, J.S.; Chong, L.-Y.; Evans, J.G. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2015, 9, CD001191. [Google Scholar] [CrossRef]
- Cummings, J.; Winblad, B. A rivastigmine patch for the treatment of Alzheimer’s disease and Parkinson’s disease dementia. Expert Rev. Neurother. 2007, 7, 1457–1463. [Google Scholar] [CrossRef]
- Yunusa, I.; Alsahali, S.; Ranes, A.; Eguale, T. Comparative Value of Cholinesterase Inhibitors and Memantine in Persons with Moderate-to-Severe Alzheimer’s Disease in the United States: A Cost-Effectiveness Analysis. J. Alzheimer’s Dis. Rep. 2021, 5, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Feldman, H.H.; Lane, R. Rivastigmine: A placebo controlled trial of twice daily and three times daily regimens in patients with Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1056–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaman, Y.; Erdoğan, F.; Köseoğlu, E.; Turan, T.; Ersoy, A. A 12-Month Study of the Efficacy of Rivastigmine in Patients with Advanced Moderate Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2005, 19, 51–56. [Google Scholar] [CrossRef] [PubMed]
- López-Pousa, S. Pilot, Multicenter, Randomized, Double-Blind, Controlled, Parallel Efficacy and Safety Study of Rivastigmine vs Placebo in the Treatment of Cognitive and Non-Cognitive Symptoms in Patients with Moderate-to-Severe Alzheimer’s Disease; IFPMA Register: Geneva, Switzerland, 2005. [Google Scholar]
- Mowla, A.; Mosavinasab, M.; Haghshenas, H.; Haghighi, A.B. Does Serotonin Augmentation Have Any Effect on Cognition and Activities of Daily Living in Alzheimer’s Dementia? A Double-Blind, Placebo-Controlled Clinical Trial. J. Clin. Psychopharmacol. 2007, 27, 484–487. [Google Scholar] [CrossRef]
- Nakamura, Y.; Imai, Y.; Shigeta, M.; Graf, A.; Shirahase, T.; Kim, H.; Fujii, A.; Mori, J.; Homma, A. A 24-Week, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Efficacy, Safety and Tolerability of the Rivastigmine Patch in Japanese Patients with Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. Extra 2011, 1, 163–179. [Google Scholar] [CrossRef]
- Winblad, B.; Grossberg, G.; Frölich, L.; Farlow, M.; Zechner, S.; Nagel, J.; Lane, R. IDEAL: A 6-month, double-blind, placebo-controlled study of the first skin patch for Alzheimer disease. Neurology 2007, 69, S14–S22. [Google Scholar] [CrossRef]
- Mimica, N.; Presecki, P. Side effects of approved antidementives. Psychiatr. Danub. 2009, 21, 108–113. [Google Scholar]
- Kuns, B.; Rosani, A.; Varghese, D. Memantine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Lo, D.; Grossberg, G.T. Use of memantine for the treatment of dementia. Expert Rev. Neurother. 2011, 11, 1359–1370. [Google Scholar] [CrossRef]
- Wong, K.H.; Riaz, M.K.; Xie, Y.; Zhang, X.; Liu, Q.; Chen, H.; Bian, Z.; Chen, X.; Lu, A.; Yang, Z. Review of Current Strategies for Delivering Alzheimer’s Disease Drugs across the Blood-Brain Barrier. Int. J. Mol. Sci. 2019, 20, E381. [Google Scholar] [CrossRef] [Green Version]
- McShane, R.; Westby, M.; Roberts, E.; Minakaran, N.; Schneider, L.; Farrimond, L.E.; Maayan, N.; Ware, J.; DeBarros, J. Memantine for dementia. Cochrane Database Syst. Rev. 2019, 3, CD003154. [Google Scholar] [CrossRef]
- Rossom, R.; Adityanjee; Dysken, M. Efficacy and tolerability of memantine in the treatment of dementia. Am. J. Geriatr. Pharmacother. 2004, 2, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, A.; King, C.; Khoury, R.; Grossberg, G.T. An evaluation of memantine ER + donepezil for the treatment of Alzheimer’s disease. Expert Opin. Pharmacother. 2018, 19, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Withdrawal Assessment Report: Memantine FGK; European Medicines Agency: Amsterdam, The Netherlands, 2012.
- Guo, J.; Wang, Z.; Liu, R.; Huang, Y.; Zhang, N.; Zhang, R. Memantine, Donepezil, or Combination Therapy—What is the best therapy for Alzheimer’s Disease? A Network Meta-Analysis. Brain Behav. 2020, 10, e01831. [Google Scholar] [CrossRef] [PubMed]
- Cappell, J.; Herrmann, N.; Cornish, S.; Lanctôt, K.L. The Pharmacoeconomics of Cognitive Enhancers in Moderate to Severe Alzheimer’s Disease. CNS Drugs 2010, 24, 909–927. [Google Scholar] [CrossRef] [PubMed]
- Knapp, M.; King, D.; Romeo, R.; Adams, J.; Baldwin, A.; Ballard, C.; Banerjee, S.; Barber, R.; Bentham, P.; Brown, R.G.; et al. Cost-effectiveness of donepezil and memantine in moderate to severe Alzheimer’s disease (the DOMINO-AD trial). Int. J. Geriatr. Psychiatry 2017, 32, 1205–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weycker, D.; Taneja, C.; Edelsberg, J.; Erder, M.H.; Schmitt, F.A.; Setyawan, J.; Oster, G. Cost-effectiveness of memantine in moderate-to-severe Alzheimer’s disease patients receiving donepezil. Curr. Med Res. Opin. 2007, 23, 1187–1197. [Google Scholar] [CrossRef]
- Hane, F.T.; Robinson, M.; Lee, B.Y.; Bai, O.; Leonenko, Z.; Albert, M.S. Recent Progress in Alzheimer’s Disease Research, Part 3: Diagnosis and Treatment. J. Alzheimer’s Dis. 2017, 57, 645–665. [Google Scholar] [CrossRef] [Green Version]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Central Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef] [Green Version]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Sun, X.; Thomas, R.G.; Aisen, P.S.; et al. A Phase 3 Trial of Semagacestat for Treatment of Alzheimer’s Disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef]
- Coric, V.; Salloway, S.; Van Dyck, C.H.; Dubois, B.; Andreasen, N.; Brody, M.; Curtis, C.; Soininen, H.; Thein, S.; Shiovitz, T.; et al. Targeting Prodromal Alzheimer Disease with Avagacestat: A Randomized Clinical Trial. JAMA Neurol. 2015, 72, 1324–1333. [Google Scholar] [CrossRef] [PubMed]
- Muntimadugu, E.; Dhommati, R.; Jain, A.; Challa, V.G.S.; Shaheen, M.; Khan, W. Intranasal delivery of nanoparticle encapsulated tarenflurbil: A potential brain targeting strategy for Alzheimer’s disease. Eur. J. Pharm. Sci. 2016, 92, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P.; Watling, M. Investigational BACE inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2019, 28, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.L.; Tariot, P.N.; Caputo, A.; Langbaum, J.B.; Liu, F.; Riviere, M.; Langlois, C.; Rouzade-Dominguez, M.; Zalesak, M.; Hendrix, S.; et al. The Alzheimer’s Prevention Initiative Generation Program: Study design of two randomized controlled trials for individuals at risk for clinical onset of Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kost, J.; Voss, T.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; Tariot, P.N.; Vellas, B.; Van Dyck, C.H.; Boada, M.; et al. Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1408–1420. [Google Scholar] [CrossRef]
- Henley, D.; Raghavan, N.; Sperling, R.; Aisen, P.; Raman, R.; Romano, G. Preliminary Results of a Trial of Atabecestat in Preclinical Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1483–1485. [Google Scholar] [CrossRef]
- Burki, T. Alzheimer’s disease research: The future of BACE inhibitors. Lancet 2018, 391, 2486. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Solfrizzi, V.; Sardone, R.; Piccininni, C.; Dibello, V.; Stallone, R.; Giannelli, G.; Bellomo, A.; Greco, A.; et al. BACE inhibitors in clinical development for the treatment of Alzheimer’s disease. Expert Rev. Neurother. 2018, 18, 847–857. [Google Scholar] [CrossRef]
- Vellas, B.; Sol, O.; Snyder, P.J.; Ousset, P.-J.; Haddad, R.; Maurin, M.; Lemarie, J.-C.; Desire, L.; Pando, M.P. EHT0202 in Alzheimer’s disease: A 3-month, randomized, placebo-controlled, double-blind study. Curr. Alzheimer Res. 2011, 8, 203–212. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Keren, R.; Porsteinsson, A.; Van Dyck, C.H.; Tariot, P.N.; Gilman, S.; Arnold, D.; Abushakra, S.; Hernandez, C.; et al. A phase 2 randomized trial of ELND005, scyllo-inositol, in mild to moderate Alzheimer disease. Neurology 2011, 77, 1253–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, T.; Lieblein, T.; Pohland, M.; Kalden, E.; Freund, P.; Zangl, R.; Grewal, R.; Heilemann, M.; Eckert, G.P.; Morgner, N.; et al. Peptidomimetics That Inhibit and Partially Reverse the Aggregation of Aβ1–42. Biochemistry 2017, 56, 4840–4849. [Google Scholar] [CrossRef] [PubMed]
- Nimmagadda, A.; Shi, Y.; Cai, J. γ-AApeptides as a New Strategy for Therapeutic Development. Curr. Med. Chem. 2019, 26, 2313–2329. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhang, M.; Sheng, R.; Ma, Y. Synthesis and biological evaluation of deferiprone-resveratrol hybrids as antioxidants, Aβ 1–42 aggregation inhibitors and metal-chelating agents for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 127, 174–186. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Bush, A.I. The essential elements of Alzheimer’s disease. J. Biol. Chem. 2020, 296, 100105. [Google Scholar] [CrossRef]
- Krishnan, H.S.; Bernard-Gauthier, V.; Placzek, M.S.; Dahl, K.; Narayanaswami, V.; Livni, E.; Chen, Z.; Yang, J.; Collier, T.L.; Ran, C.; et al. Metal Protein-Attenuating Compound for PET Neuroimaging: Synthesis and Preclinical Evaluation of [11C]PBT2. Mol. Pharm. 2018, 15, 695–702. [Google Scholar] [CrossRef]
- Wisniewski, T.; Goñi, F. Immunotherapeutic Approaches for Alzheimer’s Disease. Neuron 2015, 85, 1162–1176. [Google Scholar] [CrossRef] [Green Version]
- Folch, J.; Ettcheto, M.; Petrov, D.; Abad, S.; Pedrós, I.; Marin, M.; Olloquequi, J.; Camins, A. Una revisión de los avances en la terapéutica de la enfermedad de Alzheimer: Estrategia frente a la proteína β-amiloide. Neurología 2018, 33, 47–58. [Google Scholar] [CrossRef]
- Lacosta, A.-M.; Pascual-Lucas, M.; Pesini, P.; Casabona, D.; Perez, V.; Marcos-Campos, I.; Sarasa, L.; Canudas, J.; Badi, H.; Monleón, I.; et al. Safety, tolerability and immunogenicity of an active anti-Aβ40 vaccine (ABvac40) in patients with Alzheimer’s disease: A randomised, double-blind, placebo-controlled, phase I trial. Alzheimer’s Res. Ther. 2018, 10, 12. [Google Scholar] [CrossRef]
- Hull, M.; Sadowsky, C.; Arai, H.; Leterme, G.L.P.; Holstein, A.; Booth, K.; Peng, Y.; Yoshiyama, T.; Suzuki, H.; Ketter, N.; et al. Long-Term Extensions of Randomized Vaccination Trials of ACC-001 and QS-21 in Mild to Moderate Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 696–708. [Google Scholar] [CrossRef]
- Wang, C.Y.; Wang, P.-N.; Chiu, M.-J.; Finstad, C.L.; Lin, F.; Lynn, S.; Tai, Y.-H.; De Fang, X.; Zhao, K.; Hung, C.-H.; et al. UB-311, a novel UBITh®amyloid β peptide vaccine for mild Alzheimer’s disease. Alzheimer’s Dement. 2017, 3, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, H.; Honig, L.S.; Lin, H.; Sink, K.M.; Blondeau, K.; Quartino, A.; Dolton, M.; Carrasco-Triguero, M.; Lian, Q.; Bittner, T.; et al. Safety, Tolerability, and Pharmacokinetics of Crenezumab in Patients with Mild-to-Moderate Alzheimer’s Disease Treated with Escalating Doses for up to 133 Weeks. J. Alzheimer’s Dis. 2020, 76, 967–979. [Google Scholar] [CrossRef] [PubMed]
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimer’s Res. Ther. 2017, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef]
- Decourt, B.; Boumelhem, F.; Pope, E.D.; Shi, J.; Mari, Z.; Sabbagh, M.N. Critical Appraisal of Amyloid Lowering Agents in AD. Curr. Neurol. Neurosci. Rep. 2021, 21, 39. [Google Scholar] [CrossRef]
- Boada, M.; Anaya, F.; Ortiz, P.; Olazarán, J.; Shua-Haim, J.R.; Obisesan, T.O.; Hernández, I.; Muñoz, J.; Buendia, M.; Alegret, M.; et al. Efficacy and Safety of Plasma Exchange with 5% Albumin to Modify Cerebrospinal Fluid and Plasma Amyloid-β Concentrations and Cognition Outcomes in Alzheimer’s Disease Patients: A Multicenter, Randomized, Controlled Clinical Trial. J. Alzheimer’s Dis. 2017, 56, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Congdon, E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Hampel, H.; Lista, S.; Mango, D.; Nisticò, R.; Perry, G.; Avila, J.; Hernandez, F.; Geerts, H.; Vergallo, A. Alzheimer Precision Medicine Initiative (APMI) Lithium as a Treatment for Alzheimer’s Disease: The Systems Pharmacology Perspective. J. Alzheimer’s Dis. 2019, 69, 615–629. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Soeda, Y.; Takashima, A. New Insights Into Drug Discovery Targeting Tau Protein. Front. Mol. Neurosci. 2020, 13, 590896. [Google Scholar] [CrossRef]
- Medina, M. An Overview on the Clinical Development of Tau-Based Therapeutics. Int. J. Mol. Sci. 2018, 19, 1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atri, A. Current and Future Treatments in Alzheimer’s Disease. Semin. Neurol. 2019, 39, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, T.; De Bruyn, S.; Fadini, T.; Watanabe, S.; Germani, M.; Mesa, A.B.I.R. A Randomised, Placebo-Controlled, First-in-Human Study with a Central Tau Epitope Antibody–UCB0107. In Proceedings of the International Congress of Parkinson’s Disease and Movement Disorders, Nice, France, 22–26 September 2019. [Google Scholar]
- Barton, M.E.; Byrnes, W.; Mesa, I.R.; Bloemers, J.; Maguire, R.P.; Bouw, R.; Tesseur, I.; Ewen, C.; Scheltens, P. Design of a patient- and investigator-blind, randomized, placebo-controlled study to evaluate efficacy, safety, and tolerability of bepranemab, UCB0107, in prodromal to mild Alzheimer’s disease: The TOGETHER Study, AH0003. Alzheimer’s Dement. 2021, 17, e057586. [Google Scholar] [CrossRef]
- Taliyan, R.; Kakoty, V.; Sarathlal, K.; Kharavtekar, S.S.; Karennanavar, C.R.; Choudhary, Y.K.; Singhvi, G.; Riadi, Y.; Dubey, S.K.; Kesharwani, P. Nanocarrier mediated drug delivery as an impeccable therapeutic approach against Alzheimer’s disease. J. Control. Release 2022, 343, 528–550. [Google Scholar] [CrossRef]
- Karthivashan, G.; Ganesan, P.; Park, S.-Y.; Kim, J.-S.; Choi, D.-K. Therapeutic strategies and nano-drug delivery applications in management of ageing Alzheimer’s disease. Drug Deliv. 2018, 25, 307–320. [Google Scholar] [CrossRef] [Green Version]
- Brambilla, D.; Le Droumaguet, B.; Nicolas, J.; Hashemi, S.H.; Wu, L.; Moghimi, S.M.; Couvreur, P.; Andrieux, K. Nanotechnologies for Alzheimer’s disease: Diagnosis, therapy, and safety issues. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 521–540. [Google Scholar] [CrossRef]
- Gonzalez-Carter, D.; Liu, X.; Tockary, T.A.; Dirisala, A.; Toh, K.; Anraku, Y.; Kataoka, K. Targeting nanoparticles to the brain by exploiting the blood–brain barrier impermeability to selectively label the brain endothelium. Proc. Natl. Acad. Sci. USA 2020, 117, 19141–19150. [Google Scholar] [CrossRef]
- Xie, J.; Gonzalez-Carter, D.; Tockary, T.A.; Nakamura, N.; Xue, Y.; Nakakido, M.; Akiba, H.; Dirisala, A.; Liu, X.; Toh, K.; et al. Dual-Sensitive Nanomicelles Enhancing Systemic Delivery of Therapeutically Active Antibodies Specifically into the Brain. ACS Nano 2020, 14, 6729–6742. [Google Scholar] [CrossRef]
- Perche, F.; Uchida, S.; Akiba, H.; Lin, C.-Y.; Ikegami, M.; Dirisala, A.; Nakashima, T.; Itaka, K.; Tsumoto, K.; Kataoka, K. Improved brain expression of anti-amyloid ? scFv by complexation of mRNA including a secretion sequence with PEG-based block catiomer. Curr. Alzheimer Res. 2017, 14, 295–302. [Google Scholar] [CrossRef]
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Inflammation, Antiinflammatory Agents, and Alzheimer’s Disease: The Last 22 Years. J. Alzheimer’s Dis. 2016, 54, 853–857. [Google Scholar] [CrossRef]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellar, D.; Register, T.; Lockhart, S.N.; Aisen, P.; Raman, R.; Rissman, R.A.; Brewer, J.; Craft, S. Intranasal insulin modulates cerebrospinal fluid markers of neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A randomized trial. Sci. Rep. 2022, 12, 1346. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L. Alzheimer’s Disease, time to turn the tide. Aging 2018, 10, 2537–2538. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.N.; Maass, A.; Harrison, T.M.; Baker, S.L.; Jagust, W.J. Cortical tau deposition follows patterns of entorhinal functional connectivity in aging. eLife 2019, 8, e49132. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, R. Is Alzheimer’s Prevention Possible Today? J. Am. Geriatr. Soc. 2017, 65, 2153–2154. [Google Scholar] [CrossRef]
- Schneider, L.S.; Mangialasche, F.; Andreasen, N.; Feldman, H.; Giacobini, E.; Jones, R.; Mantua, V.; Mecocci, P.; Pani, L.; Winblad, B.; et al. Clinical trials and late-stage drug development for Alzheimer’s disease: An appraisal from 1984 to 2014. J. Intern. Med. 2014, 275, 251–283. [Google Scholar] [CrossRef]
- McGurran, H.; Glenn, J.M.; Madero, E.N.; Bott, N.T. Prevention and Treatment of Alzheimer’s Disease: Biological Mechanisms of Exercise. J. Alzheimer’s Dis. 2019, 69, 311–338. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Perry, G. Preventive and Therapeutic Strategies in Alzheimer’s Disease: Focus on Oxidative Stress, Redox Metals, and Ferroptosis. Antioxid. Redox Signal. 2021, 34, 591–610. [Google Scholar] [CrossRef]
- Zucchella, C.; Sinforiani, E.; Tamburin, S.; Federico, A.; Mantovani, E.; Bernini, S.; Casale, R.; Bartolo, M. The Multidisciplinary Approach to Alzheimer’s Disease and Dementia. A Narrative Review of Non-Pharmacological Treatment. Front. Neurol. 2018, 9, 1058. [Google Scholar] [CrossRef]
- Hötting, K.; Röder, B. Beneficial effects of physical exercise on neuroplasticity and cognition. Neurosci. Biobehav. Rev. 2013, 37, 2243–2257. [Google Scholar] [CrossRef]
- Kishimoto, Y.; Shishido, H.; Sawanishi, M.; Toyota, Y.; Ueno, M.; Kubota, T.; Kirino, Y.; Tamiya, T.; Kawai, N. Data on amyloid precursor protein accumulation, spontaneous physical activity, and motor learning after traumatic brain injury in the triple-transgenic mouse model of Alzheimer׳s disease. Data Brief 2016, 9, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Dong, Y.; Tucker, D.; Wang, R.; Ahmed, M.E.; Brann, D.; Zhang, Q. Treadmill Exercise Exerts Neuroprotection and Regulates Microglial Polarization and Oxidative Stress in a Streptozotocin-Induced Rat Model of Sporadic Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 56, 1469–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Um, H.-S.; Kang, E.-B.; Koo, J.-H.; Kim, H.-T.; Lee, J.; Kim, E.-J.; Yang, C.-H.; An, G.-Y.; Cho, I.-H.; Cho, J.-Y. Treadmill exercise represses neuronal cell death in an aged transgenic mouse model of Alzheimer’s disease. Neurosci. Res. 2011, 69, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Alkadhi, K.A.; Dao, A.T. Exercise decreases BACE and APP levels in the hippocampus of a rat model of Alzheimer’s disease. Mol. Cell. Neurosci. 2018, 86, 25–29. [Google Scholar] [CrossRef]
- Robinson, M.M.; Lowe, V.J.; Nair, K.S. Increased Brain Glucose Uptake After 12 Weeks of Aerobic High-Intensity Interval Training in Young and Older Adults. J. Clin. Endocrinol. Metab. 2018, 103, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, C.M.C.; Pereira, J.R.; de Andrade, L.; Garuffi, M.; Talib, L.L.; Forlenza, O.V.; Cancela, J.M.; Cominetti, M.R.; Stella, F. Physical Exercise in MCI Elderly Promotes Reduction of Pro-Inflammatory Cytokines and Improvements on Cognition and BDNF Peripheral Levels. Curr. Alzheimer Res. 2014, 11, 799–805. [Google Scholar] [CrossRef]
- Coelho, F.; Pereira, D.; Lustosa, L.; Silva, J.; Dias, J.; Dias, R.; Queiroz, B.; Teixeira, A.L.; Teixeira, M.; Pereira, L. Physical therapy intervention (PTI) increases plasma brain-derived neurotrophic factor (BDNF) levels in non-frail and pre-frail elderly women. Arch. Gerontol. Geriatr. 2012, 54, 415–420. [Google Scholar] [CrossRef]
- Hauer, K.; Ma, M.S.; Zieschang, T.; Essig, M.; Becker, C.; Oster, P. Physical Training Improves Motor Performance in People with Dementia: A Randomized Controlled Trial. J. Am. Geriatr. Soc. 2012, 60, 8–15. [Google Scholar] [CrossRef]
- Portugal, E.M.M.; Vasconcelos, P.G.T.; Souza, R.; Lattari, E.; Monteiro-Junior, R.S.; Machado, S.; Deslandes, A.C. Aging process, cognitive decline and Alzheimer’s disease: Can strength training modulate these responses? CNS Neurol. Disord—Drug Targets 2015, 14, 1209–1213. [Google Scholar] [CrossRef]
- Amini, Y.; Saif, N.; Greer, C.; Hristov, H.; Isaacson, R. The Role of Nutrition in Individualized Alzheimer’s Risk Reduction. Curr. Nutr. Rep. 2020, 9, 55–63. [Google Scholar] [CrossRef]
- Schelke, M.W.; Hackett, K.; Chen, J.L.; Shih, C.; Shum, J.; Montgomery, M.E.; Chiang, G.C.; Berkowitz, C.; Seifan, A.; Krikorian, R.; et al. Nutritional interventions for Alzheimer’s prevention: A clinical precision medicine approach. Ann. N. Y. Acad. Sci. 2016, 1367, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dove, A.; Shang, Y.; Xu, W.; Grande, G.; Laukka, E.J.; Fratiglioni, L.; Marseglia, A. The impact of diabetes on cognitive impairment and its progression to dementia. Alzheimer’s Dement. 2021, 17, 1769–1778. [Google Scholar] [CrossRef] [PubMed]
- Román, G.C.; Jackson, R.E.; Gadhia, R.; Román, A.N.; Reis, J. Mediterranean diet: The role of long-chain ω-3 fatty acids in fish; polyphenols in fruits, vegetables, cereals, coffee, tea, cacao and wine; probiotics and vitamins in prevention of stroke, age-related cognitive decline, and Alzheimer disease. Rev. Neurol. 2019, 175, 724–741. [Google Scholar] [CrossRef] [PubMed]
- Chainoglou, E.; Hadjipavlou-Litina, D. Curcumin in Health and Diseases: Alzheimer’s Disease and Curcumin Analogues, Derivatives, and Hybrids. Int. J. Mol. Sci. 2020, 21, E1975. [Google Scholar] [CrossRef] [Green Version]
- Lakey-Beitia, J.; Burillo, A.M.; La Penna, G.; Hegde, M.L.; Rao, K. Polyphenols as Potential Metal Chelation Compounds Against Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 82, S335–S357. [Google Scholar] [CrossRef]
- Rahman, S.; Akhtar, N.; Jamil, H.M.; Banik, R.S.; Asaduzzaman, S.M. TGF-β/BMP signaling and other molecular events: Regulation of osteoblastogenesis and bone formation. Bone Res. 2015, 3, 15005. [Google Scholar] [CrossRef] [Green Version]
- Irwin, M.R.; Vitiello, M.V. Implications of sleep disturbance and inflammation for Alzheimer’s disease dementia. Lancet Neurol. 2019, 18, 296–306. [Google Scholar] [CrossRef]
- Prodhan, A.S.U.; Cavestro, C.; Kamal, M.A.; Islam, M.A. Melatonin and Sleep Disturbances in Alzheimer’s Disease. CNS Neurol. Disord.—Drug Targets 2021, 20, 736–754. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Wan, J.; Liu, A.; Sun, J. Melatonin regulates Aβ production/clearance balance and Aβ neurotoxicity: A potential therapeutic molecule for Alzheimer’s disease. Biomed. Pharmacother. 2020, 132, 110887. [Google Scholar] [CrossRef]
- Rosales-Corral, S.A.; Acuña-Castroviejo, D.; Coto-Montes, A.; Boga, J.A.; Manchester, L.C.; Fuentes-Broto, L.; Korkmaz, A.; Ma, S.; Tan, D.X.; Reiter, R.J. Alzheimer’s disease: Pathological mechanisms and the beneficial role of melatonin. J. Pineal Res. 2012, 52, 167–202. [Google Scholar] [CrossRef]
- Vincent, B. Protective roles of melatonin against the amyloid-dependent development of Alzheimer’s disease: A critical review. Pharmacol. Res. 2018, 134, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Aguilar, M.A.; Ramírez-Salado, I.; Guevara, M.A.; Hernández-González, M.; Benitez-King, G. Melatonin Effects on EEG Activity During Sleep Onset in Mild-to-Moderate Alzheimer’s Disease: A Pilot Study. J. Alzheimer’s Dis. Rep. 2018, 2, 55–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabani, A.; Foroozanfard, F.; Kavossian, E.; Aghadavod, E.; Ostadmohammadi, V.; Reiter, R.J.; Eftekhar, T.; Asemi, Z. Effects of melatonin administration on mental health parameters, metabolic and genetic profiles in women with polycystic ovary syndrome: A randomized, double-blind, placebo-controlled trial. J. Affect. Disord. 2019, 250, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Laudon, M.; Wade, A.G.; Farmer, M.; Harari, G.; Fund, N.; Nir, T.; Frydman-Marom, A.; Zisapel, N. Add-on prolonged-release melatonin for cognitive function and sleep in mild to moderate Alzheimer’s disease: A 6-month, randomized, placebo-controlled, multicenter trial. Clin. Interv. Aging 2014, 9, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yu, H.; Sun, H.; Hu, B.; Geng, Y. Dietary Melatonin Therapy Alleviates the Lamina Cribrosa Damages in Patients with Mild Cognitive Impairments: A Double-Blinded, Randomized Controlled Study. Med. Sci. Monit. 2020, 26, e923232. [Google Scholar] [CrossRef]
- Musiek, E.S.; Holtzman, D.M. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 2016, 354, 1004–1008. [Google Scholar] [CrossRef] [Green Version]
- Dowling, G.A.; Burr, R.L.; Van Someren, E.J.W.; Ma, E.M.H.; Luxenberg, J.; Mastick, J.; Cooper, B.A. Melatonin and Bright-Light Treatment for Rest-Activity Disruption in Institutionalized Patients with Alzheimer’ Disease. J. Am. Geriatr. Soc. 2008, 56, 239–246. [Google Scholar] [CrossRef]
- Hatta, K.; Kishi, Y.; Wada, K.; Takeuchi, T.; Odawara, T.; Usui, C.; Nakamura, H. Preventive Effects of Ramelteon on Delirium: A Randomized Placebo-Controlled Trial. JAMA Psychiatry 2014, 71, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Li, H.-H.; Yao, X.-Y.; Tao, S.; Sun, X.; Li, P.-P.; Li, X.-X.; Liu, Z.-L.; Ren, C. Serotonin 2 Receptors, Agomelatine, and Behavioral and Psychological Symptoms of Dementia in Alzheimer’s Disease. Behav. Neurol. 2021, 2021, 5533827. [Google Scholar] [CrossRef]
- Ortí, J.E.D.L.R.; García-Pardo, M.P.; Iranzo, C.C.; Madrigal, J.J.C.; Castillo, S.S.; Rochina, M.J.; Gascó, V.J.P. Does Music Therapy Improve Anxiety and Depression in Alzheimer’s Patients? J. Altern. Complement. Med. 2018, 24, 33–36. [Google Scholar] [CrossRef]
- Kim, D. The Effects of a Recollection-Based Occupational Therapy Program of Alzheimer’s Disease: A Randomized Controlled Trial. Occup. Ther. Int. 2020, 2020, e6305727. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Pharmacological Treatments under Investigation | |||
|---|---|---|---|
| Mechanism of Action | Agent | ||
| Aβ pathology | γ-secretase inhibitors | Semagacestat (LY-450139) | |
| Avagacestat (BMS-708163) | |||
| Tarenflurbil | |||
| β-secretase inhibitors | Lanabecestat | ||
| Verubecestat | |||
| Atabecestat | |||
| Elenbecestat (E2609) | |||
| Umibecestat (CNP520) | |||
| α-secretase modulators | Etazolato (EHT0202) | ||
| APH-1105 | |||
| ID1201 | |||
| Aggregation inhibitors | Scyllo-inositol (ELND005) | ||
| Peptidomimetics (KLVFF, γ-AA) | |||
| Metal interfering drugs | Dyshomeotaisis (copper, iron or zinc) Deferiprona PBT2 | ||
| Drugs that enhance Aβ clearance (immunotherapy) | Active immunotherapy | CAD106 | |
| CNP520 | |||
| ABvac40 | |||
| GV1001 | |||
| ACC-001 | |||
| UB-311 | |||
| AF20513 | |||
| Passive immunotherapy | Crenezumab | ||
| Gantenerumab | |||
| LY3002813 | |||
| Pharmacological Treatments under Investigation | |||
|---|---|---|---|
| Mechanism of Action | Agent | ||
| Tau pathology | Inhibitors of tau protein hyperphosphorylation | GSK3β inhibitors | Lithium Chloride |
| Tideglusib | |||
| Tau protein aggregation inhibitors | Methylene blue | ||
| TRx0237 (LMTM) | |||
| Drugs that promote the clearance of tau (immunotherapy) | Active immunotherapy | AADvac-1 | |
| ACI-35 | |||
| Passive immunotherapy | C2N-8E12 (Tilayonemab) | ||
| Bepranemab (UCB0107) | |||
| Other anti-tau mAbs | BII076 | ||
| JNJ-63733657 | |||
| LY3303560 | |||
| Other Treatments under Investigation | |
|---|---|
| Agents | Nanomedicine strategies |
| Intravenous immunoglobulin (IVIg) | |
| Plasma exchange via albumin | |
| TNF-α inhibitors | |
| Bacterial protease inhibitors | |
| Selective tyrosine kinase inhibitors | |
| Hepatocyte growth factors | |
| Stem cells | |
| Intranasal insulin | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pardo-Moreno, T.; González-Acedo, A.; Rivas-Domínguez, A.; García-Morales, V.; García-Cozar, F.J.; Ramos-Rodríguez, J.J.; Melguizo-Rodríguez, L. Therapeutic Approach to Alzheimer’s Disease: Current Treatments and New Perspectives. Pharmaceutics 2022, 14, 1117. https://doi.org/10.3390/pharmaceutics14061117
Pardo-Moreno T, González-Acedo A, Rivas-Domínguez A, García-Morales V, García-Cozar FJ, Ramos-Rodríguez JJ, Melguizo-Rodríguez L. Therapeutic Approach to Alzheimer’s Disease: Current Treatments and New Perspectives. Pharmaceutics. 2022; 14(6):1117. https://doi.org/10.3390/pharmaceutics14061117
Chicago/Turabian StylePardo-Moreno, Teresa, Anabel González-Acedo, Antonio Rivas-Domínguez, Victoria García-Morales, Francisco Jose García-Cozar, Juan Jose Ramos-Rodríguez, and Lucía Melguizo-Rodríguez. 2022. "Therapeutic Approach to Alzheimer’s Disease: Current Treatments and New Perspectives" Pharmaceutics 14, no. 6: 1117. https://doi.org/10.3390/pharmaceutics14061117